Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models



Abstract
1. Introduction
2. PE Based Immunotoxins
3. Inhibition of Protein Synthesis as a Possible Trigger of Immunity
4. The Restrained Power of Anti-Tumor Immunity
5. Clinical Observations Suggesting that PE Immunotoxins Induce Anti-Tumor Immunity
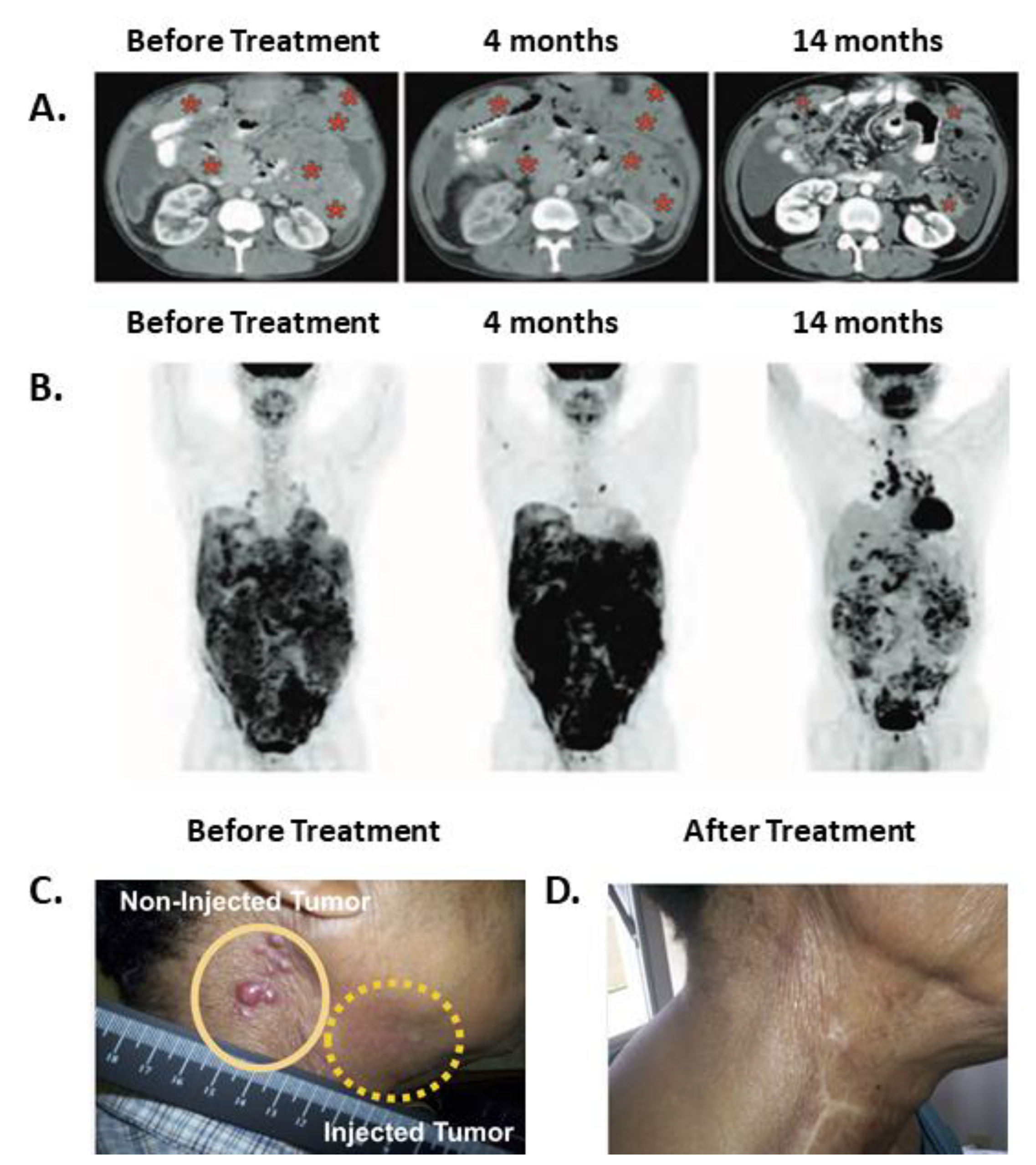
5.1. Systemically Administered SS1P in Combination with Immune Modulating Chemotherapies
5.2. Intra-Tumoral PE Immunotoxins in Patients with Epithelial Cancers
5.3. Intra-Tumoral PE Immunotoxins to Treat Brain Tumors
6. Anti-Tumor Immunity Achieved by PE Immunotoxins in Preclinical Murine Models
7. Synergy between Local Anti-Mesothelin Immunotoxins and Systemic Anti-CTLA-4 in Murine Cancer Models
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Onda, M.; Pastan, I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 2016, 270, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; King, E.M.; Pastan, I. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am. J. Pathol. 2018, 188, 1736–1743. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Hassan, R.; Alewine, C.; Pastan, I. New life for immunotoxin cancer therapy. Clin. Cancer Res. 2016, 22, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Dyda, F.; Benhar, I.; Pastan, I.; Davies, D.R. Crystal structure of the catalytic domain of Pseudomonas exotoxin A complexed with a nicotinamide adenine dinucleotide analog: Implications for the activation process and for ADP ribosylation. Proc. Natl. Acad. Sci. USA 1996, 93, 6902–6906. [Google Scholar] [CrossRef]
- Chang, J.H.; Kwon, H.Y. Expression of 14-3-3delta, cdc2 and cyclin B proteins related to exotoxin A-induced apoptosis in HeLa S3 cells. Int. Immunopharmacol. 2007, 7, 1185–1191. [Google Scholar] [CrossRef]
- Andersson, Y.; Juell, S.; Fodstad, O. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin a immunotoxin. Int. J. Cancer 2004, 112, 475–483. [Google Scholar] [CrossRef]
- Iglewski, B.H.; Kabat, D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar] [CrossRef]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.E.; le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Hairy Cell Leukemia Treatment Approved. Cancer Discov. 2018. [CrossRef]
- Johannes, L.; Romer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef]
- McCusker, K.T.; Braaten, B.A.; Cho, M.W.; Low, D.A. Legionella pneumophila inhibits protein synthesis in Chinese hamster ovary cells. Infect. Immun. 1991, 59, 240–246. [Google Scholar] [PubMed]
- Dunbar, T.L.; Yan, Z.; Balla, K.M.; Smelkinson, M.G.; Troemel, E.R. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 2012, 11, 375–386. [Google Scholar] [CrossRef]
- McEwan, D.L.; Kirienko, N.V.; Ausubel, F.M. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 2012, 11, 364–374. [Google Scholar] [CrossRef]
- Fontana, M.F.; Shin, S.; Vance, R.E. Activation of host mitogen-activated protein kinases by secreted Legionella pneumophila effectors that inhibit host protein translation. Infect. Immun. 2012, 80, 3570–3575. [Google Scholar] [CrossRef]
- Fontana, M.F.; Banga, S.; Barry, K.C.; Shen, X.; Tan, Y.; Luo, Z.Q.; Vance, R.E. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 2011, 7, e1001289. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with Nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 blockade with Pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T cell co-receptors for cancer therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef]
- Hoos, A. Evolution of end points for cancer immunotherapy trials. Ann. Oncol. 2012, 23 (Suppl. 8), viii47–viii52. [Google Scholar] [CrossRef] [PubMed]
- Chiou, V.L.; Burotto, M. Pseudoprogression and immune-related response in solid tumors. J. Clin. Oncol. 2015, 33, 3541–3543. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Tomas, M.B.; Tronco, G.G.; Palestro, C.J. FDG PET of infection and inflammation. Radiographics 2005, 25, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Benz, M.R.; Allen-Auerbach, M.S.; Radu, C.; Chmielowski, B.; Seja, E.; Williams, J.L.; Gomez-Navarro, J.; McCarthy, T.; Czernin, J. Imaging of CTLA4 blockade-induced cell replication with (18)F-FLT PET in patients with advanced melanoma treated with tremelimumab. J. Nucl. Med. 2010, 51, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Lipson, E.J.; Im, H.J.; Rowe, S.P.; Gonzalez, E.M.; Blackford, A.; Chirindel, A.; Pardoll, D.M.; Topalian, S.L.; Wahl, R.L. Prediction of response to immune checkpoint inhibitor therapy using early-time-point (18)F-FDG PET/CT imaging in patients with advanced melanoma. J. Nucl. Med. 2017, 58, 1421–1428. [Google Scholar] [CrossRef]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H.; et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med. 2013, 5, 208ra147. [Google Scholar] [CrossRef]
- MacDonald, G.C.; Rasamoelisolo, M.; Entwistle, J.; Cizeau, J.; Bosc, D.; Cuthbert, W.; Kowalski, M.; Spearman, M.; Glover, N. A phase I clinical study of VB4-845: Weekly intratumoral administration of an anti-EpCAM recombinant fusion protein in patients with squamous cell carcinoma of the head and neck. Drug Des. Dev. Ther. 2009, 2, 105–114. [Google Scholar]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Andersson, Y.; Engebraaten, O.; Juell, S.; Aamdal, S.; Brunsvig, P.; Fodstad, O.; Dueland, S. Phase I trial of EpCAM-targeting immunotoxin MOC31PE, alone and in combination with cyclosporin. Br. J. Cancer 2015, 113, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Azemar, M.; Djahansouzi, S.; Jager, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; von Minckwitz, G.; Dall, P.; et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res. Treat. 2003, 82, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Chacko, A.M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: Crossing the blood-brain barrier divide. Expert Opin. Drug Deliv. 2013, 10, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; et al. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J. Neurooncol. 2003, 64, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Rainov, N.G.; Heidecke, V. Long term survival in a patient with recurrent malignant glioma treated with intratumoral infusion of an IL4-targeted toxin (NBI-3001). J. Neurooncol. 2004, 66, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Park, J.K.; Hodges, T.; Arko, L.; Shen, M.; Dello Iacono, D.; McNabb, A.; Olsen Bailey, N.; Kreisl, T.N.; Iwamoto, F.M.; Sul, J.; et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J. Clin. Oncol. 2010, 28, 3838–3843. [Google Scholar] [CrossRef]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; Kunwar, S.; Marcus, S.; et al. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003, 65, 27–35. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E., 2nd; Kunwar, S.; et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008, 10, 320–329. [Google Scholar] [CrossRef]
- Sampson, J.H.; Reardon, D.A.; Friedman, A.H.; Friedman, H.S.; Coleman, R.E.; McLendon, R.E.; Pastan, I.; Bigner, D.D. Sustained radiographic and clinical response in patient with bifrontal recurrent glioblastoma multiforme with intracerebral infusion of the recombinant targeted toxin TP-38: Case study. Neuro Oncol. 2005, 7, 90–96. [Google Scholar] [CrossRef]
- Ochiai, H.; Archer, G.E.; Herndon, J.E., 2nd; Kuan, C.T.; Mitchell, D.A.; Bigner, D.D.; Pastan, I.H.; Sampson, J.H. EGFRvIII-targeted immunotoxin induces antitumor immunity that is inhibited in the absence of CD4+ and CD8+ T cells. Cancer Immunol. Immunother. 2008, 57, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Terabe, M.; Kioi, M.; Berzofsky, J.A.; Puri, R.K. Intratumoral therapy with IL13-PE38 results in effective CTL-mediated suppression of IL-13Ralpha2-expressing contralateral tumors. Clin. Cancer Res. 2006, 12, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Leshem, Y.; O’Brien, J.; Liu, X.; Bera, T.K.; Terabe, M.; Berzofsky, J.A.; Bossenmaier, B.; Niederfellner, G.; Tai, C.H.; Reiter, Y.; et al. Combining local immunotoxins targeting mesothelin with CTLA-4 blockade synergistically eradicates murine cancer by promoting anticancer immunity. Cancer Immunol. Res. 2017, 5, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Leshem, Y.; King, E.M.; Mazor, R.; Reiter, Y.; Pastan, I. SS1P Immunotoxin induces markers of immunogenic cell death and enhances the effect of the CTLA-4 blockade in AE17M mouse mesothelioma tumors. Toxins 2018, 10, 470. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; King, E.; Pastan, I. Anti-drug antibodies to LMB-100 are enhanced by mAbs targeting OX40 and CTLA4 but not by mAbs targeting PD1 or PDL-1. Cell. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; King, E.M.; Onda, M.; Cuburu, N.; Addissie, S.; Crown, D.; Liu, X.F.; Kishimoto, T.K.; Pastan, I. Tolerogenic nanoparticles restore the antitumor activity of recombinant immunotoxins by mitigating immunogenicity. Proc. Natl. Acad. Sci. USA 2018, 115, E733–E742. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Keir, S.T.; Nair, S.K.; Pastan, I.; Bigner, D.D.; Chandramohan, V. A combinatorial immunotherapy for malignant brain tumors: D2C7 immunotoxin and immune checkpoint inhibitors. J. Clin. Oncol. 2017, 35, 102. [Google Scholar] [CrossRef]


{kind=link}
| Cancer Type | Drug and References | Target | Route | Other Drugs | Patients (n) | Clinical Effect | Delayed Effect | Findings in Support of Anti-Tumor Immunity |
|---|---|---|---|---|---|---|---|---|
| Hairy cell leukemia | Moxetumomab Pasudotox [12] | CD22 | IV | None | 80 | 41% CR, 75% OR | 5 pts | Delayed clinical effects |
| Mesothelioma | SS1P [38] | MSLN | IV | CP + P | 10 | 0 CR, 3 PR, 3 SD, 4 PD | 2 pts | Delayed clinical effects; maintenance of tumor control; disease regression accompanied by an increased signal on PET-CT. |
| SCCHN | VB4-845 [39] | Ep-Cam | IT | None | 20 | Injected site: 4 CR, 6 PR, 4 SD | Not reported | Local redness, edema and pain. Systemic fever in 4 patients; three patients showed regressions of non-injected tumor sites. |
| Cutaneous metastases | ScFv(FRP5)-ETA [42] | ErbB2 | IT | None | 11 | Injected site: 4 CR, 3 PR | Not reported | Local inflammation at injection site (symptoms not specified); systemic fever in 2 patients. |
| Brain tumors | TP-38 [49,50] | EGFR | IT | None | 15 residual disease | 1 near CR, 1 durable PR | 2 pts | Slowly occurring clinical effects; maintenance of tumor control. |
| 5 NED | 4 PD, 1 NED | N/A | A case of contrast enhanced area that appeared at 9 weeks and then disappeared spontaneously. | |||||
| Brain tumors | NBI-3001 [44,45] | IL-4 R | IT | None | 31 | Radiographic signs of tumor necrosis in 71% of pts | Not reported | Typically, MRI contrast enhancement decreased after the infusion, then increased at 4 weeks, and then again decreased gradually. One patient had durable partial regression lasting for 3 years. |
| Effect | Contributing Evidence | Murine Models | Toxins | Ref. # |
|---|---|---|---|---|
| Anti-tumor effect of immunotoxins is partially mediated by the immune system | Depletion of CD8+ cells with or without depletion of CD4+ cells reduces survival | Malignant astrocytoma, Melanoma | MR1-1, IL13-PE38 | [51,52] |
| Immunotoxins render tumors more sensitive to immune checkpoint inhibitors | Increased survival and number of tumor infiltrating CD8+ cells demonstrated in the combination treated mice. | Breast cancer, Mesothelioma, Glioma | SS1P, LMB-100, D2C7 | [53,54,57] |
| Immunotoxins induce markers of immunogenic cell death | Increased ATP secretion and calreticulin surface expression. | Mesothelioma | SS1P, LMB-100 | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leshem, Y.; Pastan, I. Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models. Toxins 2019, 11, 20. https://doi.org/10.3390/toxins11010020
Leshem Y, Pastan I. Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models. Toxins. 2019; 11(1):20. https://doi.org/10.3390/toxins11010020
Chicago/Turabian StyleLeshem, Yasmin, and Ira Pastan. 2019. "Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models" Toxins 11, no. 1: 20. https://doi.org/10.3390/toxins11010020
APA StyleLeshem, Y., & Pastan, I. (2019). Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models. Toxins, 11(1), 20. https://doi.org/10.3390/toxins11010020