
Is microRNA-33 an Appropriate Target in the Treatment of Atherosclerosis?

{kind=link}
{kind=link}
Abstract
1. Introduction
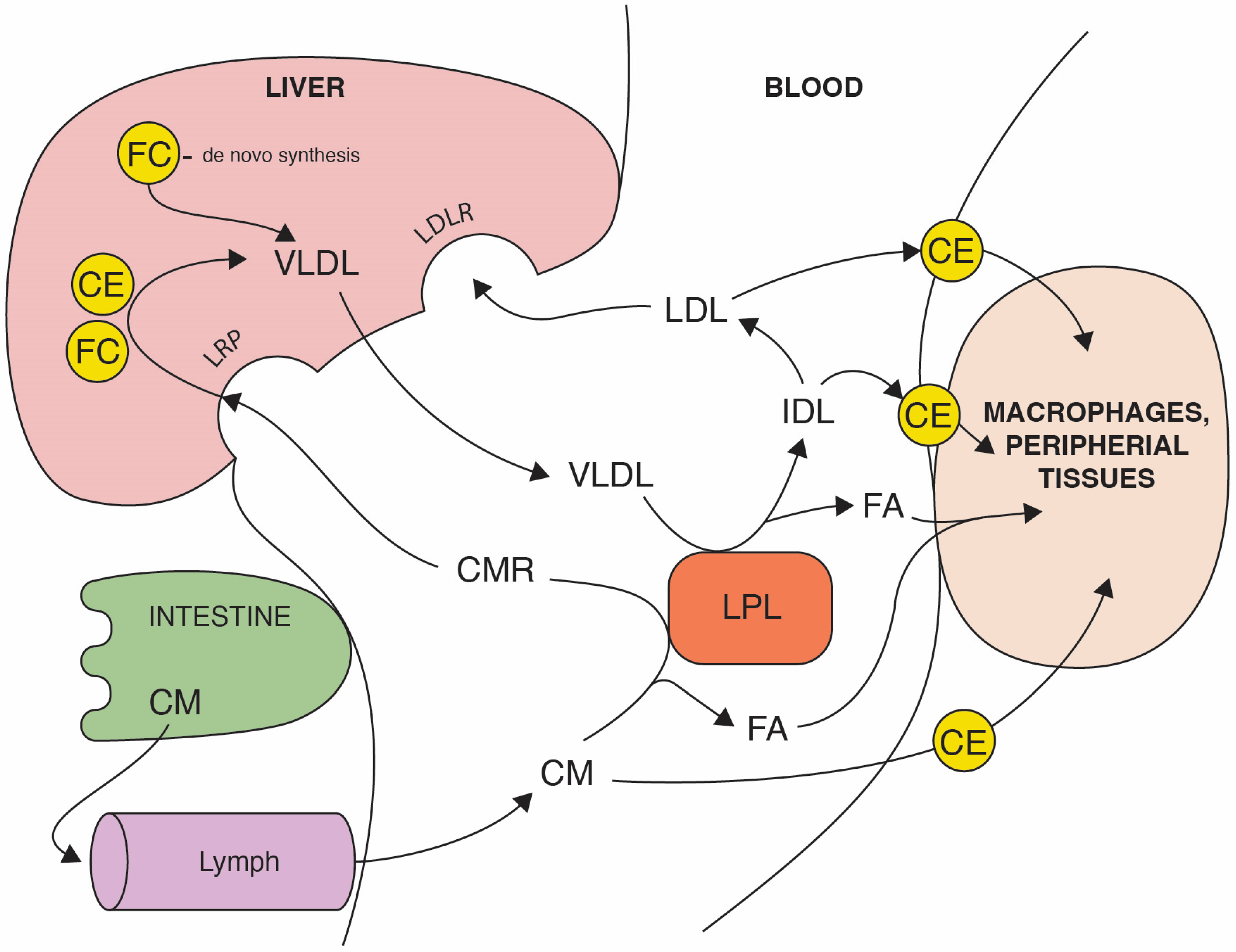
2. How Can microRNA-33 Regulate Cholesterol Metabolism?
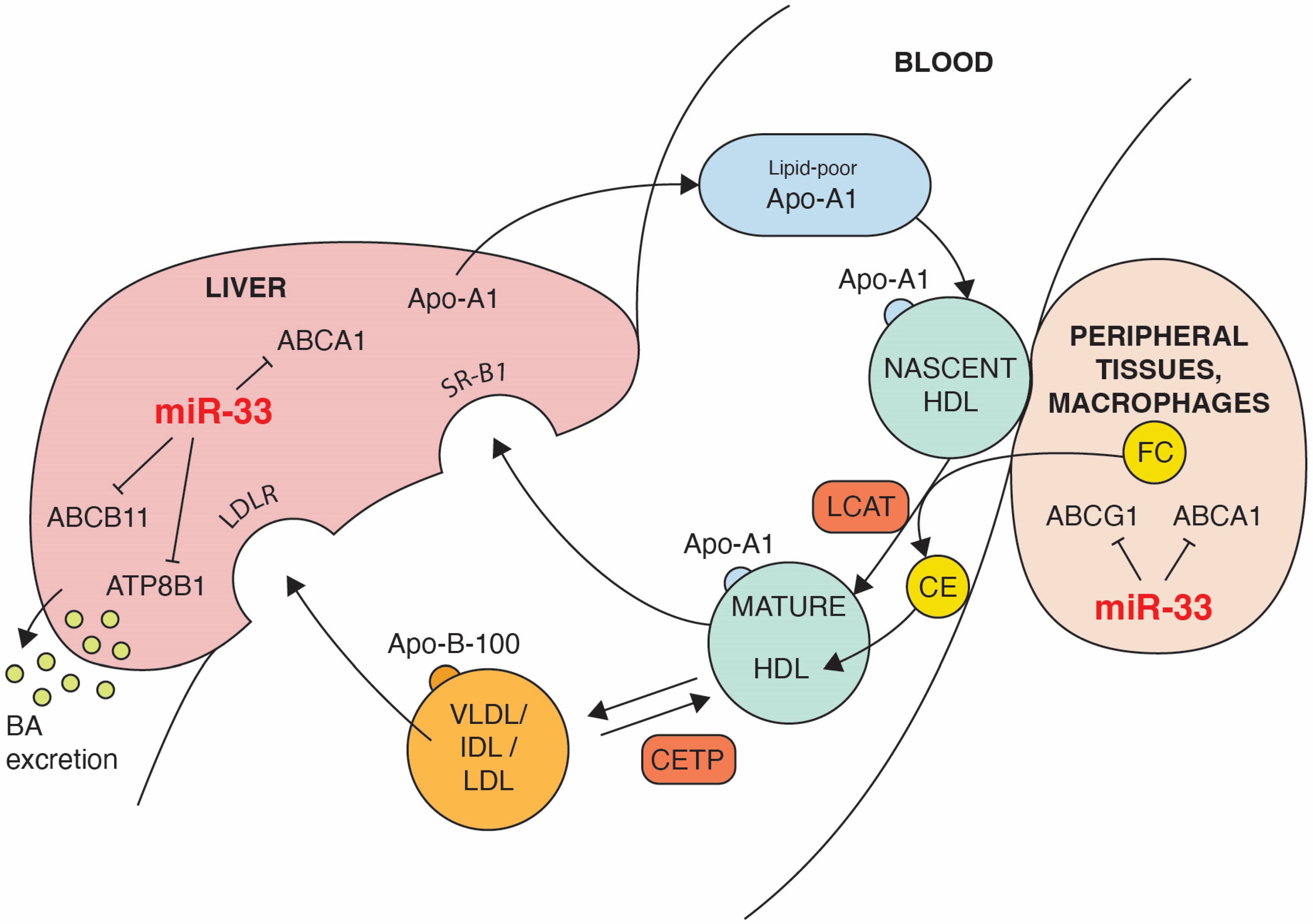
3. HDL Formation and microRNA-33 Influence
4. The Impact of microRNA-33 on Reverse Cholesterol Transport
5. Bile Formation and Secretion Can Be Deregulated by microRNA-33
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barquera, S.; Pedroza-Tobías, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Hofland, J., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2021. [Google Scholar]
- Zhou, R.; Stouffer, G.A.; Frishman, W.H. Cholesterol Paradigm and Beyond in Atherosclerotic Cardiovascular Disease: Cholesterol, Sterol Regulatory Element-Binding Protein, Inflammation, and Vascular Cell Mobilization in Vasculopathy. Cardiol. Rev. 2022, 30, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.M.; Zhang, X.; Bandyopadhyay, C.; Rotllan, N.; Sugiyama, M.G.; Aryal, B.; Liu, X.; He, S.; Kraehling, J.R.; Ulrich, V.; et al. Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 2019, 140, 225–239. [Google Scholar] [CrossRef]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef]
- Zhang, Y.; Zanotti, I.; Reilly, M.P.; Glick, J.M.; Rothblat, G.H.; Rader, D.J. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 2003, 108, 661–663. [Google Scholar] [CrossRef]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef]
- Mao, M.; Lei, H.; Liu, Q.; Chen, Y.; Zhao, L.; Li, Q.; Luo, S.; Zuo, Z.; He, Q.; Huang, W.; et al. Effects of miR-33a-5P on ABCA1/G1-mediated cholesterol efflux under inflammatory stress in THP-1 macrophages. PLoS ONE 2014, 9, e109722. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Venkateswaran, A.; Tarr, P.T.; Xenarios, I.; Kudoh, J.; Shimizu, N.; Edwards, P.A. Characterization of the human ABCG1 gene: Liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J. Biol. Chem. 2001, 276, 39438–39447, Erratum in J. Biol. Chem. 2002, 277, 17375. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Horiguchi, M.; Nishi, H.; Nakamura, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Marusawa, H.; Iwanaga, Y.; et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17321–17326. [Google Scholar] [CrossRef]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Näär, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef]
- Rotllan, N.; Ramírez, C.M.; Aryal, B.; Esau, C.C.; Fernández-Hernando, C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1973–1977. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Yang, Z.; Cappello, T.; Wang, L. Emerging role of microRNAs in lipid metabolism. Acta Pharm. Sin. B 2015, 5, 145–150. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Hueso, M.; Griñán, R.; Mallen, A.; Navarro, E.; Purqueras, E.; Gomá, M.; Sbraga, F.; Blasco-Lucas, A.; Revilla, G.; Santos, D.; et al. MiR-125b downregulates macrophage scavenger receptor type B1 and reverse cholesterol transport. Biomed. Pharmacother. 2022, 146, 112596. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.S.; Anderson, N.N.; Wagschal, A.; de Cabo, R.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef]
- Dong, J.; He, M.; Li, J.; Pessentheiner, A.; Wang, C.; Zhang, J.; Sun, Y.; Wang, W.T.; Zhang, Y.; Liu, J.; et al. microRNA-483 ameliorates hypercholesterolemia by inhibiting PCSK9 production. JCI Insight 2020, 5, e143812. [Google Scholar] [CrossRef]
- Zhang, X.; Price, N.L.; Fernández-Hernando, C. Non-coding RNAs in lipid metabolism. Vascul. Pharmacol. 2019, 114, 93–102. [Google Scholar] [CrossRef]
- Fernández-Tussy, P.; Ruz-Maldonado, I.; Fernández-Hernando, C. MicroRNAs and Circular RNAs in Lipoprotein Metabolism. Curr. Atheroscler. Rep. 2021, 23, 33. [Google Scholar] [CrossRef]
- Hennessy, E.J.; van Solingen, C.; Scacalossi, K.R.; Ouimet, M.; Afonso, M.S.; Prins, J.; Koelwyn, G.J.; Sharma, M.; Ramkhelawon, B.; Carpenter, S.; et al. The long noncoding RNA CHROME regulates cholesterol homeostasis in primate. Nat. Metab. 2019, 1, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gulshan, K.; Brubaker, G.; Hazen, S.L.; Smith, J.D. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xiong, G.; Hua, L.; Hu, Y.; Guo, X.; Peng, X. APOA1 mRNA and protein in kidney renal clear cell carcinoma correlate with the disease outcome. Sci. Rep. 2022, 12, 12406. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Albavera, L.; Domínguez-Pérez, M.; Medina-Leyte, D.J.; González-Garrido, A.; Villarreal-Molina, T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int. J. Mol. Sci. 2021, 22, 1593. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.M.; Lee, J.Y.; Boudyguina, E.; Kluckman, K.D.; Brunham, L.R.; Mulya, A.; Gebre, A.K.; Coutinho, J.M.; Colvin, P.L.; Smith, T.L.; et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Investig. 2005, 115, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Iqbal, J.; Fievet, C.; Timmins, J.M.; Pape, T.D.; Coburn, B.A.; Bissada, N.; Staels, B.; Groen, A.K.; et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Investig. 2006, 116, 1052–1062. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldán, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef]
- Ouimet, M.; Marcel, Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef]
- Zaiou, M.; Bakillah, A. Epigenetic Regulation of ATP-Binding Cassette Protein A1 (ABCA1) Gene Expression: A New Era to Alleviate Atherosclerotic Cardiovascular Disease. Diseases 2018, 6, 34. [Google Scholar] [CrossRef]
- Czarnecka, H.; Yokoyama, S. Regulation of cellular cholesterol efflux by lecithin:cholesterol acyltransferase reaction through nonspecific lipid exchange. J. Biol. Chem. 1996, 271, 2023–2028. [Google Scholar] [CrossRef]
- Ji, Y.; Jian, B.; Wang, N.; Sun, Y.; Moya, M.L.; Phillips, M.C.; Rothblat, G.H.; Swaney, J.B.; Tall, A.R. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J. Biol. Chem. 1997, 272, 20982–20985. [Google Scholar] [CrossRef] [PubMed]
- Jessup, W.; Gelissen, I.C.; Gaus, K.; Kritharides, L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr. Opin. Lipidol. 2006, 17, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The roles of different pathways in the release of cholesterol from macrophages. J. Lipid Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Azzam, K.M.; Lin, W.C.; Rai, P.; Lowe, J.M.; Gabor, K.A.; Madenspacher, J.H.; Aloor, J.J.; Parks, J.S.; Näär, A.M.; et al. MicroRNA-33 Regulates the Innate Immune Response via ATP Binding Cassette Transporter-mediated Remodeling of Membrane Microdomains. J. Biol. Chem. 2016, 291, 19651–19660. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, H.; Nohara, A.; Inazu, A. Cholesteryl ester transfer protein (CETP) deficiency and CETP inhibitors. Mol. Cells 2014, 37, 777–784. [Google Scholar] [CrossRef]
- Goedeke, L.; Salerno, A.; Ramírez, C.M.; Guo, L.; Allen, R.M.; Yin, X.; Langley, S.R.; Esau, C.; Wanschel, A.; Fisher, E.A.; et al. Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol. Med. 2014, 6, 1133–1141. [Google Scholar] [CrossRef]
- Price, N.L.; Zhang, X.; Fernández-Tussy, P.; Singh, A.K.; Burnap, S.A.; Rotllan, N.; Goedeke, L.; Sun, J.; Canfrán-Duque, A.; Aryal, B.; et al. Loss of hepatic miR-33 improves metabolic homeostasis and liver function without altering body weight or atherosclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2006478118. [Google Scholar] [CrossRef]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Trauner, M.; Claudel, T.; Fickert, P.; Moustafa, T.; Wagner, M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig. Dis. 2010, 28, 220–224. [Google Scholar] [CrossRef]
- Claudel, T.; Zollner, G.; Wagner, M.; Trauner, M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim. Biophys. Acta 2011, 1812, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Edwards, P.A. FXR signaling in metabolic disease. FEBS Lett. 2008, 582, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Li, T.; Francl, J.M.; Boehme, S.; Chiang, J.Y. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7α-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology 2013, 58, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Morotti, R.A.; Suchy, F.J.; Magid, M.S. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: A review of the liver pathology findings. Semin. Liver Dis. 2011, 31, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hammer, R.E.; Li-Hawkins, J.; Von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA 2002, 99, 16237–16242. [Google Scholar] [CrossRef]
- Allen, R.M.; Marquart, T.J.; Albert, C.J.; Suchy, F.J.; Wang, D.Q.; Ananthanarayanan, M.; Ford, D.A.; Baldán, A. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol. Med. 2012, 4, 882–895. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Linton, M.F.; Fisher, E.A.; Guyton, J.R. JCL roundtable: Lipids and inflammation in atherosclerosis. J. Clin. Lipidol. 2021, 15, 3–17. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorkiewicz, M. Is microRNA-33 an Appropriate Target in the Treatment of Atherosclerosis? Nutrients 2023, 15, 902. https://doi.org/10.3390/nu15040902
Sidorkiewicz M. Is microRNA-33 an Appropriate Target in the Treatment of Atherosclerosis? Nutrients. 2023; 15(4):902. https://doi.org/10.3390/nu15040902
Chicago/Turabian StyleSidorkiewicz, Malgorzata. 2023. "Is microRNA-33 an Appropriate Target in the Treatment of Atherosclerosis?" Nutrients 15, no. 4: 902. https://doi.org/10.3390/nu15040902
APA StyleSidorkiewicz, M. (2023). Is microRNA-33 an Appropriate Target in the Treatment of Atherosclerosis? Nutrients, 15(4), 902. https://doi.org/10.3390/nu15040902