
Neuroprotective Effects of Nicotinamide (Vitamin B3) on Neurodegeneration in Diabetic Rat Retinas


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
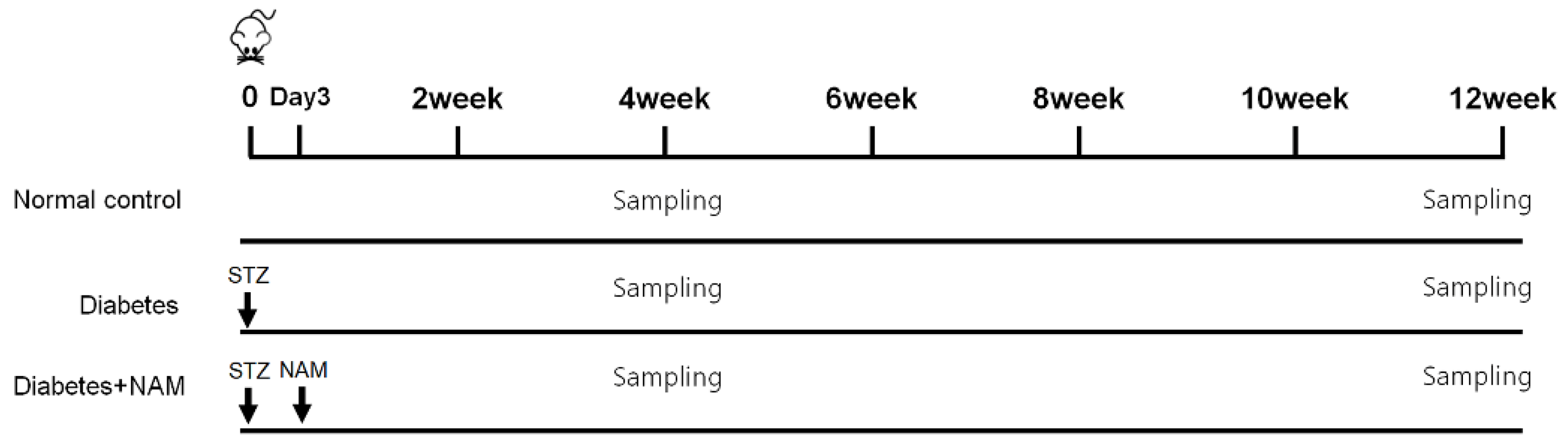
2.2. Induction of Diabetes
2.3. Allocation of Groups and Drug Treatments
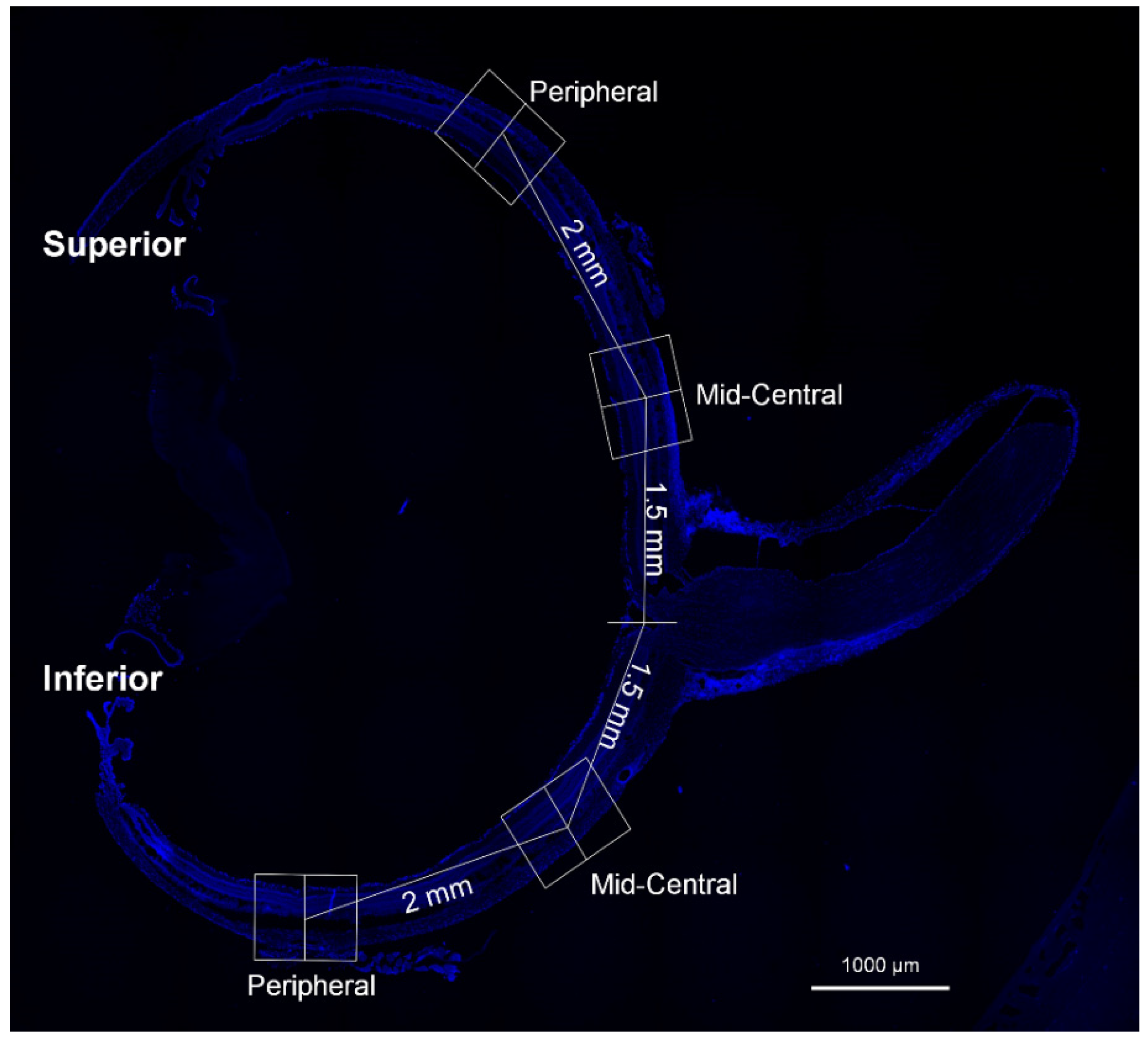
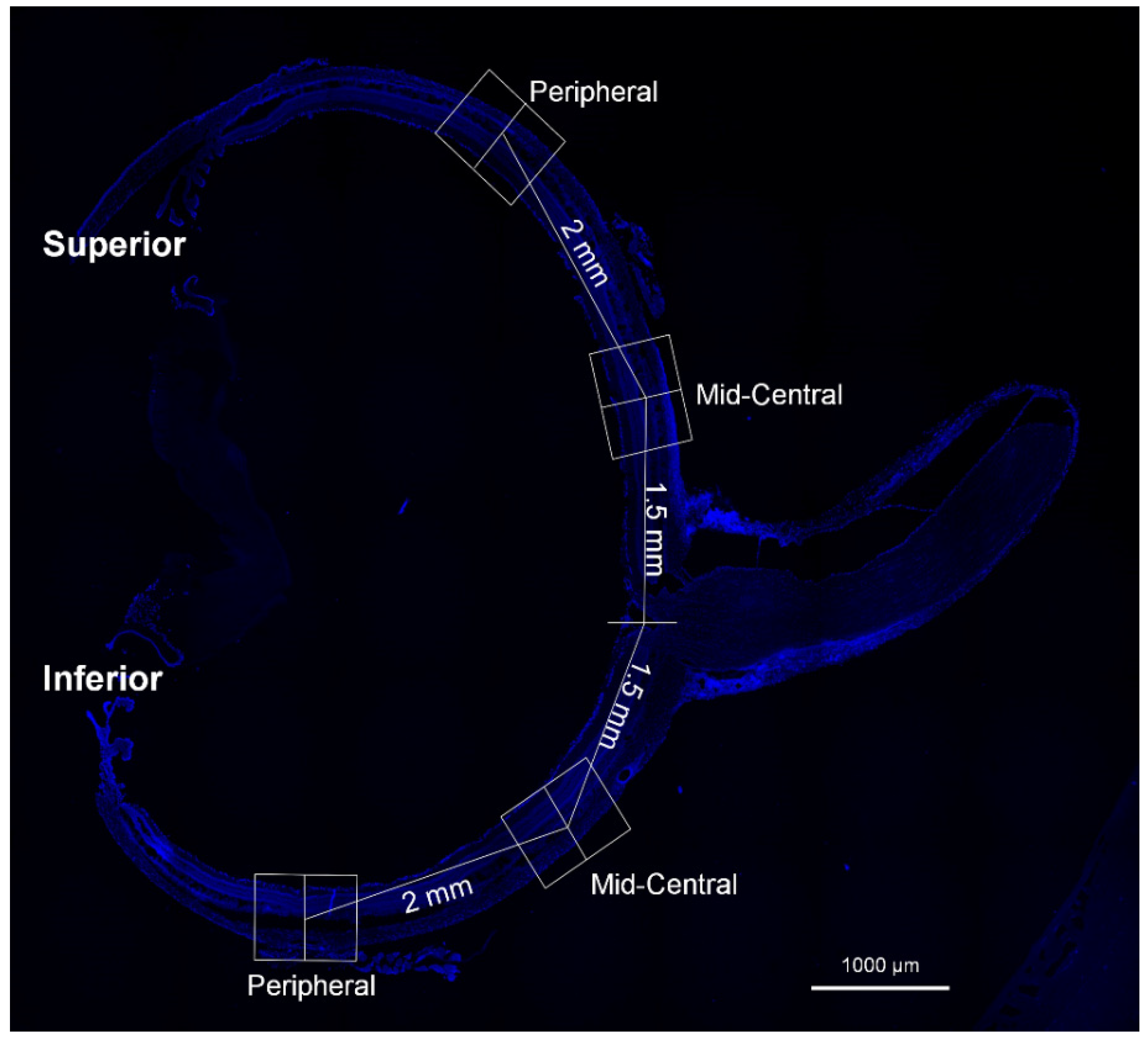
2.4. Immunofluorescence Staining
2.5. Measurement of Reactive Oxygen Species
2.6. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling
2.7. Western Blot Analysis
2.8. Data Analysis
3. Results
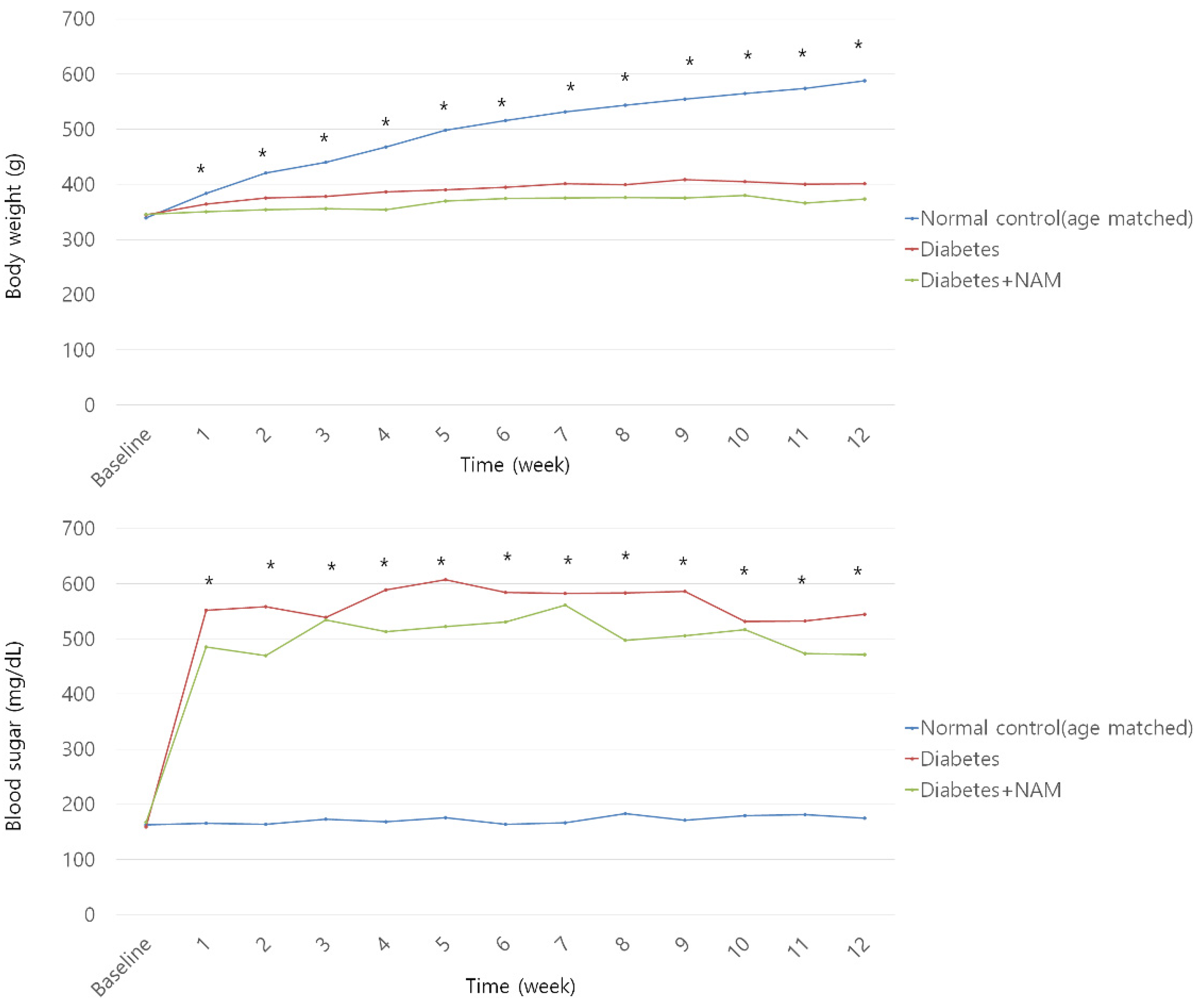
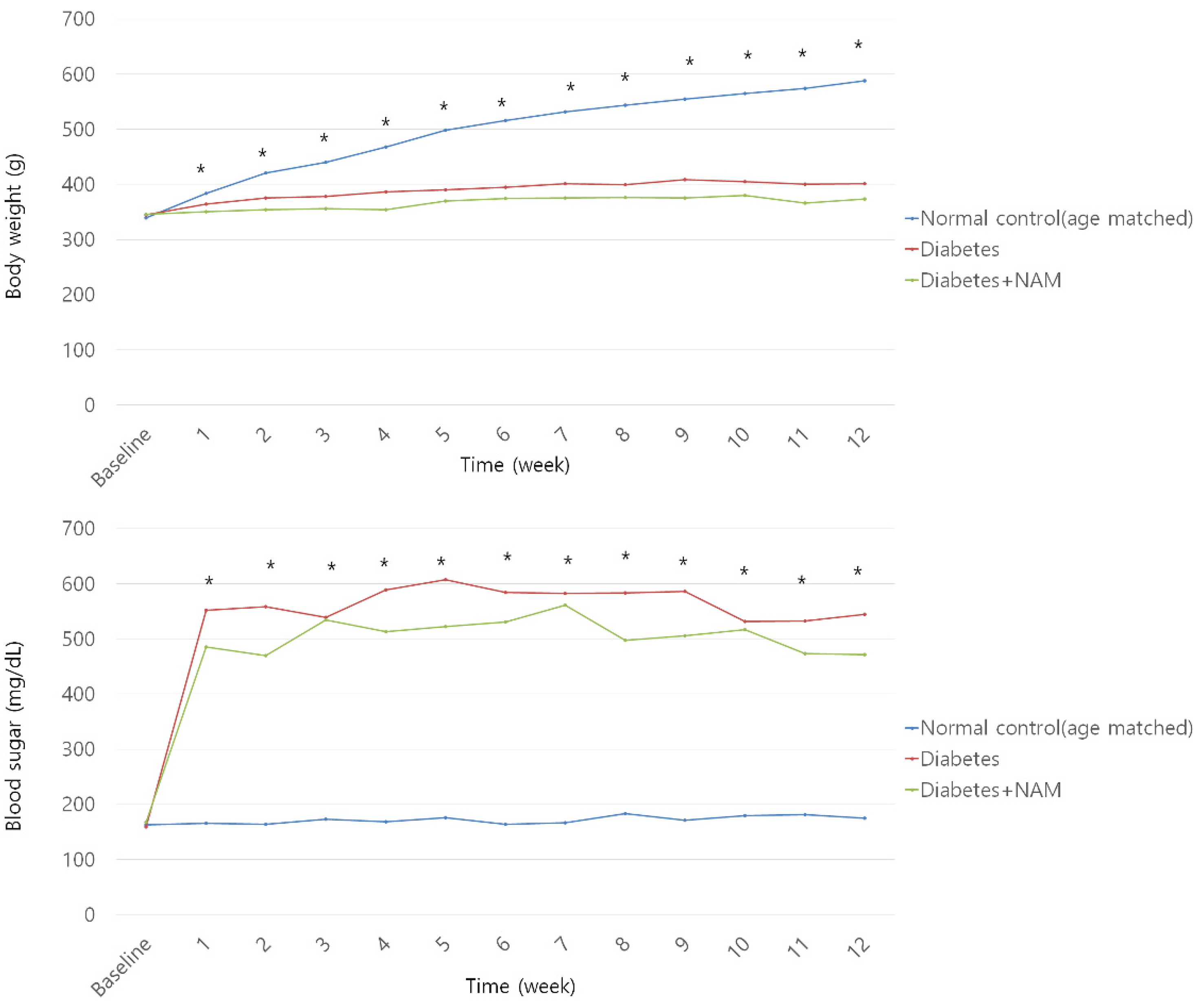
3.1. Body Weight and Blood Glucose
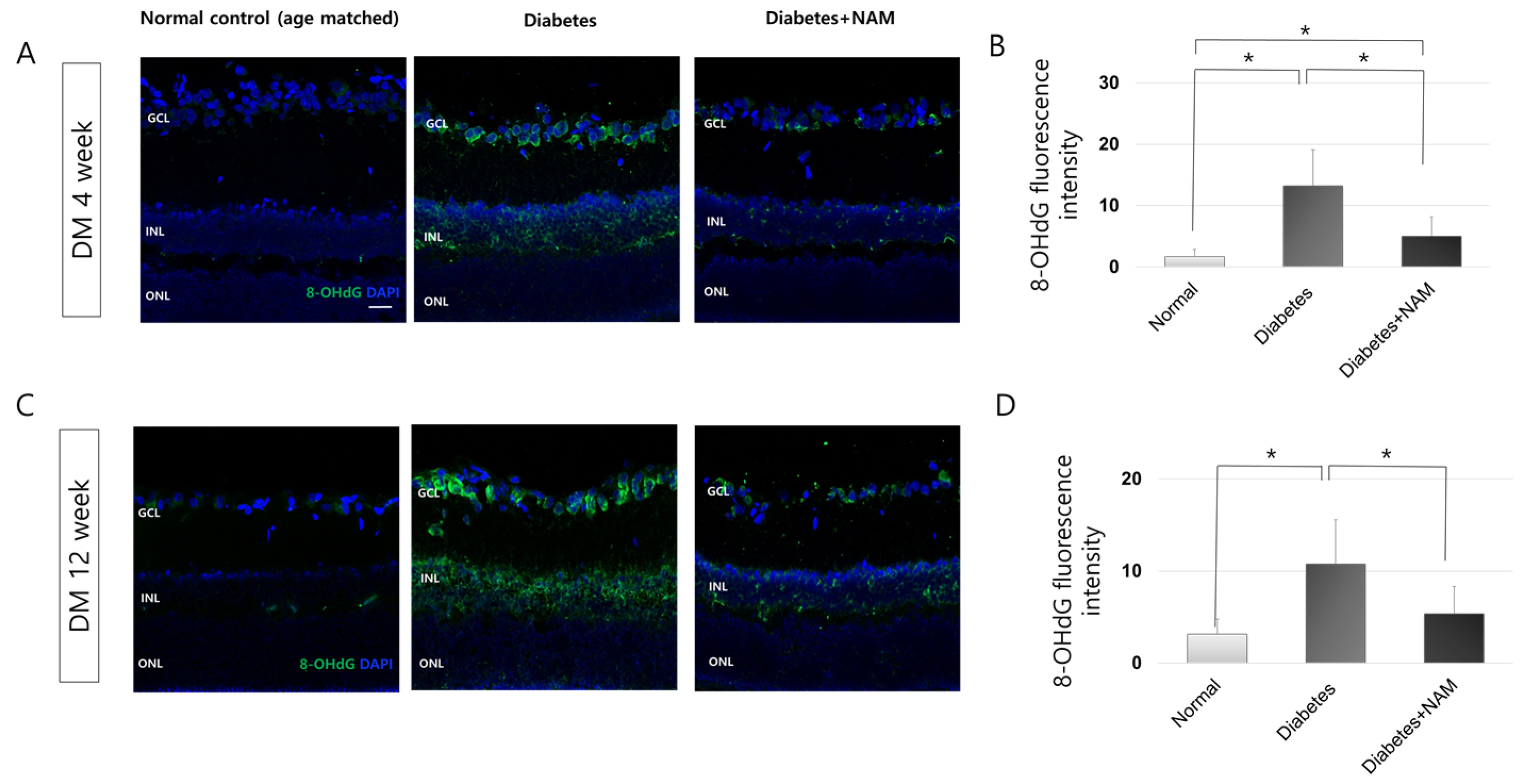
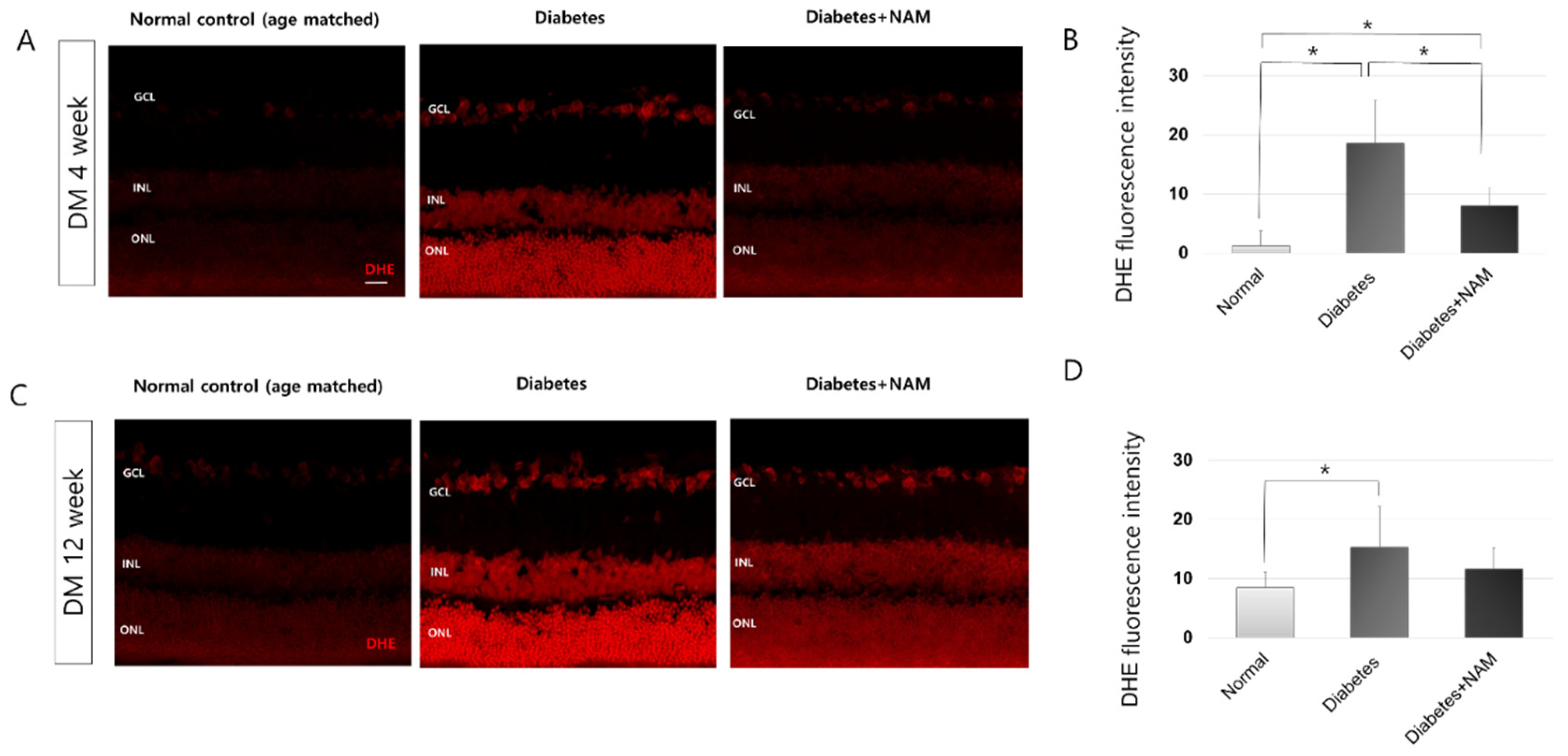
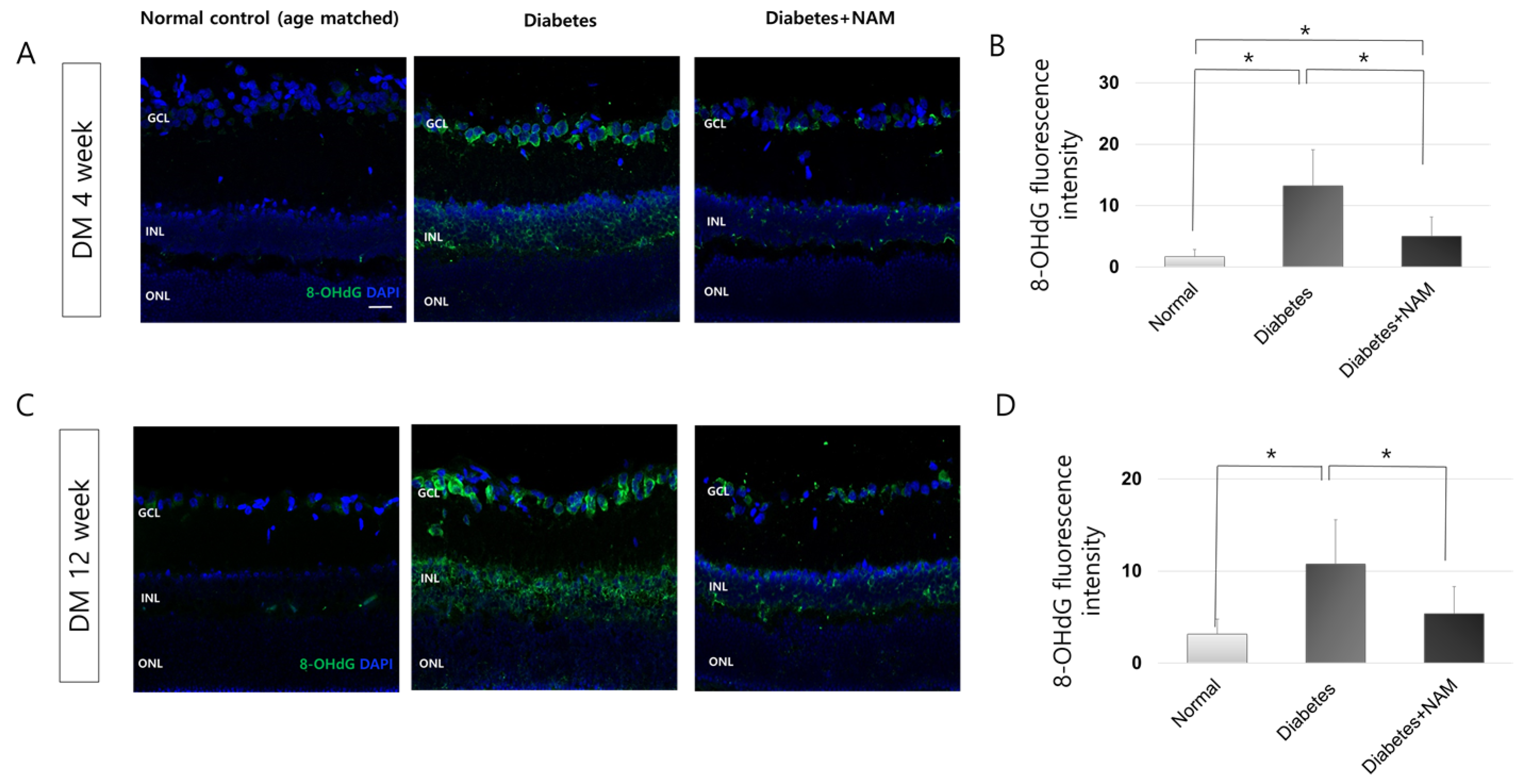
3.2. Oxidative Stress and Oxidative DNA Damage
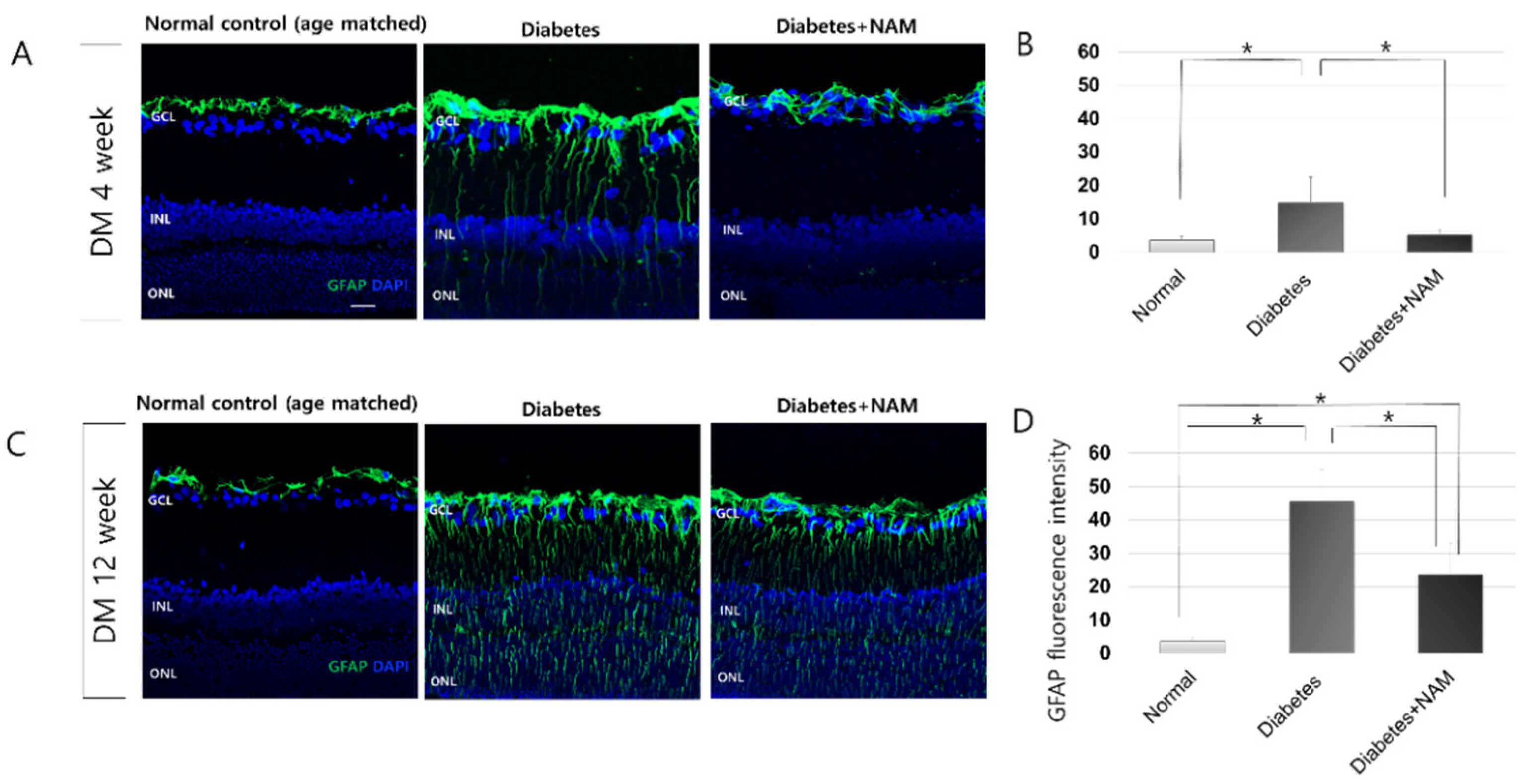
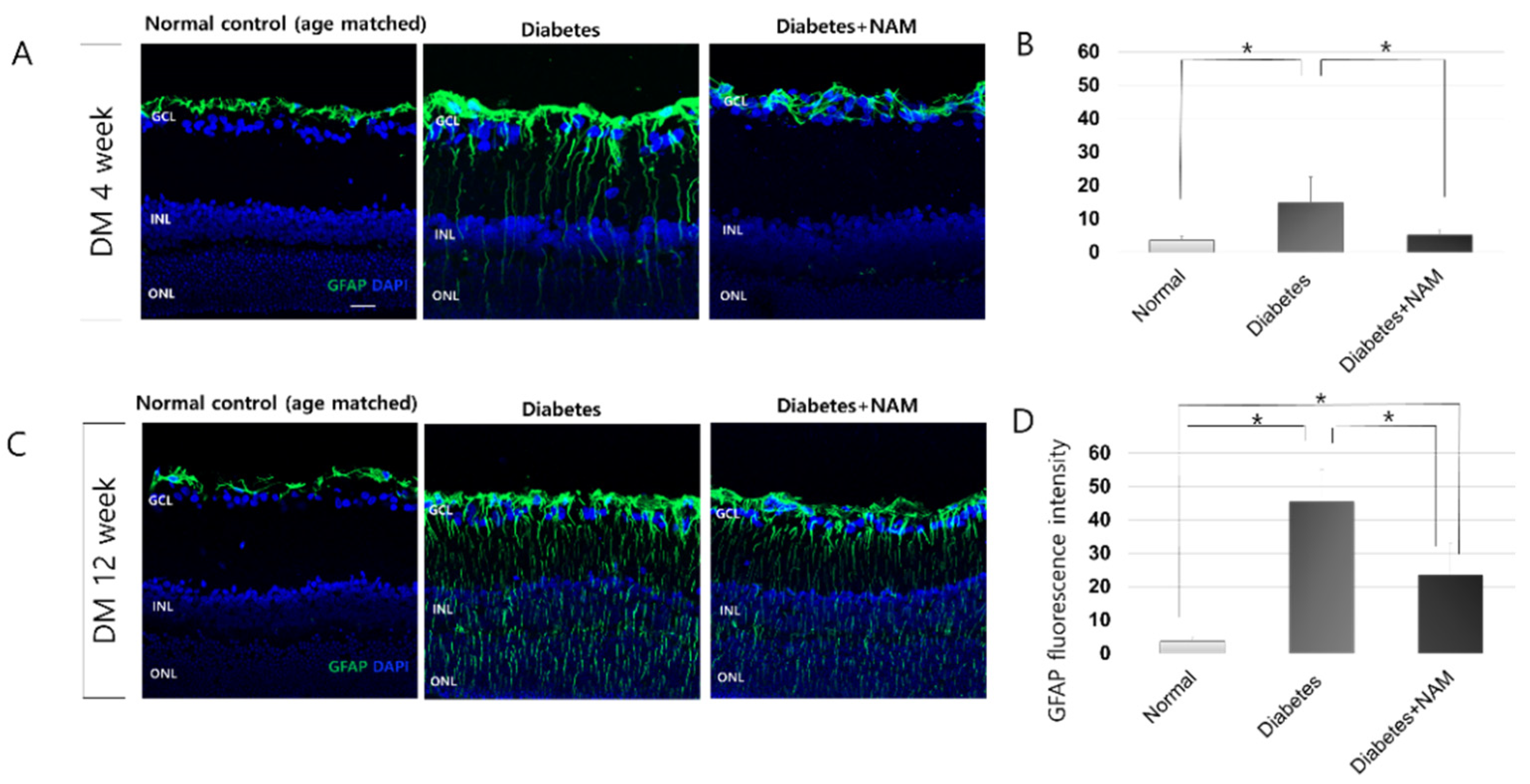
3.3. Glial Activation in the Retina
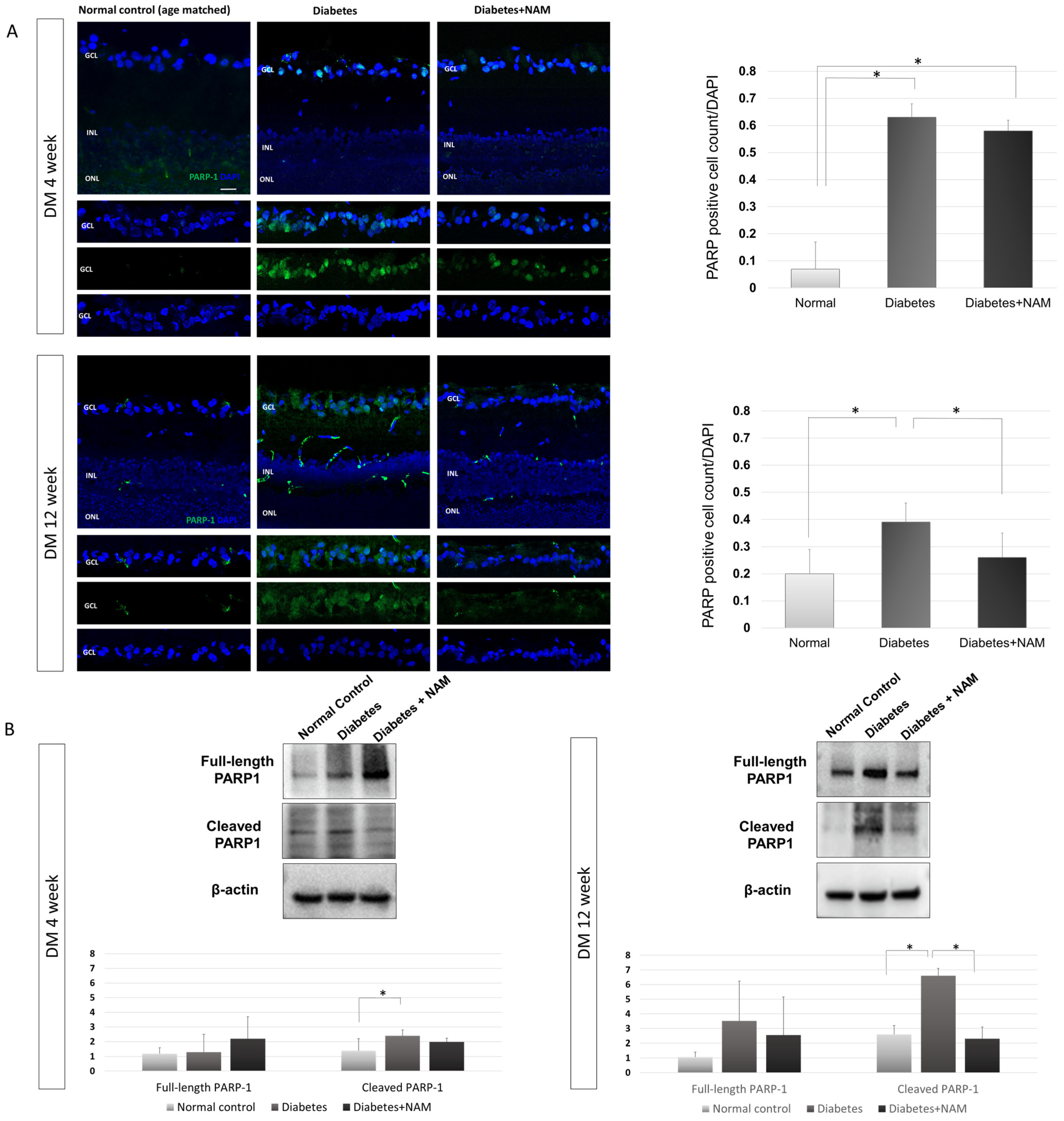
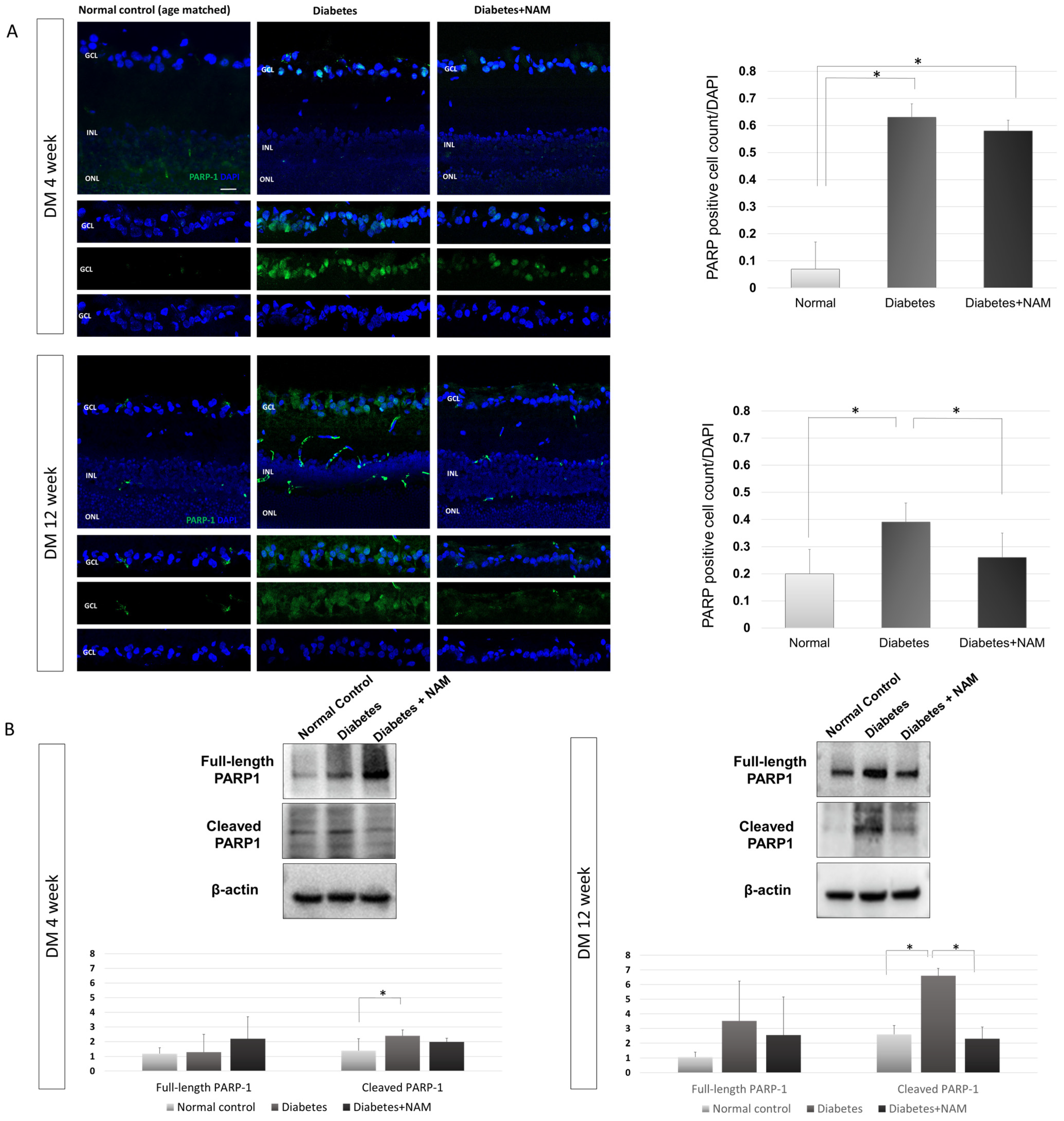
3.4. PARP-1
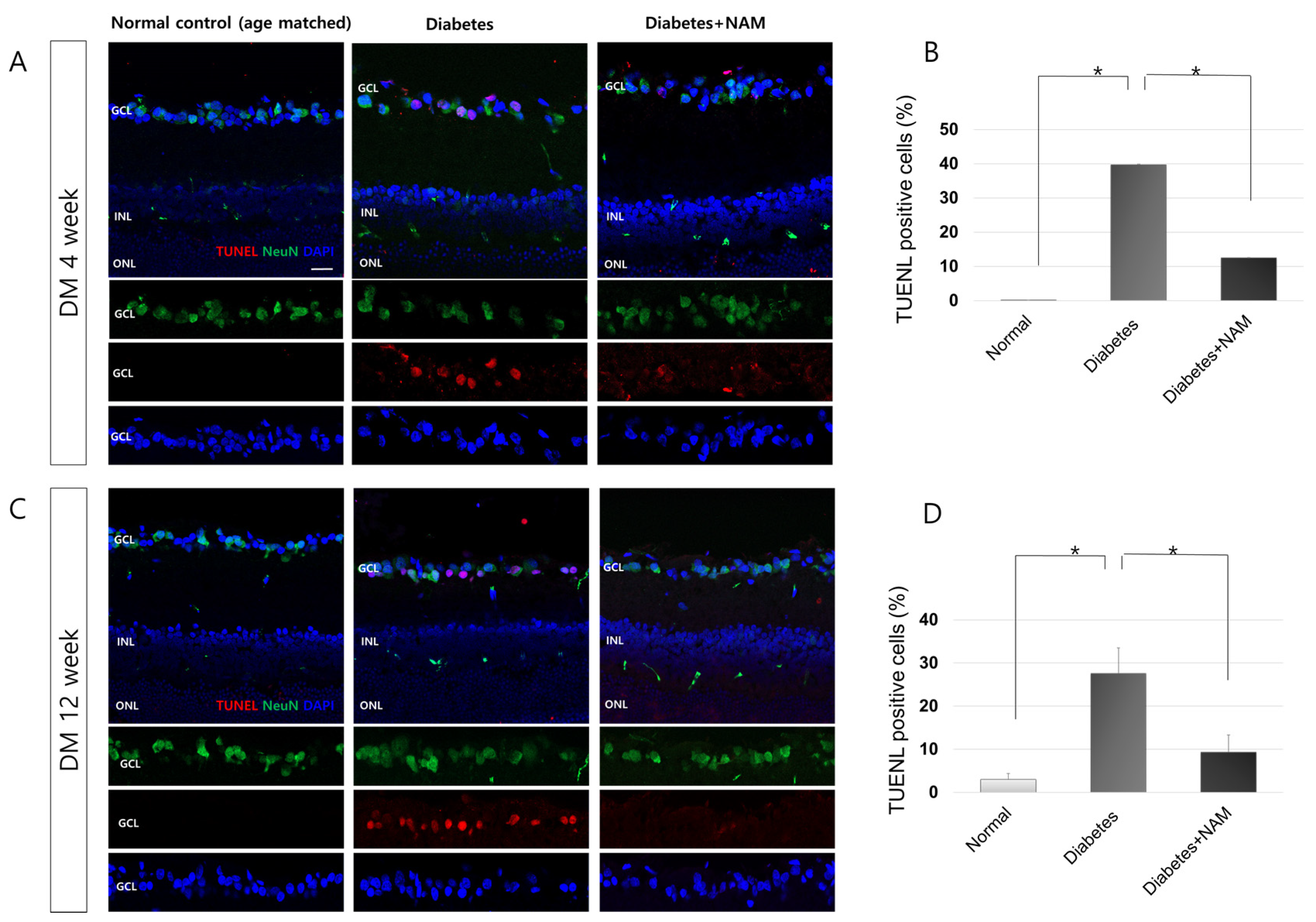
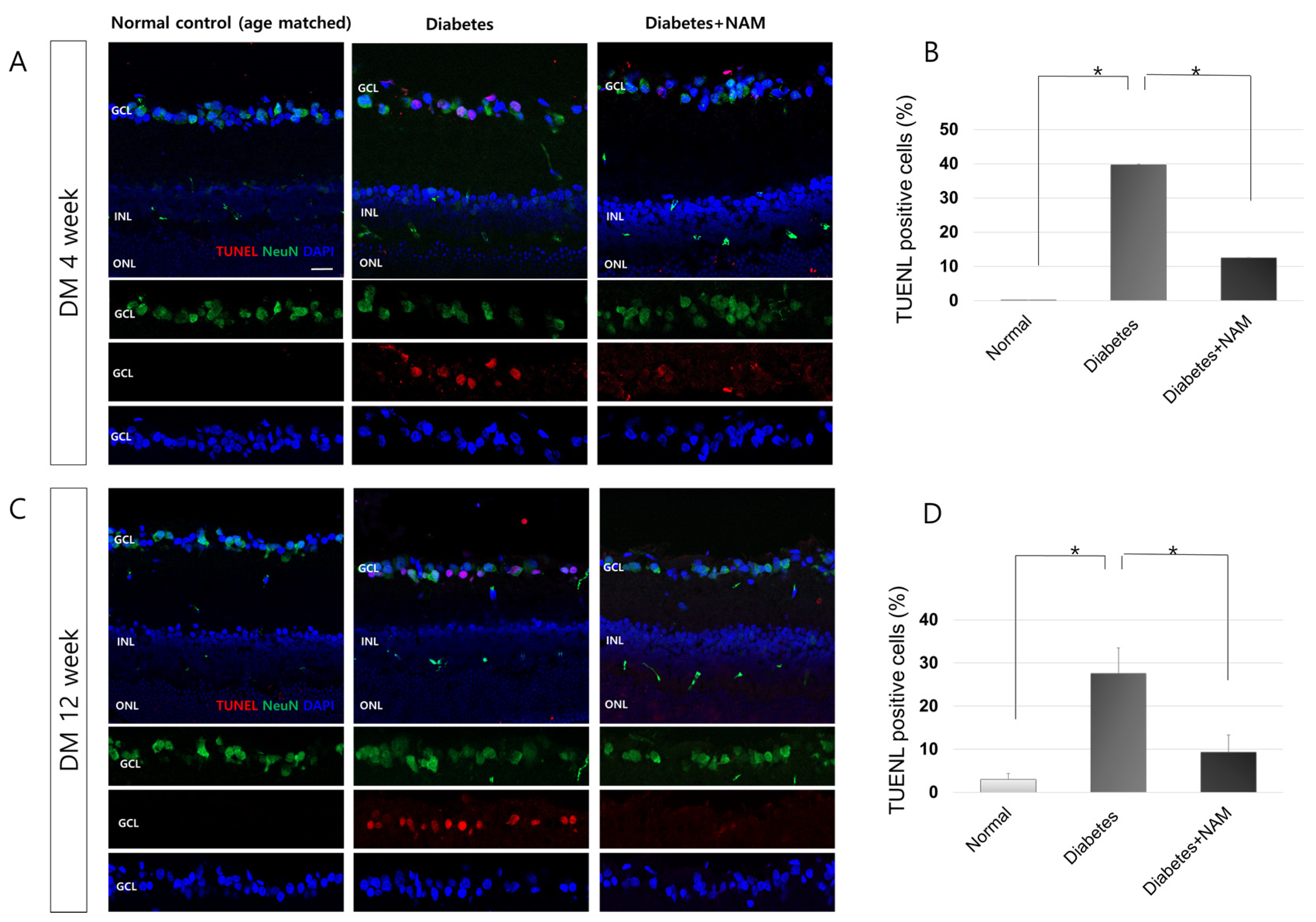
3.5. Apoptotic Retinal Ganglion Cell Death
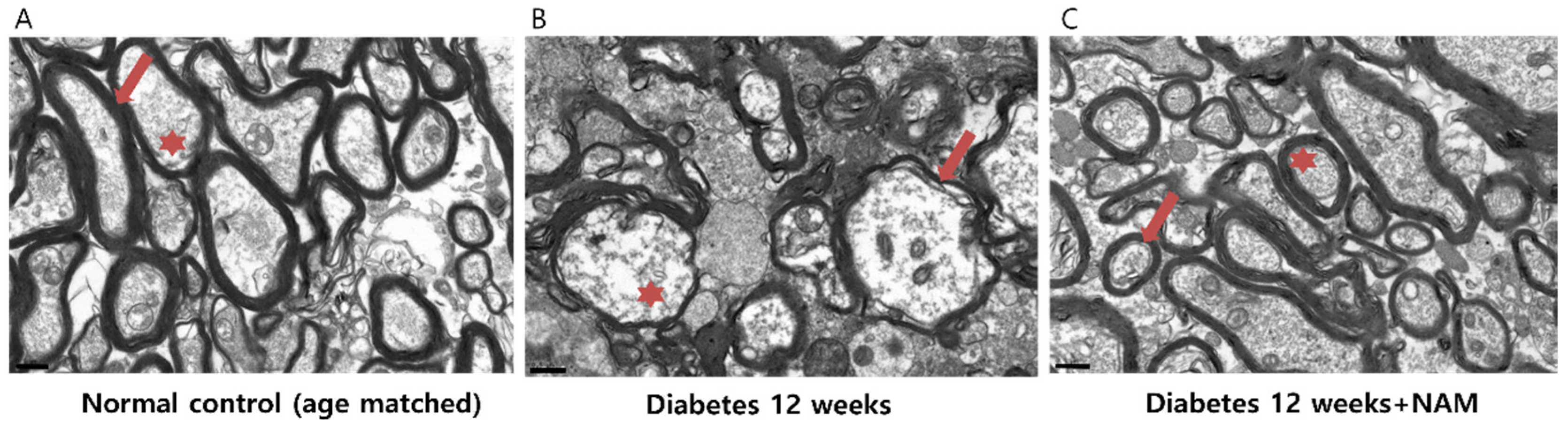
3.6. Ultrastructural Features of the Optic Nerve
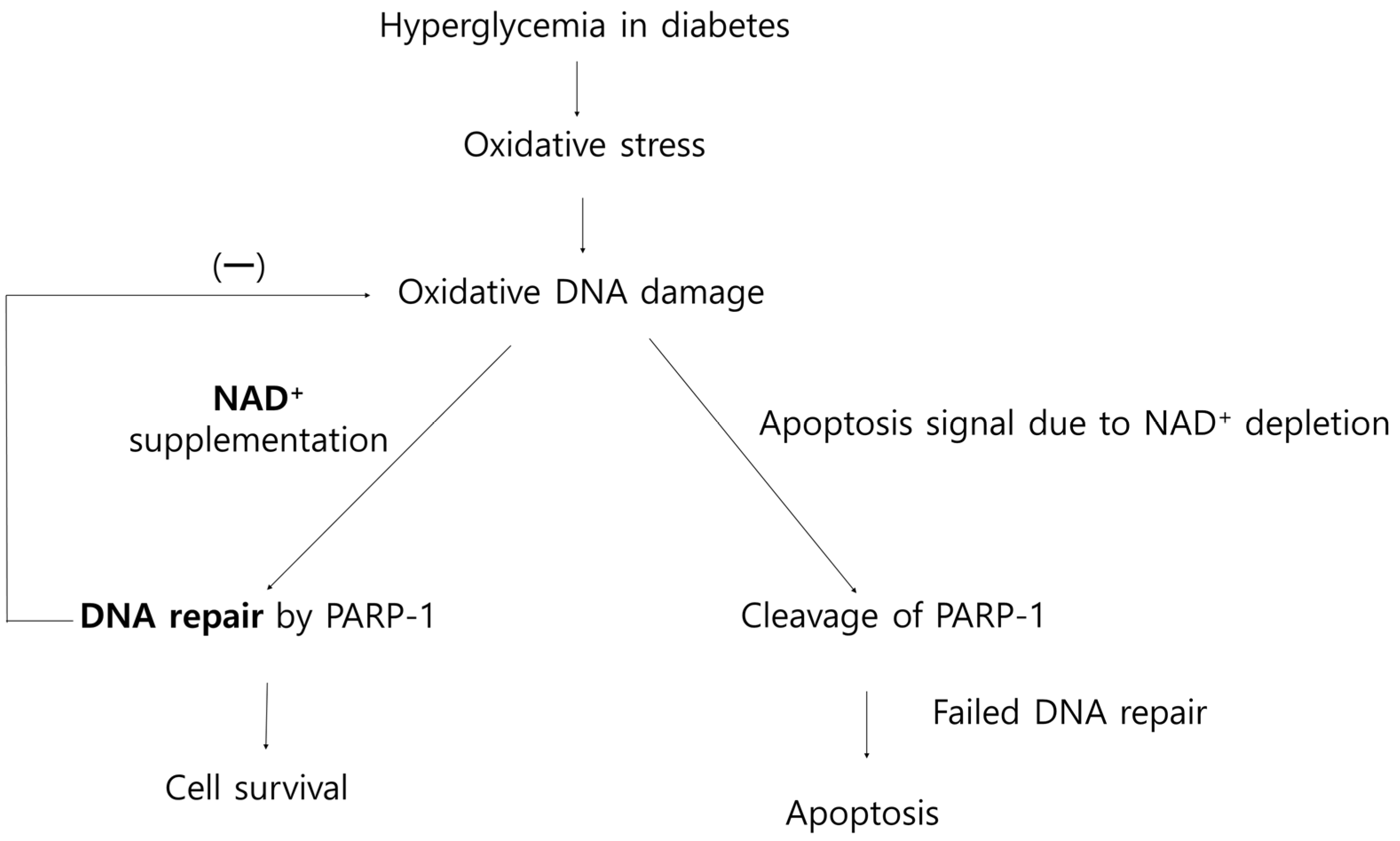
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Simo, R.; Simo-Servat, O.; Bogdanov, P.; Hernandez, C. Neurovascular Unit: A New Target for Treating Early Stages of Diabetic Retinopathy. Pharmaceutics 2021, 13, 1320. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, I.T.; Park, C.K. Early diabetic changes in the nerve fibre layer at the macula detected by spectral domain optical coherence tomography. Br. J. Ophthalmol. 2011, 95, 1223–1228. [Google Scholar] [CrossRef]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes 2003, 52, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef]
- Simo, R.; Hernandez, C.; European Consortium for the Early Treatment of Diabetic Retinopathy. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Bikbova, G.; Oshitari, T.; Baba, T.; Yamamoto, S. Neurotrophic factors for retinal ganglion cell neuropathy—With a special reference to diabetic neuropathy in the retina. Curr. Diabetes Rev. 2014, 10, 166–176. [Google Scholar] [CrossRef]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M. Oxidative stress in developmental brain disorders. Neuropathology 2009, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat. Res. 2007, 614, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.Y.; Jin, J.; Lu, G.; Kang, X.L. Astaxanthin attenuates the apoptosis of retinal ganglion cells in db/db mice by inhibition of oxidative stress. Mar. Drugs 2013, 11, 960–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arden, G.B.; Sidman, R.L.; Arap, W.; Schlingemann, R.O. Spare the rod and spoil the eye. Br. J. Ophthalmol. 2005, 89, 764–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 2020, 37, 101779. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146-148, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Sairanen, T.; Szepesi, R.; Karjalainen-Lindsberg, M.L.; Saksi, J.; Paetau, A.; Lindsberg, P.J. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009, 118, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Strom, C.E.; Helleday, T. Strategies for the Use of Poly(adenosine diphosphate ribose) Polymerase (PARP) Inhibitors in Cancer Therapy. Biomolecules 2012, 2, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzyk, M.M.; Tykhomyrov, A.A.; Nedzvetsky, V.S.; Prischepa, I.V.; Grinenko, T.V.; Yanitska, L.V.; Kuchmerovska, T.M. Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors Reduce Reactive Gliosis and Improve Angiostatin Levels in Retina of Diabetic Rats. Neurochem. Res. 2016, 41, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Siddiquei, M.M.; Abu El-Asrar, A.M. Poly (ADP-ribose) polymerase mediates diabetes-induced retinal neuropathy. Mediat. Inflamm. 2013, 2013, 510451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Cacicedo, J.M.; Ido, Y. Impaired nicotinamide adenine dinucleotide (NAD+) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. J. Diabetes Investig 2020, 11, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Ido, Y.; Williamson, J.R. Hyperglycemic cytosolic reductive stress ‘pseudohypoxia’: Implications for diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1467–1470. [Google Scholar]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Ozawa, Y.; Kurihara, T.; Kubota, S.; Yuki, K.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K. Neurodegenerative influence of oxidative stress in the retina of a murine model of diabetes. Diabetologia 2010, 53, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Laspas, P.; Zhutdieva, M.B.; Brochhausen, C.; Musayeva, A.; Zadeh, J.K.; Pfeiffer, N.; Xia, N.; Li, H.; Wess, J.; Gericke, A. The M1 muscarinic acetylcholine receptor subtype is important for retinal neuron survival in aging mice. Sci. Rep. 2019, 9, 5222. [Google Scholar] [CrossRef]
- Jung, K.I.; Kim, J.H.; Park, H.Y.; Park, C.K. Neuroprotective effects of cilostazol on retinal ganglion cell damage in diabetic rats. J. Pharmacol. Exp. 2013, 345, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Kino, K.; Hirao-Suzuki, M.; Morikawa, M.; Sakaga, A.; Miyazawa, H. Generation, repair and replication of guanine oxidation products. Genes Environ. 2017, 39, 21. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.C.; Surjana, D.; Halliday, G.M.; Damian, D.L. Nicotinamide enhances repair of ultraviolet radiation-induced DNA damage in primary melanocytes. Exp. Dermatol. 2014, 23, 509–511. [Google Scholar] [CrossRef]
- Wu, M.F.; Yin, J.H.; Hwang, C.S.; Tang, C.M.; Yang, D.I. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef]
- Massudi, H.; Grant, R.; Guillemin, G.J.; Braidy, N. NAD+ metabolism and oxidative stress: The golden nucleotide on a crown of thorns. Redox Rep. 2012, 17, 28–46. [Google Scholar] [CrossRef]
- Bringmann, A.; Wiedemann, P. Muller glial cells in retinal disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Woo, J.E.; Park, C.K. Intraocular pressure fluctuation and neurodegeneration in the diabetic rat retina. Br. J. Pharm. 2020, 177, 3046–3059. [Google Scholar] [CrossRef]
- Nian, S.; Lo, A.C.Y.; Mi, Y.; Ren, K.; Yang, D. Neurovascular unit in diabetic retinopathy: Pathophysiological roles and potential therapeutical targets. Eye Vis. 2021, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.; Chen, L. The cells involved in the pathological process of diabetic retinopathy. Biomed. Pharmacother. 2020, 132, 110818. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Muller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.A.; Tan, A.A.; Smith, D.C.; Hoane, M.R. Nicotinamide treatment provides acute neuroprotection and GFAP regulation following fluid percussion injury. J. Neurotrauma 2008, 25, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Shear, D.A.; Dixon, C.E.; Bramlett, H.M.; Mondello, S.; Dietrich, W.D.; Deng-Bryant, Y.; Schmid, K.E.; Wang, K.K.; Hayes, R.L.; Povlishock, J.T.; et al. Nicotinamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Szanto, M.; Brunyanszki, A.; Kiss, B.; Nagy, L.; Gergely, P.; Virag, L.; Bai, P. Poly(ADP-ribose) polymerase-2: Emerging transcriptional roles of a DNA-repair protein. Cell Mol. Life Sci. 2012, 69, 4079–4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kergoat, H.; Herard, M.E.; Lemay, M. RGC sensitivity to mild systemic hypoxia. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5423–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol. Dis. 2000, 7, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Moskowitz, M.A. Cell biology. PARP-1—A perpetrator of apoptotic cell death? Science 2002, 297, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.A.; Bingley, P.J.; Emmett, C.L.; Collier, T.; European Nicotinamide Diabetes Intervention Trial (ENDIT) Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): A randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004, 363, 925–931. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Kim, Y.C.; Park, C.K. Dietary Niacin and Open-Angle Glaucoma: The Korean National Health and Nutrition Examination Survey. Nutrients 2018, 10, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A.; European Nicotinamide Diabetes Intervention Trial Group. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.C.; Martin, A.J.; Choy, B.; Fernandez-Penas, P.; Dalziell, R.A.; McKenzie, C.A.; Scolyer, R.A.; Dhillon, H.M.; Vardy, J.L.; Kricker, A.; et al. A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N. Engl. J. Med. 2015, 373, 1618–1626. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.L.; Boyer, J.L. Hepatic toxicity from large doses of vitamin B3 (nicotinamide). N. Engl. J. Med. 1973, 289, 1180–1182. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, K.I.; Han, J.-S.; Park, C.K. Neuroprotective Effects of Nicotinamide (Vitamin B3) on Neurodegeneration in Diabetic Rat Retinas. Nutrients 2022, 14, 1162. https://doi.org/10.3390/nu14061162
Jung KI, Han J-S, Park CK. Neuroprotective Effects of Nicotinamide (Vitamin B3) on Neurodegeneration in Diabetic Rat Retinas. Nutrients. 2022; 14(6):1162. https://doi.org/10.3390/nu14061162
Chicago/Turabian StyleJung, Kyoung In, Jeong-Sun Han, and Chan Kee Park. 2022. "Neuroprotective Effects of Nicotinamide (Vitamin B3) on Neurodegeneration in Diabetic Rat Retinas" Nutrients 14, no. 6: 1162. https://doi.org/10.3390/nu14061162
APA StyleJung, K. I., Han, J.-S., & Park, C. K. (2022). Neuroprotective Effects of Nicotinamide (Vitamin B3) on Neurodegeneration in Diabetic Rat Retinas. Nutrients, 14(6), 1162. https://doi.org/10.3390/nu14061162