Ganoderma lucidum Extract Reduces Insulin Resistance by Enhancing AMPK Activation in High-Fat Diet-Induced Obese Mice


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. G. Lucidum Extract Powder
2.2. Animals
2.3. Tissue Weight Measurement
2.4. Histopathological Observation
2.5. Measurement of Blood Lipid Concentration
2.6. Measurement of Blood Glucose Concentration
2.7. Glucose and Insulin Tolerance Tests (GTTs and ITTs)
2.8. Measurement of Insulin, Leptin, and Adiponectin Levels
2.9. Measurement of 2-Deoxy-Glucose Uptake Following In Vivo Insulin Stimulation
2.10. qRT-PCR of Lipogenesis Genes in Adipose and Liver Tissues
2.11. Quantification of Glucose and Lipid Metabolism-Related Protein Expression in WATs
2.12. Statistical Analysis
3. Results
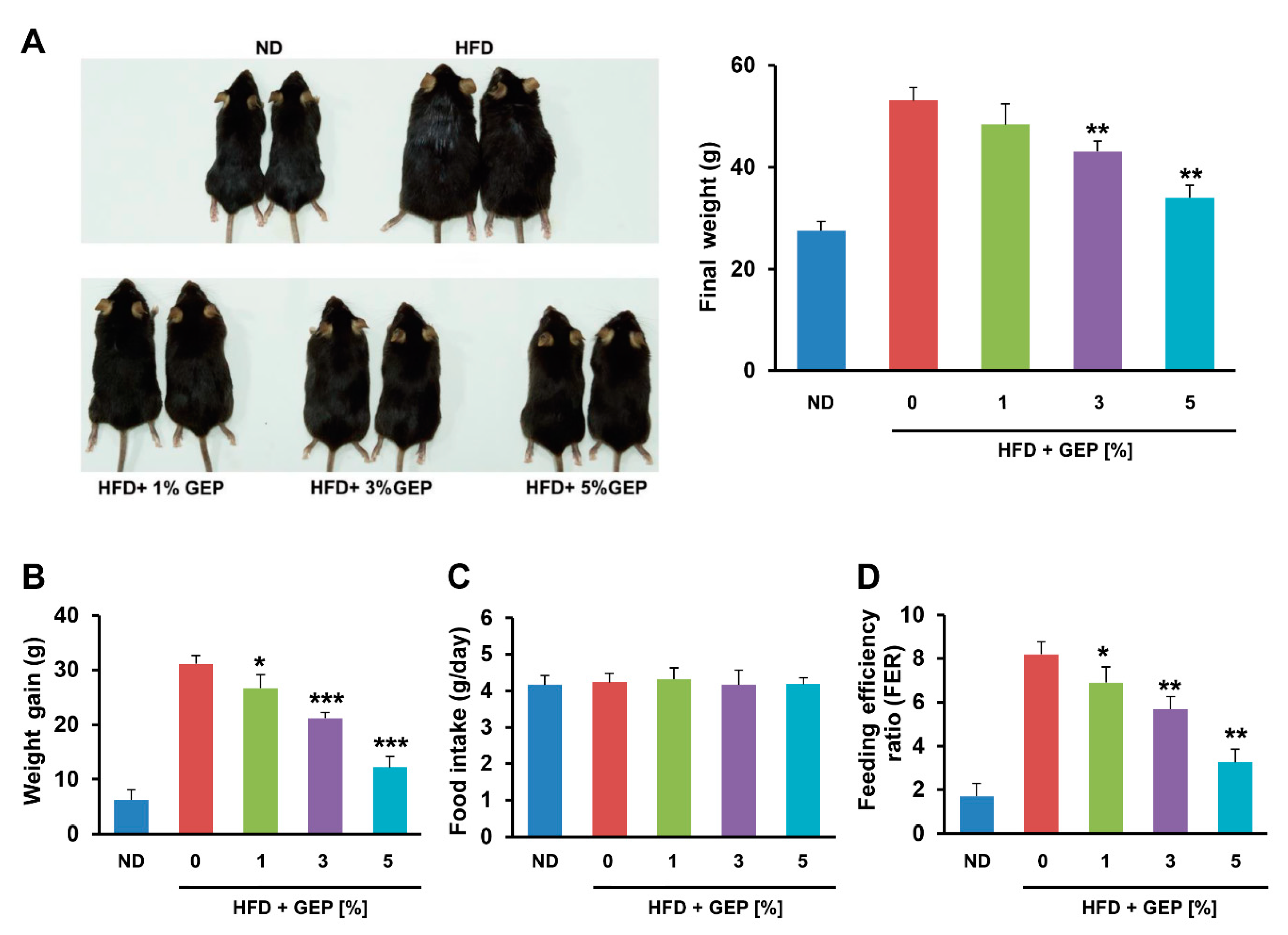
3.1. Effect of G. Lucidum Extract on Obesity
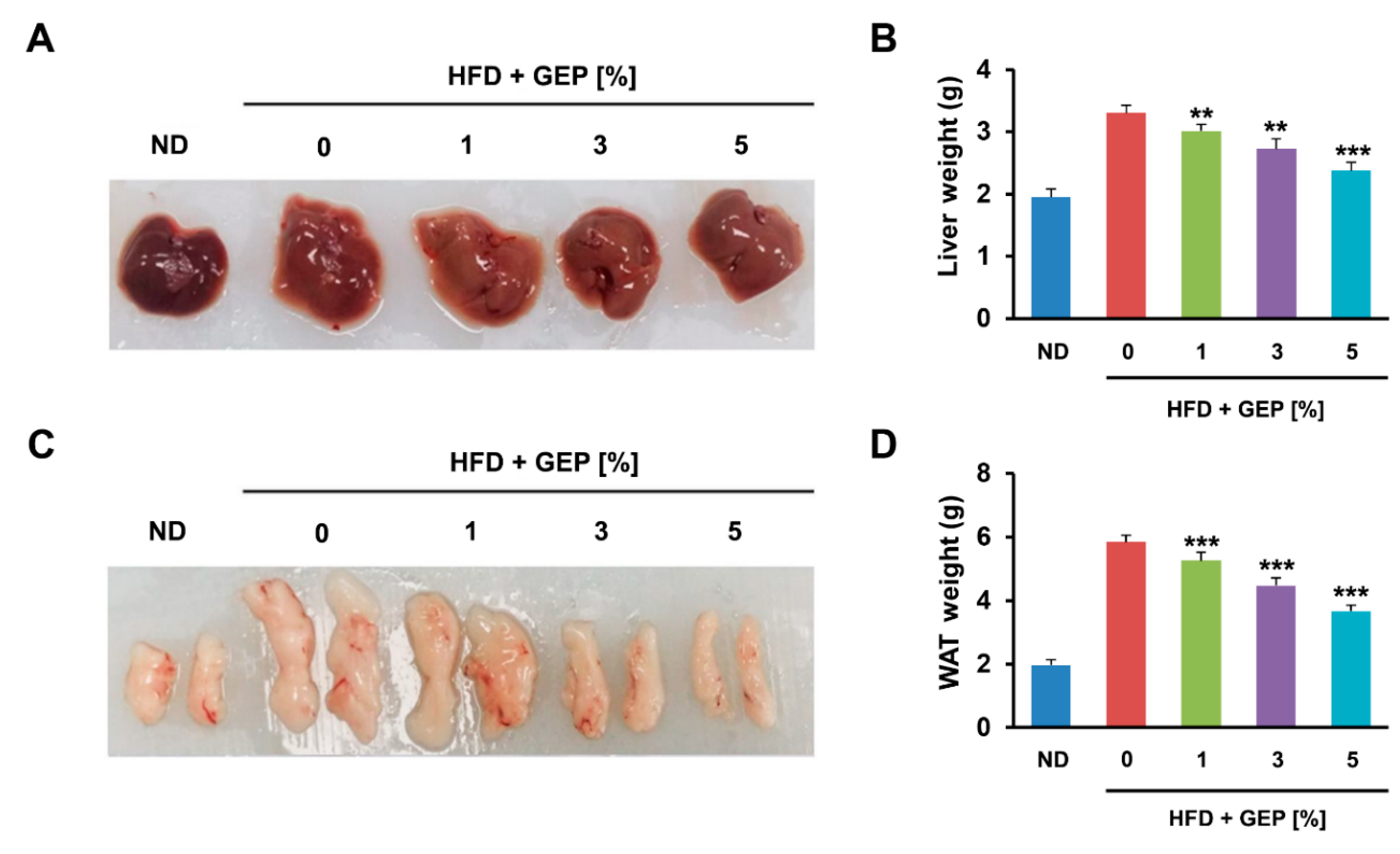
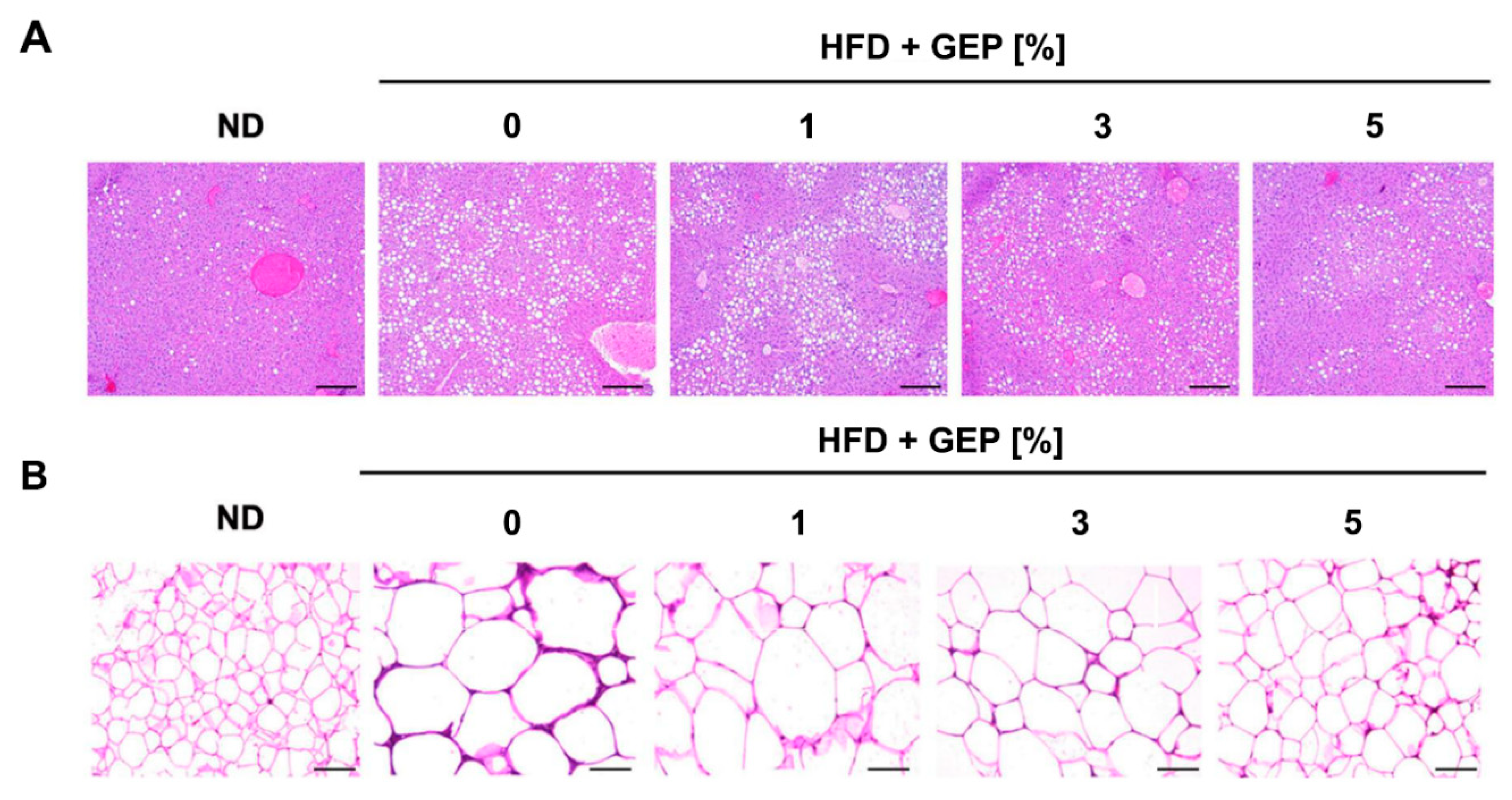
3.2. Effect of G. Lucidum Extract on the Liver and Adipose Tissue
3.3. Effect of G. Lucidum on Serum Lipid Levels
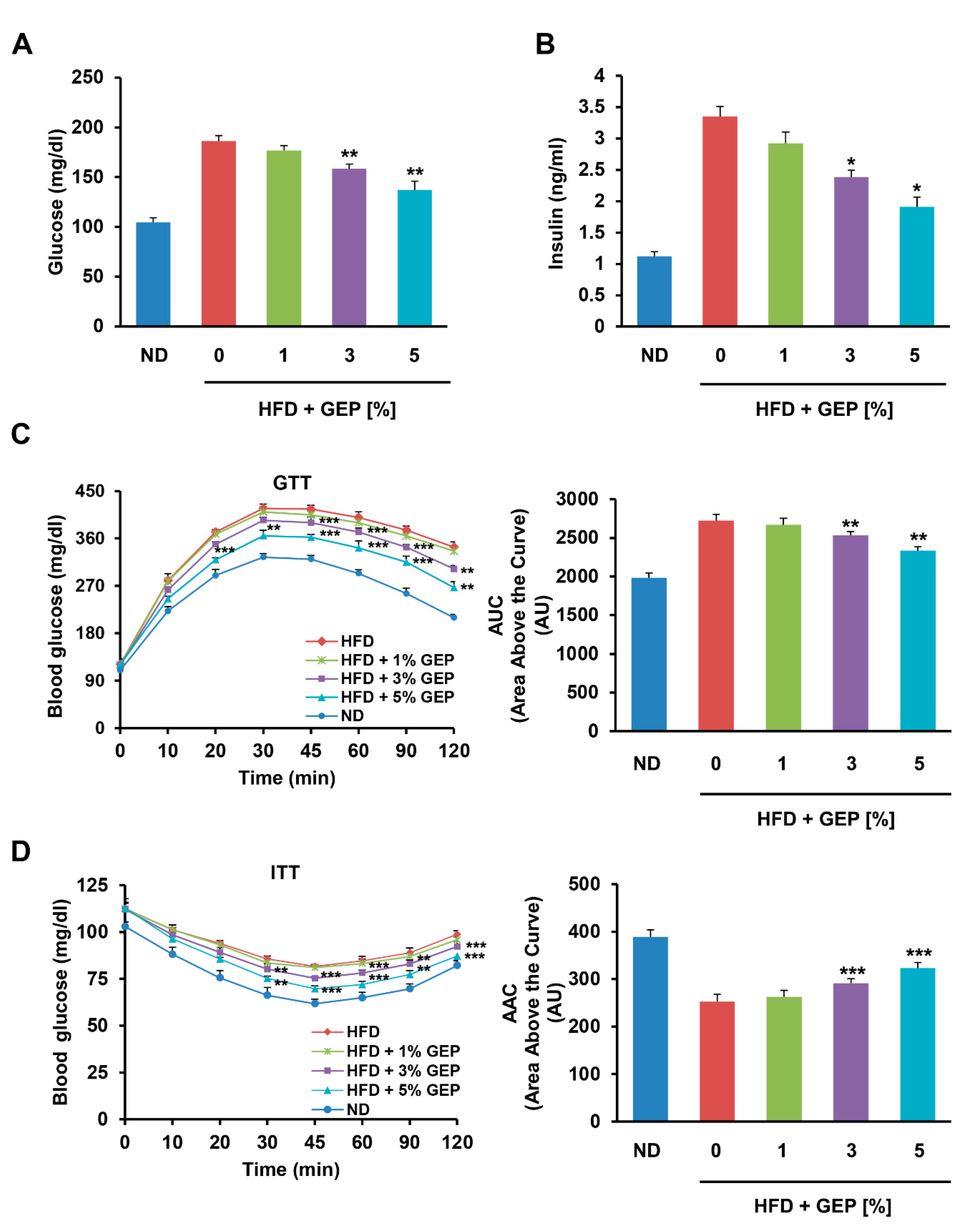
3.4. Effect of G. Lucidum Extract on Glucose Uptake, Insulin Tolerance, and Glucose Tolerance
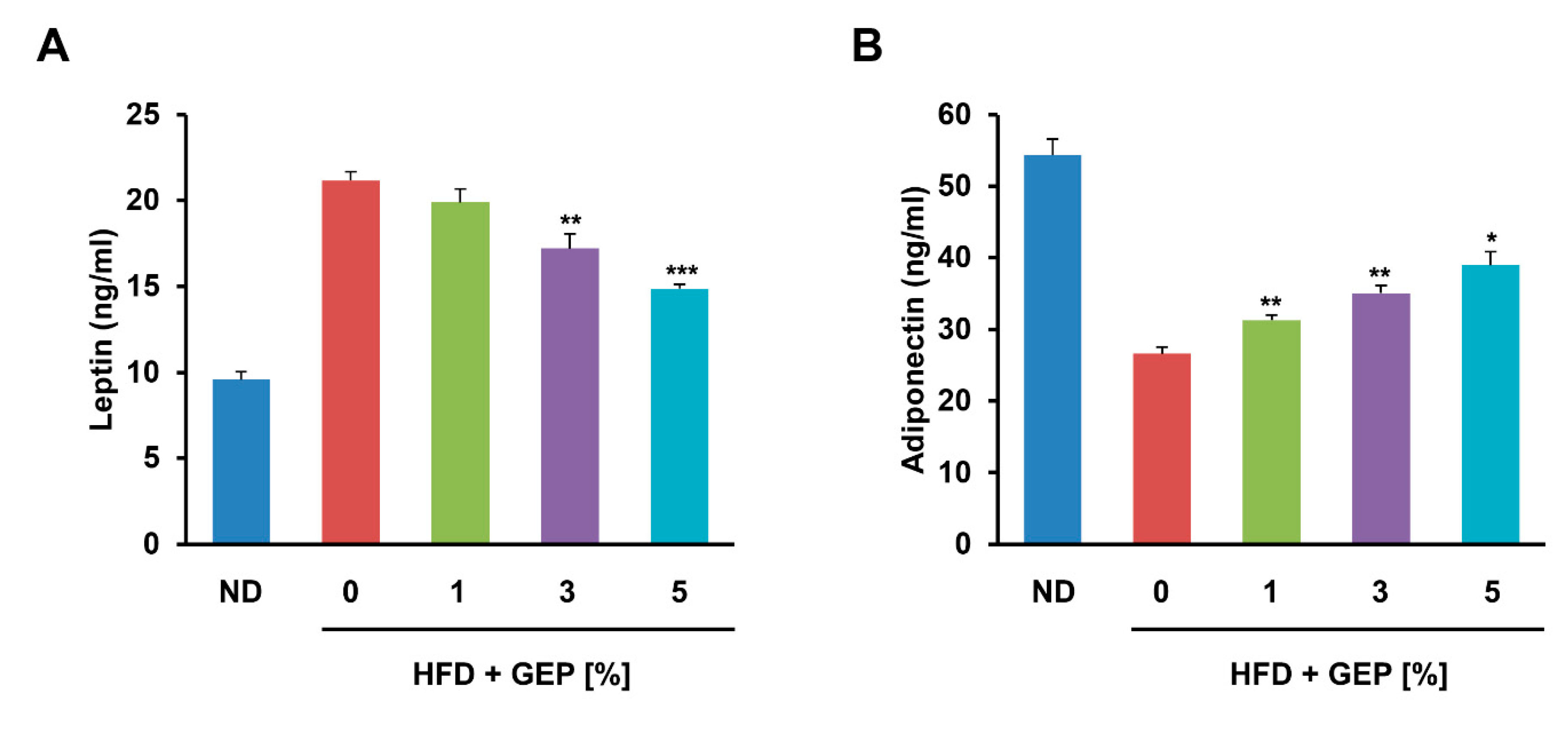
3.5. Effect of G. Lucidum on Adiponectin and Leptin Levels
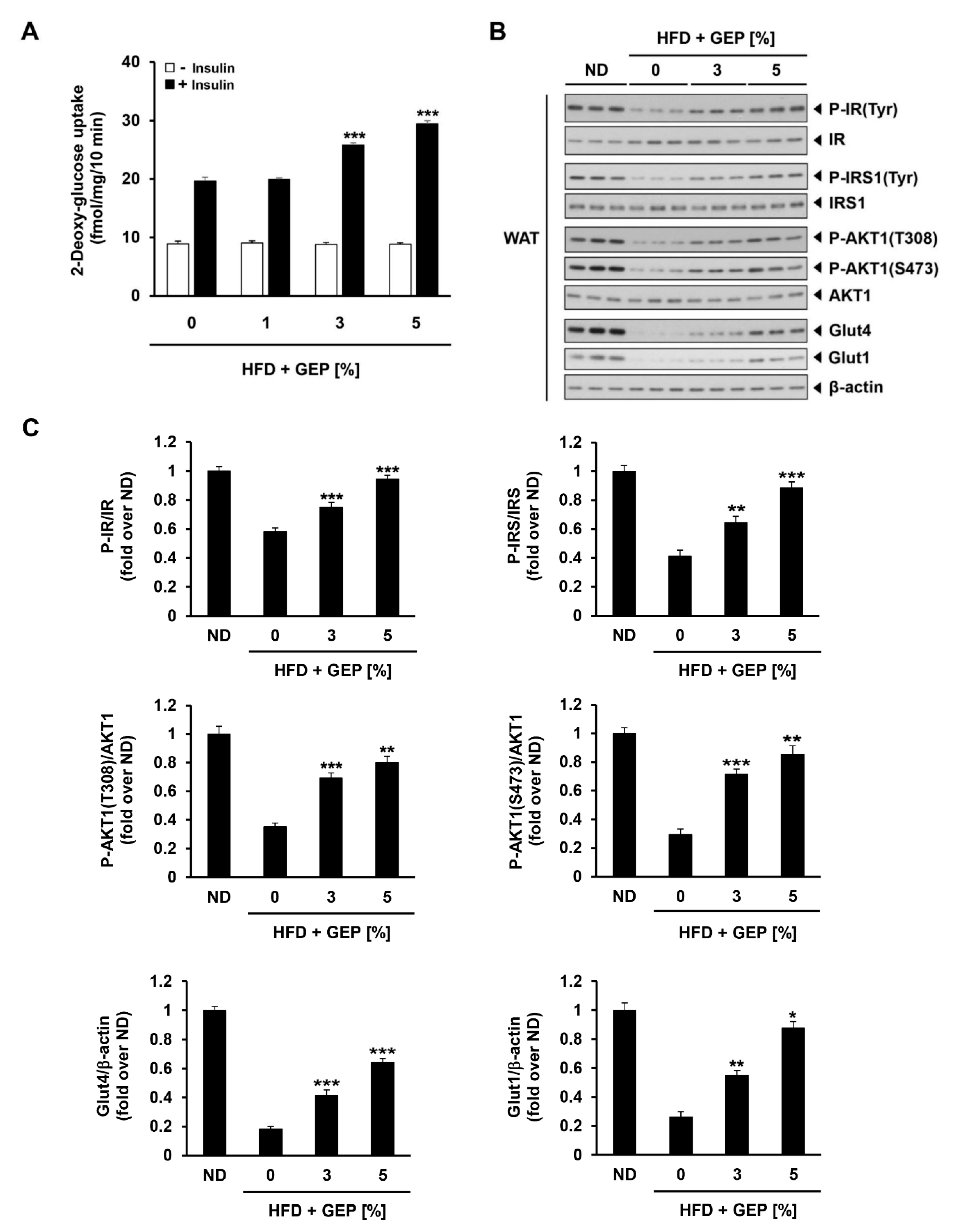
3.6. Effect of G. Lucidum on Glucose Uptake and Glucose Metabolism-Related Proteins
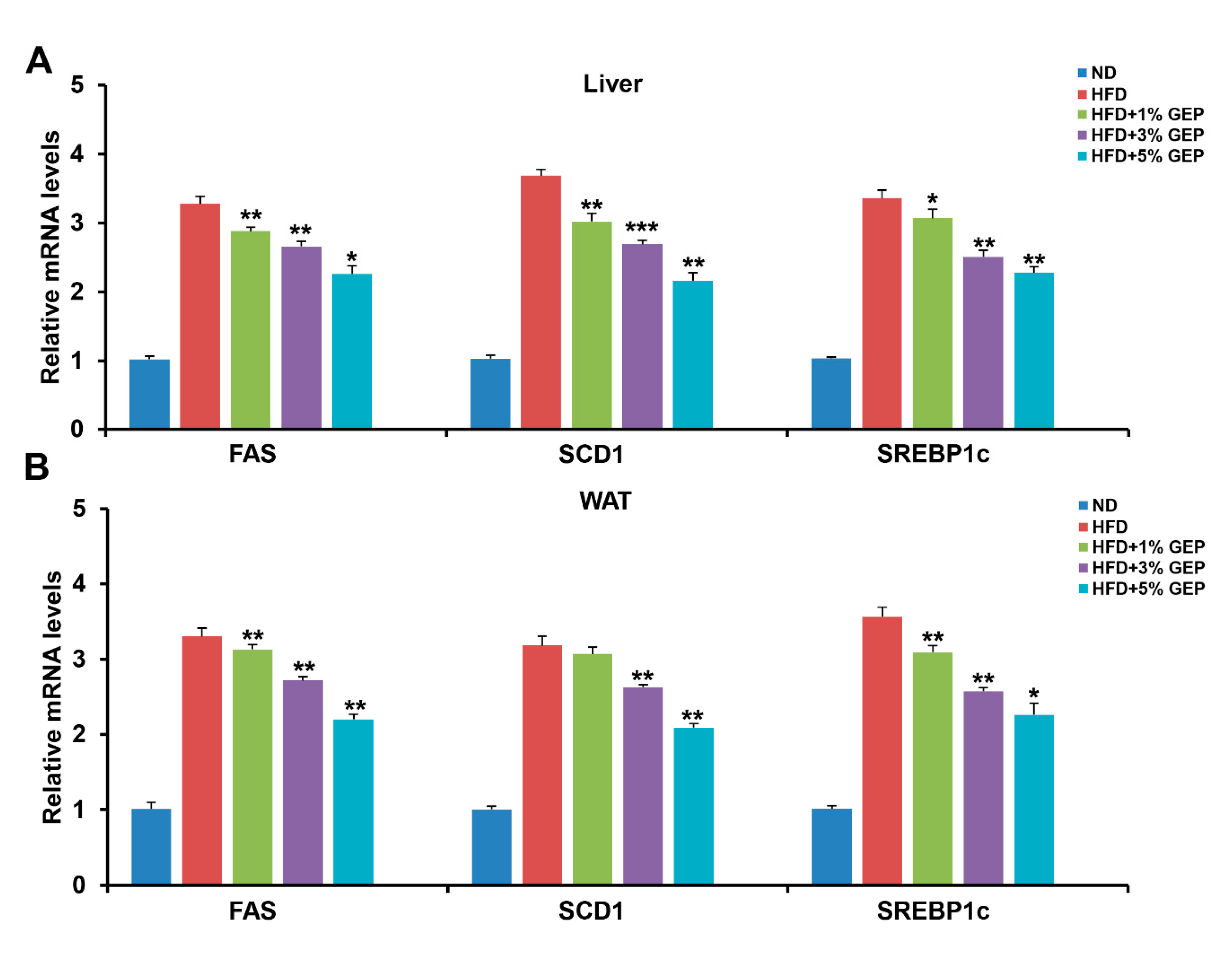
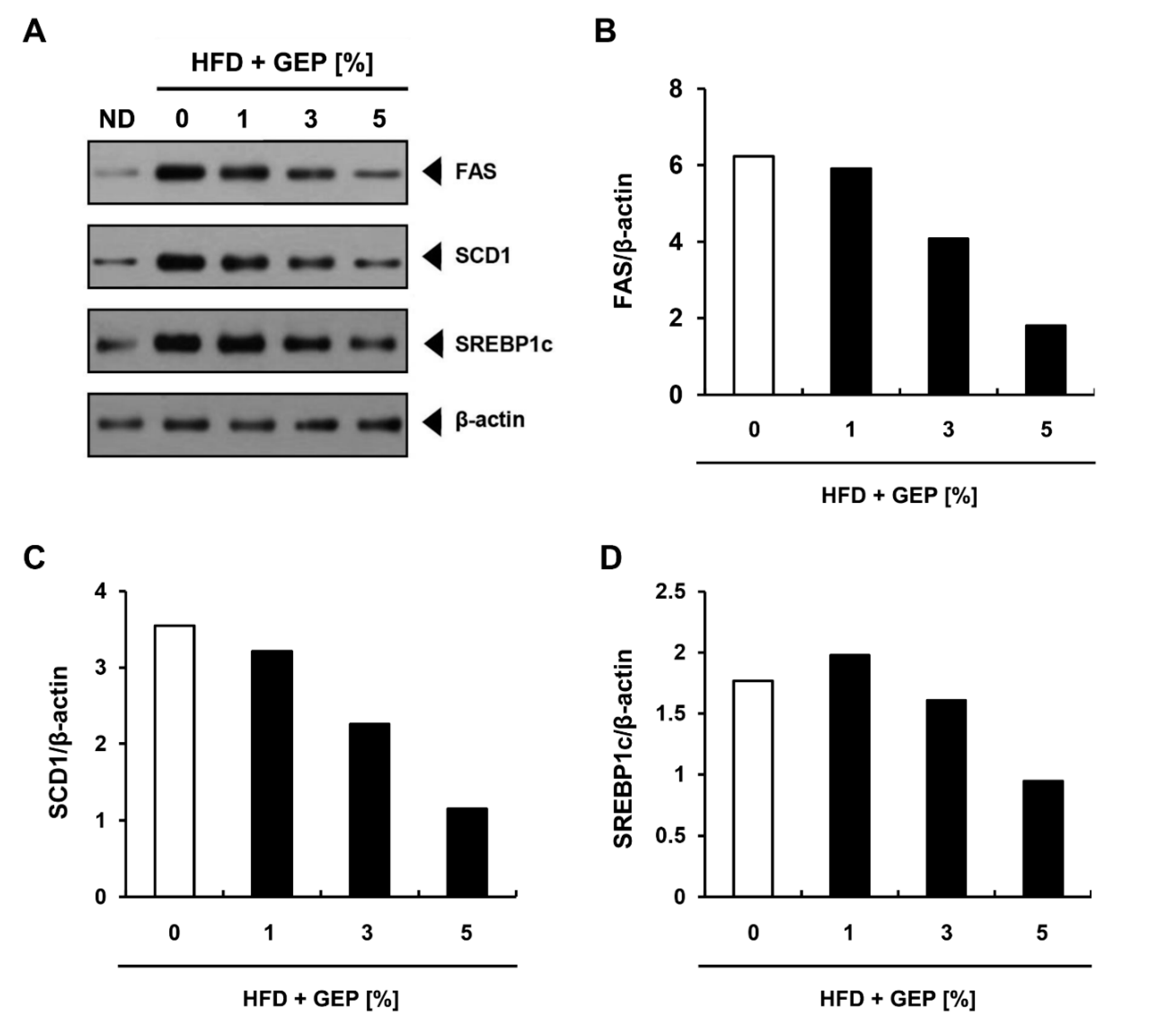
3.7. Effect of G. Lucidum Extract on Lipogenesis-Related Genes and Proteins in WAT and the Liver
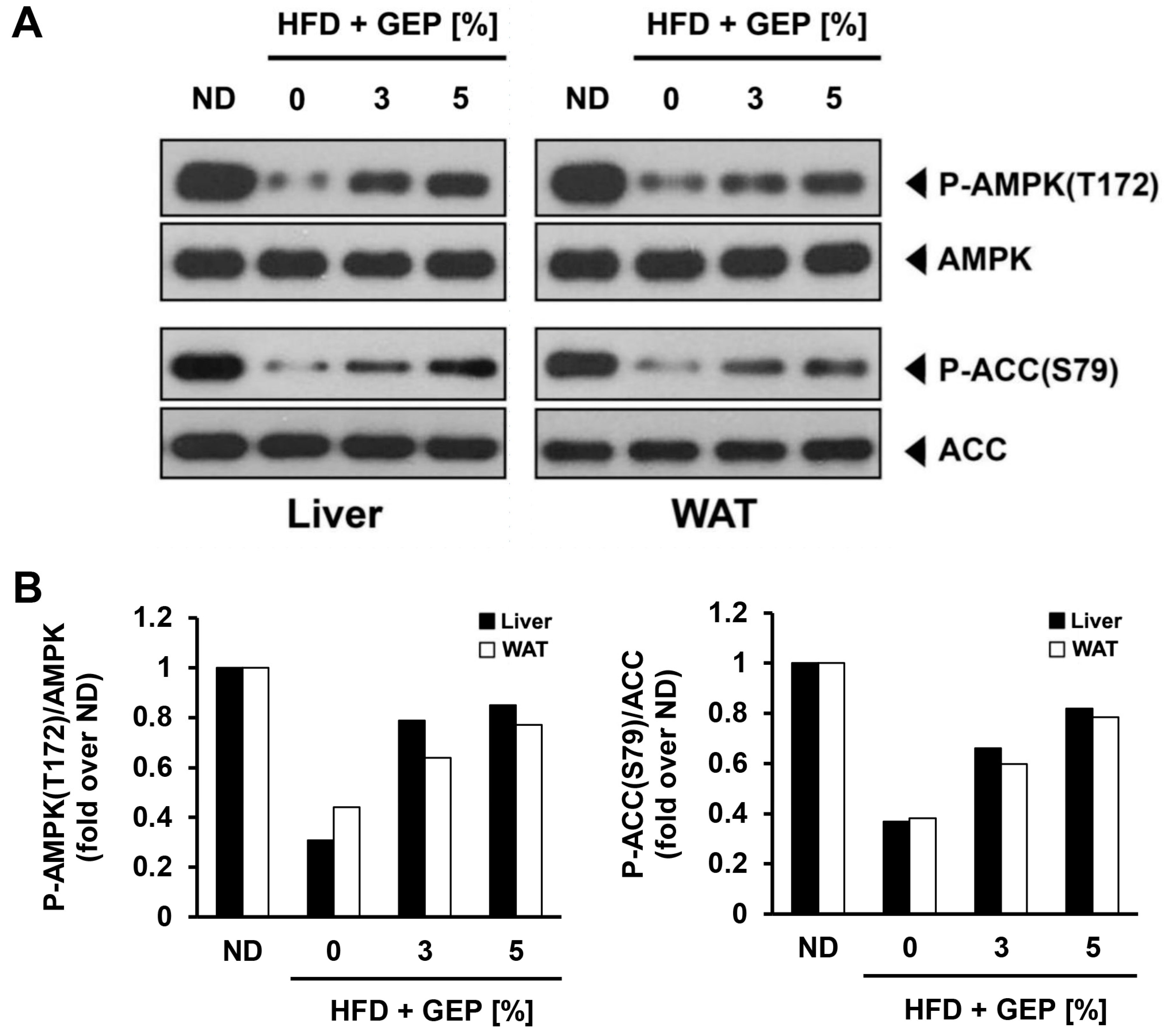
3.8. Effect of G. Lucidum Extract on AMPK and ACC
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 18s rRNA | 18s ribosomal RNA |
| AAC | Area above the curve |
| ACC | Acetyl-CoA carboxylase |
| AKT1 | AKT serine/threonine kinase 1 |
| AMPK | 5′ AMP-activated protein kinase |
| AS160 | Akt substrate of 160 kDa |
| AUC | Area under the curve |
| C/EBPα | CCAAT-enhancer-binding protein α |
| CPT1 | Carnitine palmitoyltransferase I |
| DAG | Diacylglycerol |
| FAS | Fatty acid synthase |
| FER | Feeding efficiency ratio |
| FFA | Free fatty acid |
| G6Pase | Glucose 6-phosphatase |
| GEP | Ganoderma lucidum extract powder |
| GLUT1 | Glucose transporter type 1 |
| GLUT4 | Glucose transporter type 4 |
| GTT | Glucose tolerance test |
| HDL | High-density lipoprotein-cholesterol |
| HFD | High-fat diet |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| IR | Insulin receptor |
| IRS1 | Insulin receptor substrate 1 |
| ITT | Insulin tolerance test |
| LDL | Low-density lipoprotein-cholesterol |
| mTOR | Mammalian target of rapamycin |
| ND | Normal diet |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PI3K | Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptors γ |
| PTP1B | Protein tyrosine phosphatase 1B |
| S6K1 | Ribosomal protein S6 kinase beta-1 |
| SCD 1 | Stearoyl-CoA desaturase 1 |
| SD | Standard deviation |
| SREBP1c | Sterol regulatory element-binding protein-1c |
| TBC1D4 | TBC1 Domain Family Member 4 |
| TC | Total cholesterol |
| TG | Triglyceride |
| TORC2 | mTOR Complex 2 |
| WAT | White adipose tissue |
References
- Tremblay, A.; Bouchard, L.; Bouchard, C.; Després, J.P.; Drapeau, V.; Pérusse, L. Long-term adiposity changes are related to a glucocorticoid receptor polymorphism in young females. J. Clin. Endocrinol. Metab. 2003, 88, 3141–3145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Grune, T.; Speckmann, B. The two faces of reactive oxygen species (ROS) in adipocyte function and dysfunction. Biol. Chem. 2016, 397, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Ryu, R.; Kwon, E.Y.; Choi, J.Y.; Shon, J.C.; Liu, K.H.; Choi, M.S. Chrysanthemum leaf ethanol extract prevents obesity and metabolic disease in diet-induced obese mice via lipid mobilization in white adipose tissue. Nutrients 2019, 11, 1347. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Viollet, B.; Lantier, L.; Devin-Leclerc, J.; Hebrard, S.; Amouyal, C.; Mounier, R.; Marc, F.; Fabrizio, A. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front. Biosci. 2009, 3380–3400. [Google Scholar] [CrossRef]
- Thyagarajan-sahu, A.; Lane, B.; Sliva, D. ReishiMax, mushroom based dietary supplement, inhibits adipocyte differentiation, stimulates glucose uptake and activates AMPK. BMC Complement. Altern. Med. 2011, 11, 74. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Wang, Q.; Yang, Y. Crude extracts from Lycium barbarum suppress SREBP-1c expression and prevent diet-induced fatty liver through AMPK activation. Biomed Res. Int. 2014, 2014, 196198. [Google Scholar] [CrossRef]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting-and cold-induced appetite but not thermogenesis. eLife 2018, 7, e32656. [Google Scholar] [CrossRef] [PubMed]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Seto, S.W.; Lam, T.Y.; Tam, H.L.; Au, A.L.S.; Chan, S.W.; Wu, J.H.; Yu, P.H.F.; Leung, G.P.H.; Ngai, S.M.; Yeung, J.H.K.; et al. Novel hypoglycemic effects of Ganoderma lucidum water-extract in obese/diabetic (+db/+db) mice. Phytomedicine 2009, 16, 426–436. [Google Scholar] [CrossRef]
- Taylor, R. Insulin resistance and type 2 diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef]
- Lyu, K.; Zhang, D.; Nozaki, Y.; Zhang, Y.; Bhanot, S.; Cline, G.; Samuel, V.; Shulman, G.I. Membrane sn-1,2 Diacylglycerol Mediates Lipid-Induced Hepatic Insulin Resistance In Vivo. Diabetes 2018, 67, 243. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Sanodiya, B.; Thakur, G.; Baghel, R.; Prasad, G.; Bisen, P. Ganoderma lucidum: A Potent Pharmacological Macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-L.; Liu, B.-Y.; Ma, K.-W. Traditional Chinese medicine. Lancet 2008, 372, 1938–1940. [Google Scholar] [CrossRef]
- Teng, B.S.; Wang, C.D.; Zhang, D.; Wu, J.S.; Pan, D.; Pan, L.F.; Yang, H.J.; Zhou, P. Hypoglycemic effect and mechanism of a proteoglycan from ganoderma lucidum on streptozotocin-induced type 2 diabetic rats. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 166–175. [Google Scholar]
- Xiao, C.; Wu, Q.; Zhang, J.; Xie, Y.; Cai, W.; Tan, J. Antidiabetic activity of Ganoderma lucidum polysaccharides F31 down-regulated hepatic glucose regulatory enzymes in diabetic mice. J. Ethnopharmacol. 2017, 196, 47–57. [Google Scholar] [CrossRef]
- Chang, C.; Lin, C.; Lu, C.; Martel, J.; Ko, Y.; Ojcius, D.M.; Tseng, S.; Wu, T.; Chen, Y.M.; Young, J.D.; et al. Ganoderma lucidum Reduces Obesity in Mice by Modulating the Composition of the Gut Microbiota. Nat. Commun. 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hirotani, M.; Ino, C.; Furuya, T.; Shiro, M. Ganoderic acids T, S and R, new triterpenoids from the cultured mycelia of ganoderma lucidum. Chem. Pharm. Bull. 1986, 34, 2282–2285. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, G.; Dai, X.; Ye, J.; Zhou, S. A Phase I/II Study of Ling Zhi Mushroom Ganoderma lucidum (W.Curt.:Fr.) Lloyd (Aphyllophoromycetideae) Extract in Patients with Coronary Heart Disease. Int. J. Med. Mushrooms 2004, 6, 327–334. [Google Scholar] [CrossRef]
- Lin, Z.-B.; Wang, M.-Y.; Liu, Q.; Che, Q.-M. Effects of Total Triterpenoids Extract from Ganoderma lucidum (Curt.: Fr.) P. Karst. (Reishi Mushroom) on Experimental Liver Injury Models Induced by Carbon Tetrachloride or D-Galactosamine in Mice. Int. J. Med. Mushrooms 2002, 4, 6. [Google Scholar] [CrossRef]
- Chen, R.Y.; Yu, D.Q. Studies on the triterpenoid constituents of the spores from Ganoderma lucidum karst. Yao Xue Xue Bao 1991, 26, 267–273. [Google Scholar] [CrossRef]
- Wasser, S.P. Current findings, future trends, and unsolved problems in studies of medicinal mushrooms. Appl. Microbiol. Biotechnol. 2011, 89, 1323–1332. [Google Scholar] [CrossRef]
- Li, B.; Lee, D.S.; Kang, Y.; Yao, N.Q.; An, R.B.; Kim, Y.C. Protective effect of ganodermanondiol isolated from the Lingzhi mushroom against tert-butyl hydroperoxide-induced hepatotoxicity through Nrf2-mediated antioxidant enzymes. Food Chem. Toxicol. 2013, 53, 317–324. [Google Scholar] [CrossRef]
- Wińska, K.; MacZka, W.; Gabryelska, K.; Grabarczyk, M. Mushrooms of the genus ganoderma used to treat diabetes and insulin resistance. Molecules 2019, 24, 4075. [Google Scholar] [CrossRef]
- Seong, H.A.; Ha, H. Thr55 phosphorylation of p21 by MPK38/MELK ameliorates defects in glucose, lipid, and energy metabolism in diet-induced obese mice. Cell Death Dis. 2019, 10, 380. [Google Scholar] [CrossRef]
- Jung, S.; Son, H.; Hwang, C.; Cho, K.; Park, S.; Kim, H. Ganoderma lucidum Ameliorates Non-Alcoholic Steatosis by Upregulating Energy Metabolizing Enzymes in the Liver. J. Clin. Med. 2018, 7, 152. [Google Scholar] [CrossRef]
- Meneses, M.E.; Martínez-Carrera, D.; Torres, N.; Sánchez-Tapia, M.; Aguilar-López, M.; Morales, P.; Sobal, M.; Bernabé, T.; Escudero, H.; Granados-Portillo, O.; et al. Hypocholesterolemic properties and prebiotic effects of Mexican Ganoderma lucidum in C57BL/6 mice. PLoS ONE 2016, 11, e0159631. [Google Scholar] [CrossRef] [PubMed]
- Bowe, J.E.; Franklin, Z.J.; Hauge-Evans, A.C.; King, A.J.; Persaud, S.J.; Jones, P.M. Metabolic phenotyping guidelines: Assessing glucose homeostasis in rodent models. J. Endocrinol. 2014, 222, G13–G25. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Lin, C.S.; Lu, C.C.; Martel, J.; Ko, Y.F.; Ojcius, D.M.; Tseng, S.F.; Wu, T.R.; Chen, Y.Y.M.; Young, J.D.; et al. Corrigendum: Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 2017, 8, 16130. [Google Scholar] [CrossRef] [PubMed]
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed]
- Mîinea, C.P.; Sano, H.; Kane, S.; Sano, E.; Fukuda, M.; Peränen, J.; Lane, W.S.; Lienhard, G.E. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 2005, 391, 87–93. [Google Scholar] [CrossRef]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and Indirect Mechanisms for Regulation of Fatty Acid Synthase Gene Expression by Liver X Receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef]
- Payne, V.; Au, W.; Lowe, C.; Rahman, S.; Friedman, J.; O’rahilly, S.; Rochford, J.J. C/EBP transcription factors regulate SREBP1c gene expression during adipogenesis. Biochem. J. 2010, 425, 215–223. [Google Scholar] [CrossRef]
- Beech, I.; Bergel, A.; Mollica, A.; Flemming, H.; Scotto, V.; Sand, W. Simple methods for the investigation of of the role of biofilms in corrosion. Brite Euram Themat. Netw. MIC Ind. Mater. 2000, 1–27. [Google Scholar]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Singla, P. Metabolic effects of obesity: A review. World J. Diabetes 2010, 1, 76. [Google Scholar] [CrossRef]
- Roland, A.V.; Moenter, S.M. Regulation of gonadotropin-releasing hormone neurons by glucose. Trends Endocrinol. Metab. 2011, 22, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 2003, 111, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Haluzík, M.M.; Haluzík, M. PPAR-alpha and insulin sensitivity. Physiol. Res. 2006, 55, 115–122. [Google Scholar] [PubMed]
- Viollet, B.; Andreelli, F. AMP-Activated protein kinase and metabolic control. Handb. Exp. Pharmacol. 2015, 203, 303–330. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 7th ed.; W.H. Freeman: Basingstoke, UK, 2012; pp. 634–635. ISBN 13: 978-1429276351. [Google Scholar]
- Ruderman, N.; Prentki, M. AMP kinase and malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat. Rev. Drug Discov. 2004, 3, 340–351. [Google Scholar] [CrossRef]
- Long, S.D.; Pekala, P.H. Lipid mediators of insulin resistance: Ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem. J. 1996, 319, 179–184. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef]
- Mukherjee, B.; Hossain, C.M.; Mondal, L.; Paul, P.; Ghosh, M.K. Obesity and insulin resistance: An abridged molecular ccorrelation. Lipid Insights 2013, 6, 1–11. [Google Scholar] [CrossRef]
- Pan, D.; Zhang, D.; Wu, J.; Chen, C.; Xu, Z.; Yang, H.; Zhou, P. Antidiabetic, antihyperlipidemic and antioxidant activities of a novel proteoglycan from ganoderma lucidum fruiting bodies on db/db mice and the possible mechanism. PLoS ONE 2013, 8, e68332. [Google Scholar] [CrossRef]
- Guo, W.L.; Guo, J.B.; Liu, B.Y.; Lu, J.Q.; Chen, M.; Liu, B.; Bai, W.D.; Rao, P.F.; Ni, L.; Lv, X.C. Ganoderic acid A from Ganoderma lucidum ameliorates lipid metabolism and alters gut microbiota composition in hyperlipidemic mice fed a high-fat diet. Food Funct. 2020, 11, 6818–6833. [Google Scholar] [CrossRef]
- Wu, Y.S.; Ho, S.Y.; Nan, F.H.; Chen, S.N. Ganoderma lucidum beta 1,3/1,6 glucan as an immunomodulator in inflammation induced by a high-cholesterol diet. BMC Complement. Altern. Med. 2016, 16, 500. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Contents (%) |
|---|---|
| Ganoderic Acid | |
| A | 2.74 |
| B | 3.68 |
| C1 | 0.69 |
| C2 | 0.17 |
| C6 | 10.39 |
| D | 0.95 |
| F | 1.09 |
| G | 2.93 |
| H | 6.98 |
| I | 1.1 |
| K | 2.3 |
| L | 0.57 |
| LM2 | 5.43 |
| M | 12 |
| N | 1.15 |
| T-Q | 1.1 |
| Total ganoderic acid | 53.27 |
| Glucan | |
| α-glucan | 3.40 ± 0.19 |
| β-glucan | 23.93 ± 1.51 |
| Total glucan | 27.32 ± 1.70 |
| Ingredient (g/kg) | Experimental Group | ||||
|---|---|---|---|---|---|
| ND | HFD + GEP [%] | ||||
| 0 | 1 | 3 | 5 | ||
| Powdered GEP | 0 | 0 | 10 | 30 | 50 |
| Casein | 200 | 200 | 200 | 200 | 200 |
| Corn starch | 457 | 260 | 250 | 230 | 210 |
| Sucrose | 200 | 200 | 200 | 200 | 200 |
| Cellulose | 50 | 50 | 50 | 50 | 50 |
| Soybean oil | 43 | 25 | 25 | 25 | 25 |
| L-Cysteine | 3 | 3 | 3 | 3 | 3 |
| Choline bitartrate | 2 | 2 | 2 | 2 | 2 |
| Lard | 0 | 215 | 215 | 215 | 215 |
| Mineral mix | 35 | 35 | 35 | 35 | 35 |
| Vitamin mix | 10 | 10 | 10 | 10 | 10 |
| Total grams (g) | 1000 | 1000 | 1000 | 1000 | 1000 |
| Calories from fat (%) | 10 | 45 | 45 | 45 | 45 |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| FAS | TGCTCCAGGGATAACAGC | CCAAATCCAACATGGGACA |
| SCD1 | ACCTGCCTCTTCGGGATTTT | GTCGGCGTGTGTTTCTGAGA |
| SREBP1c | AGCTGCGTGGTTTCCAACA | CCTCATGTAGGAATACCCTCCTCAT |
| 18s rRNA | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Group | Subcutaneous WAT (g) | Epididymal WAT (g) | Mesenteric WAT (g) |
|---|---|---|---|
| ND | 0.99 ± 0.05 | 0.8 ± 0.12 | 0.17 ± 0.05 |
| HFD | 3.4 ± 0.15 | 1.81 ± 0.04 | 0.62 ± 0.07 |
| HFD + 1% GEP | 3.07 ± 0.18 | 1.75 ± 0.04 | 0.43 ± 0.11 |
| HFD + 3% GEP | 2.77 ± 0.12 | 1.53 ± 0.06 | 0.17 ± 0.12 |
| HFD + 5% GEP | 2.29 ± 0.09 | 1.31 ± 0.11 | 0.06 ± 0.03 |
| Group. | TG (mg/dL) | TC (mg/dL) | HDL (mg/dL) | LDL (mg/dL) | FFA (mEq/dL) |
|---|---|---|---|---|---|
| ND | 73.78 ± 1.5 | 110.72 ± 2.79 | 97.29 ± 3.83 | 35.92 ± 2.96 | 0.98 ± 0.04 |
| HFD | 123.04 ± 3.12 | 248.63 ± 8.19 | 160.65 ± 3.42 | 80.65 ± 3.00 | 1.98 ± 0.11 |
| HFD + 1% GEP | 118.91 ± 3.04 | 224.45 ± 6.83 | 150.14 ± 1.85 | 70.08 ± 1.83 | 1.93 ± 0.11 |
| HFD + 3% GEP | 104.09 ± 3.66 | 197.19 ± 8.61 | 139.23 ± 3.8 | 60.19 ± 3.71 | 1.66 ± 0.06 |
| HFD + 5% GEP | 88.83 ± 1.83 | 171.37 ± 9.62 | 119.38 ± 4.55 | 53.39 ± 3.74 | 1.39 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.A.; Cho, J.-H.; Afinanisa, Q.; An, G.-H.; Han, J.-G.; Kang, H.J.; Choi, S.H.; Seong, H.-A. Ganoderma lucidum Extract Reduces Insulin Resistance by Enhancing AMPK Activation in High-Fat Diet-Induced Obese Mice. Nutrients 2020, 12, 3338. https://doi.org/10.3390/nu12113338
Lee HA, Cho J-H, Afinanisa Q, An G-H, Han J-G, Kang HJ, Choi SH, Seong H-A. Ganoderma lucidum Extract Reduces Insulin Resistance by Enhancing AMPK Activation in High-Fat Diet-Induced Obese Mice. Nutrients. 2020; 12(11):3338. https://doi.org/10.3390/nu12113338
Chicago/Turabian StyleLee, Hyeon A, Jae-Han Cho, Qonita Afinanisa, Gi-Hong An, Jae-Gu Han, Hyo Jeung Kang, Seong Ho Choi, and Hyun-A Seong. 2020. "Ganoderma lucidum Extract Reduces Insulin Resistance by Enhancing AMPK Activation in High-Fat Diet-Induced Obese Mice" Nutrients 12, no. 11: 3338. https://doi.org/10.3390/nu12113338
APA StyleLee, H. A., Cho, J.-H., Afinanisa, Q., An, G.-H., Han, J.-G., Kang, H. J., Choi, S. H., & Seong, H.-A. (2020). Ganoderma lucidum Extract Reduces Insulin Resistance by Enhancing AMPK Activation in High-Fat Diet-Induced Obese Mice. Nutrients, 12(11), 3338. https://doi.org/10.3390/nu12113338