Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and Biochemical Parameters
2.2. Ultrasound Evaluation
- Normal liver echotexture (absence of steatosis).
- Mild hyperechoic liver tissue (compared with the adjacent kidney cortex) was considered as mild steatosis.
- Diffuse increase of echo intensity with decreased beam penetration (with slightly decreased visualization of the diaphragm) associated with a decrease in the visualization of silhouetting of the portal vein borders was considered as moderate steatosis.
- The marked increase in echoes intensity with no visualization of portal vein border, obscured diaphragm and posterior portion of the right lobe was considered as severe steatosis.
2.3. Assessment of Diet
2.4. Transferrin Isoelectric Focusing
2.5. ALDOB Molecular Analysis and Genotype-Phenotype Correlation
2.6. Statistical Analysis
3. Results
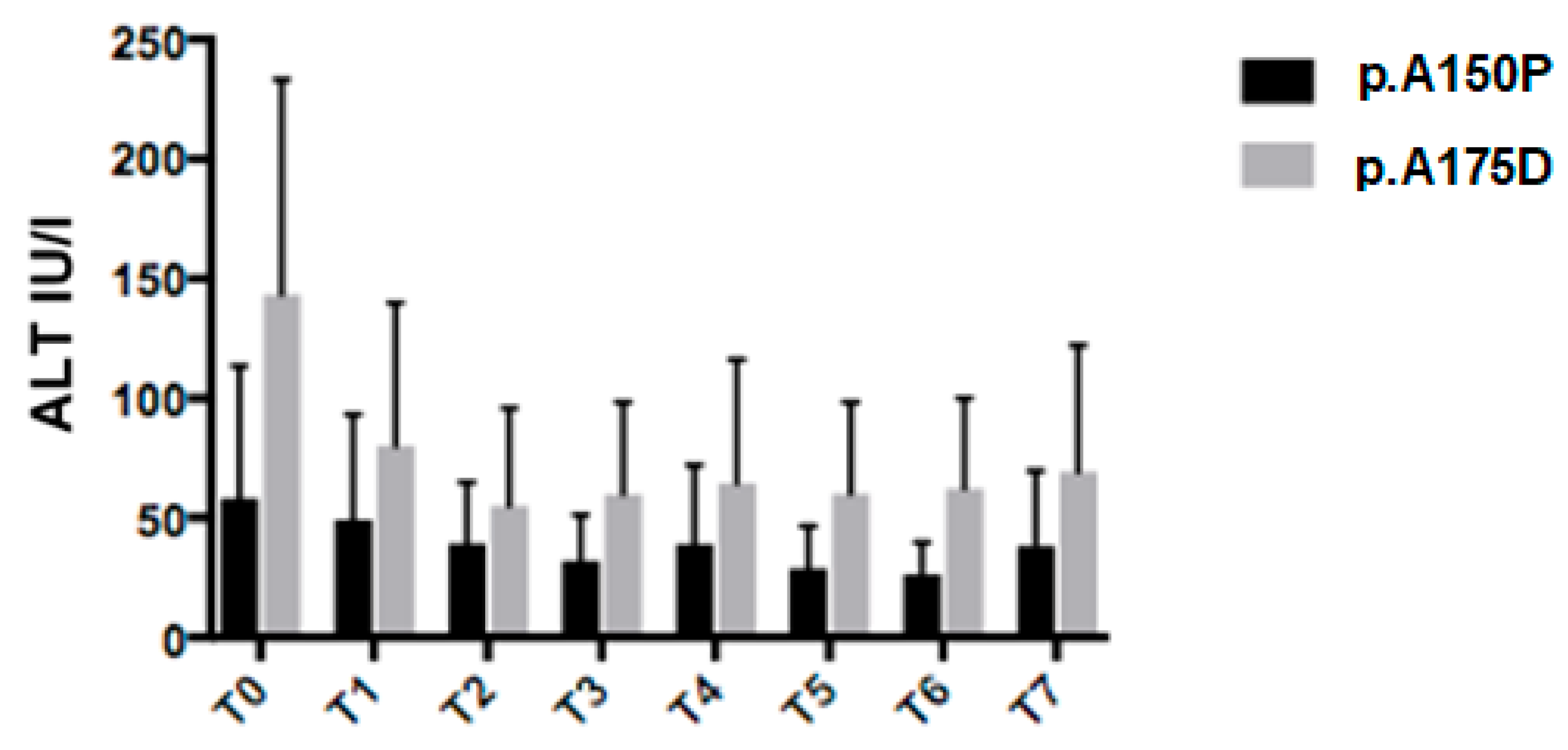
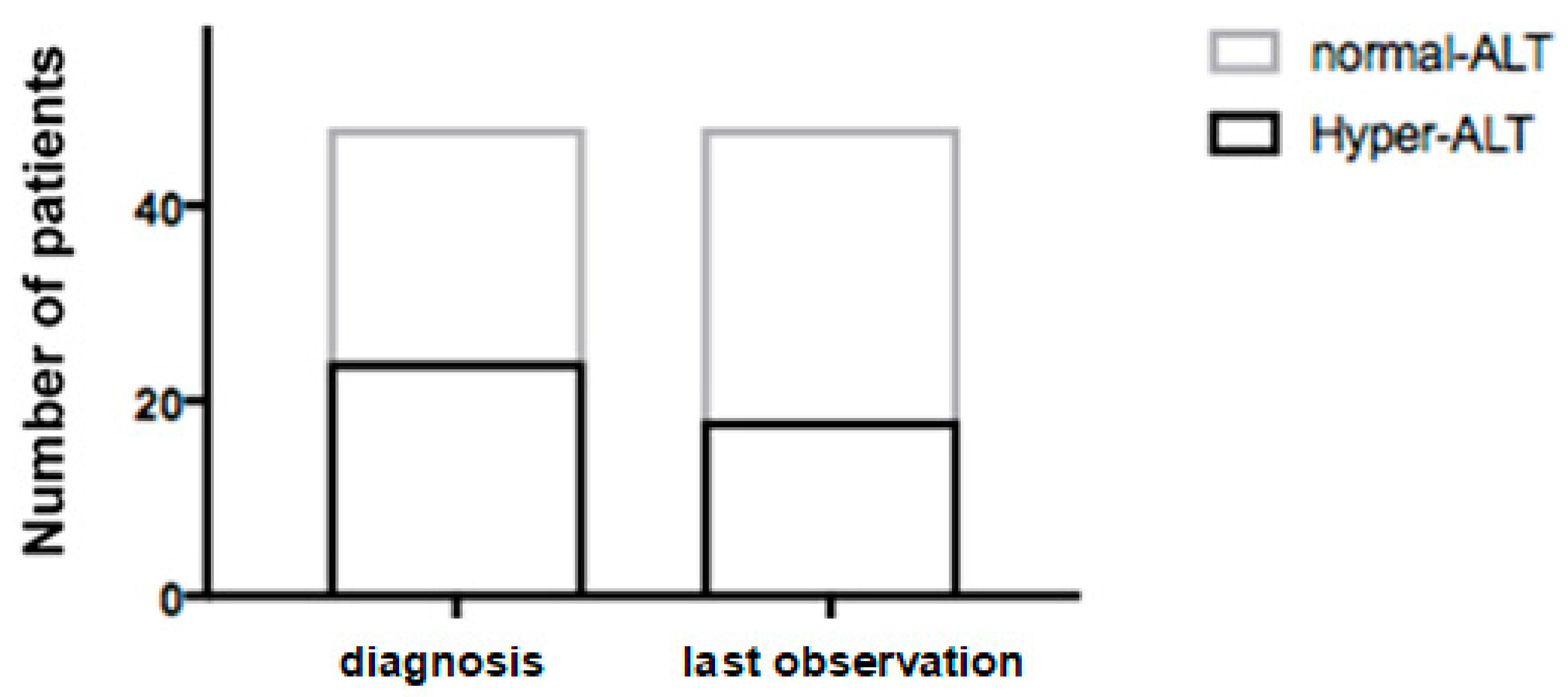
3.1. Clinical and Biochemical Parameters
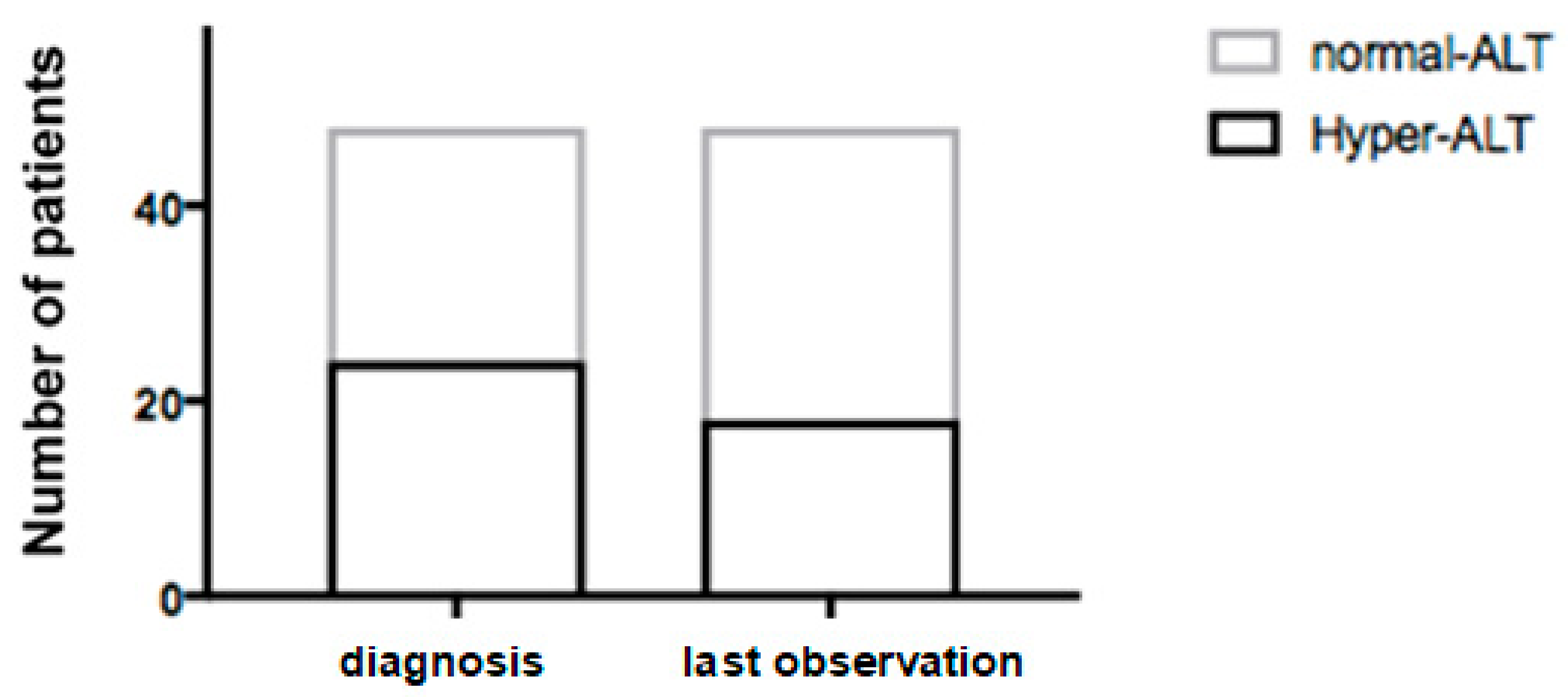
3.2. Ultrasound Evaluation
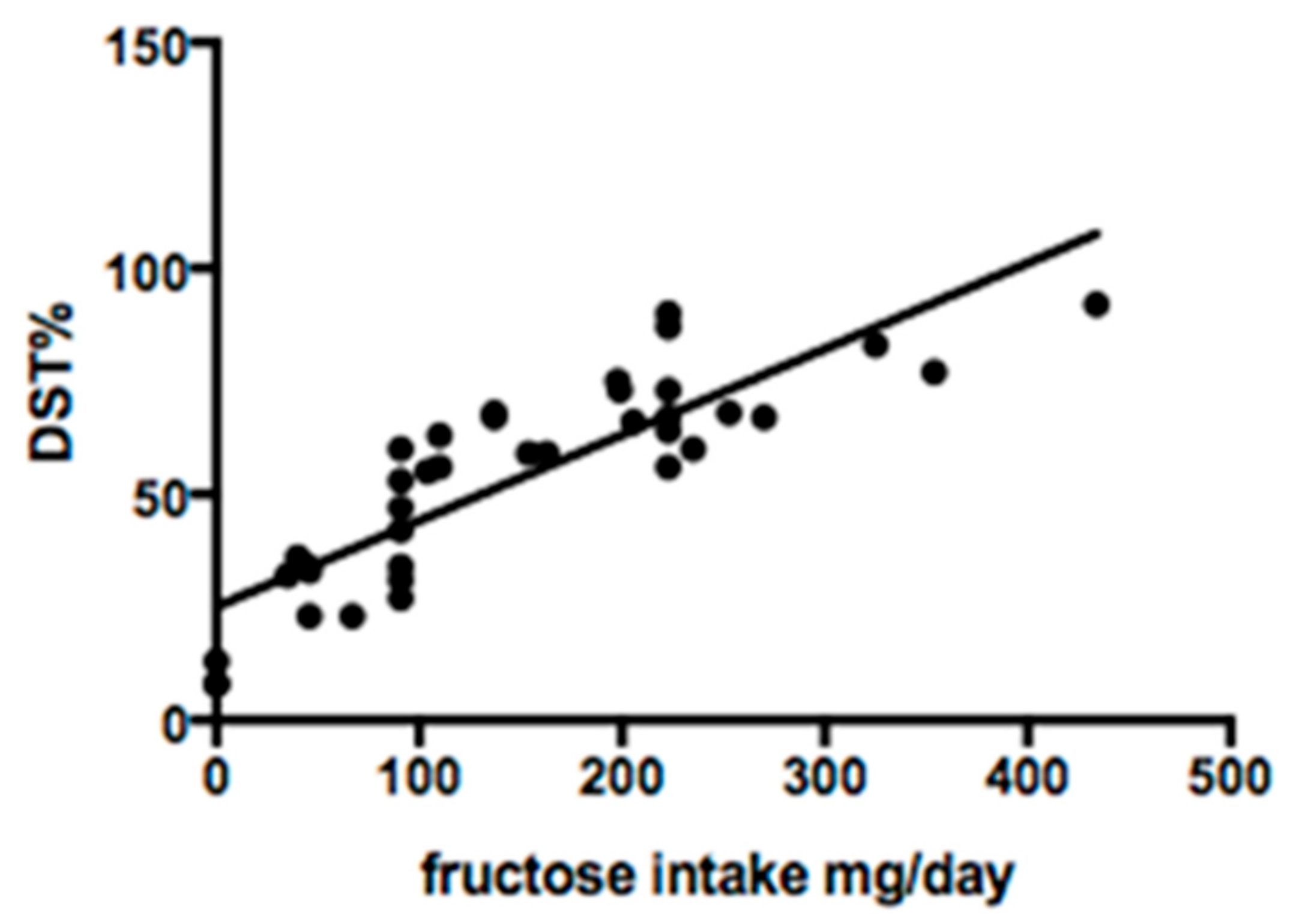
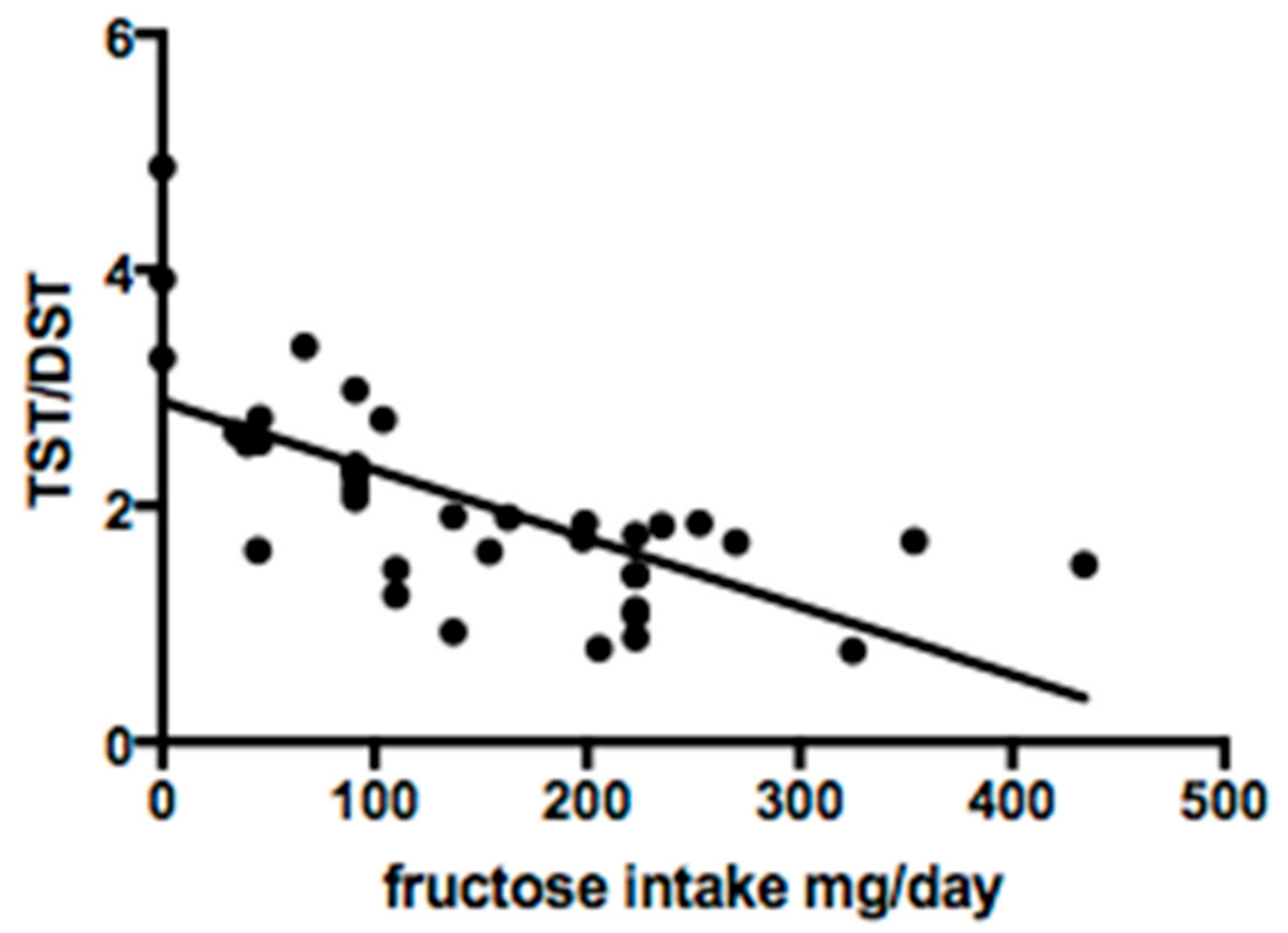
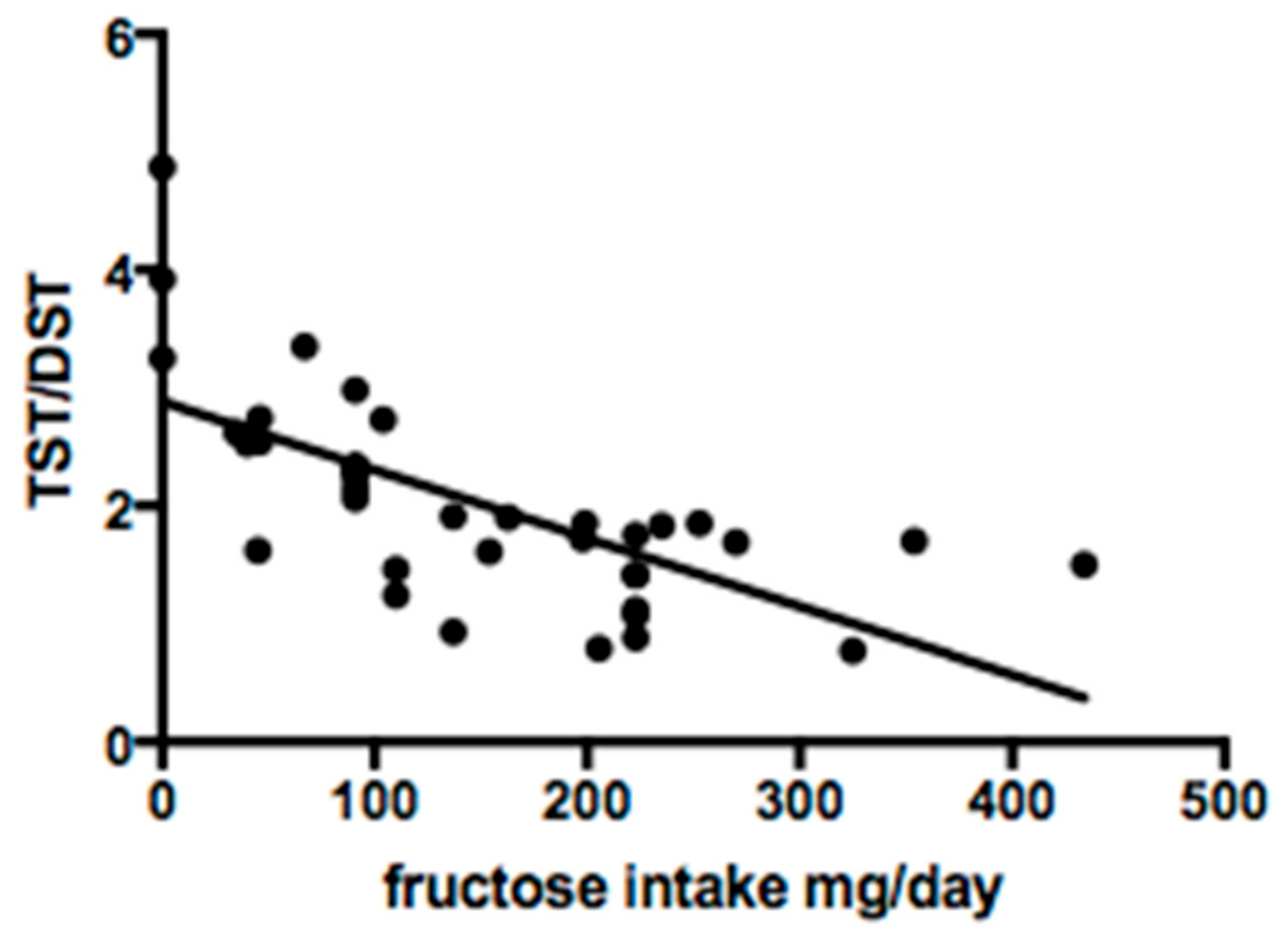
3.3. Fructose Intake
3.4. Serum Sialotransferrin Isoelectric Focusing
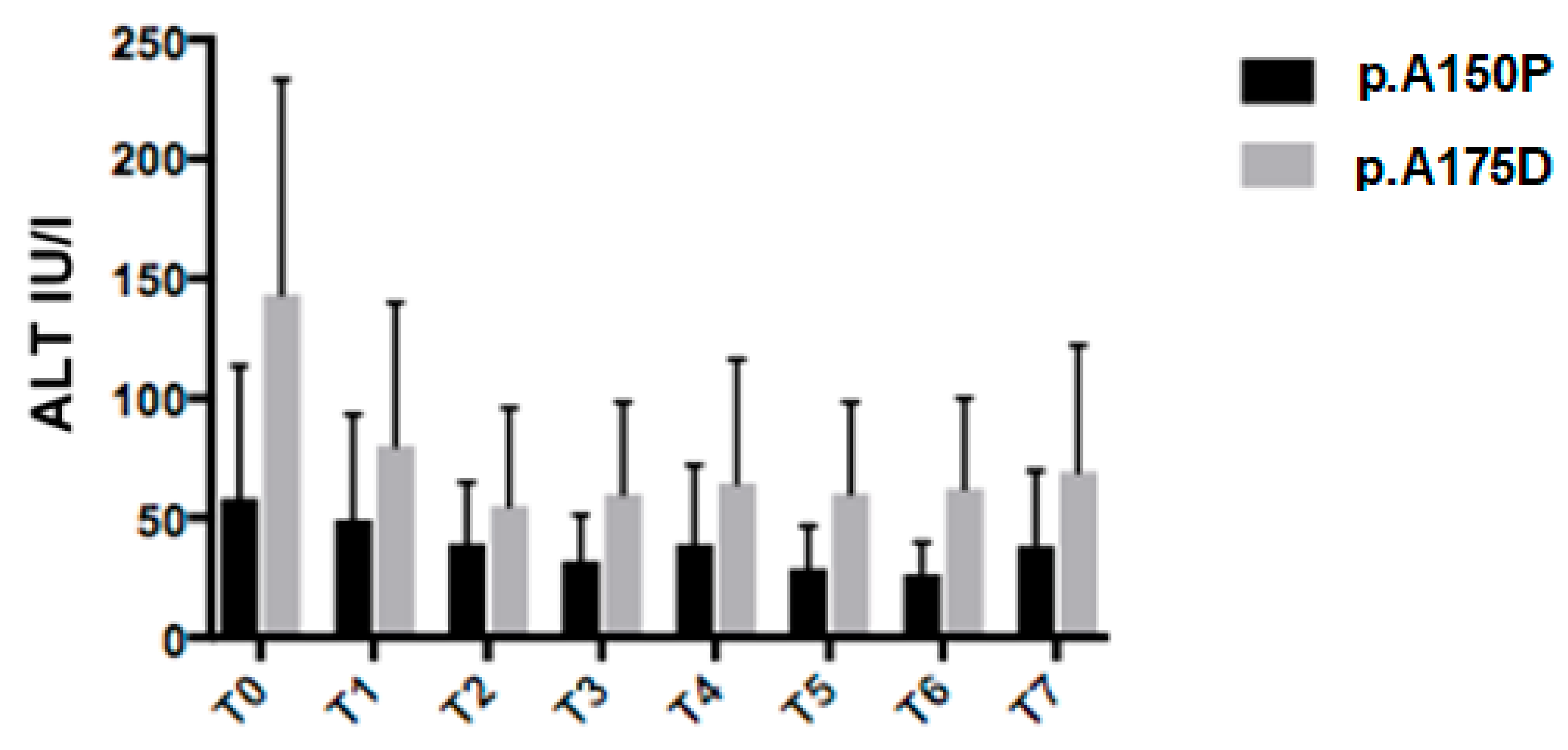
3.5. Genotype Analysis
3.6. Multivariate Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ali, M.; Rellos, P.; Cox, T.M. Hereditary fructose intolerance. J. Med. Genet. 1998, 35, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Odièvre, M.; Gentil, C.; Gautier, M.; Alagille, D. Hereditary fructose intolerance in childhood. Diagnosis, management, and course in 55 patients. Am. J. Dis. Child. 1978, 132, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Bouteldja, N.; Timson, D.J. The biochemical basis of hereditary fructose intolerance. J. Inherit. Metab. Dis. 2010, 33, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Mock, D.M.; Perman, J.A.; Thaler, M.; Morris, R.C. Chronic fructose intoxication after infancy in children with hereditary fructose intolerance. A cause of growth retardation. N. Engl. J. Med. 1983, 309, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; Sherwood, W.G. Current practices and improved recommendations for treating hereditary fructose intolerance. J. Am. Diet. Assoc. 1987, 87, 721–728. [Google Scholar] [PubMed]
- Adamowicz, M.; Pronicka, E. Carbohydrate deficient glycoprotein syndrome—Like transferrin isoelectric focusing pattern in untreated fructosaemia. Eur. J. Pediatr. 1996, 155, 347–348. [Google Scholar] [PubMed]
- Jaeken, J.; Pirard, M.; Adamowicz, M.; Pronicka, E.; van Schaftingen, E. Inhibition of phosphomannose isomerase by fructose 1-phosphate: An explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr. Res. 1996, 40, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Pronicka, E.; Adamowicz, M.; Kowalik, A.; Płoski, R.; Radomyska, B.; Rogaszewska, M.; Rokicki, D.; Sykut-Cegielska, J. Elevated carbohydrate-deficient transferrin (CDT) and its normalization on dietary treatment as a useful biochemical test for hereditary fructose intolerance and galactosemia. Pediatr. Res. 2007, 62, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Imperato, M.R.; Ieno, L.; Sorvillo, R.; Benigno, V.; Parenti, G.; Parini, R.; Vitagliano, L.; Zagari, A.; Salvatore, F. Hereditary fructose intolerance: Functional study of two novel ALDOB natural variants and characterization of a partial gene deletion. Hum. Mutat. 2010, 31, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Santamaria, R.; Vitagliano, L.; Ieno, L.; Viola, A.; Fiori, L.; Parenti, G.; Zancan, L.; Zagari, A.; Salvatore, F. Six novel alleles identified in Italian hereditary fructose intolerance patients enlarge the mutation spectrum of the aldolase B gene. Hum. Mutat. 2004, 24, 534. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Ardouin, S.; Burrows, T. The validity of dietary assessment methods to accurately measure energy intake in children and adolescents who are overweight or obese: A systematic review. Eur. J. Clin. Nutr. 2018, 72, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Sepe, A.; Giannattasio, A.; Cirillo, F.; Vegnente, A. Hypertransaminasemia in childhood as a marker of genetic liver disorders. J. Gastroenterol. 2005, 40, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Aldámiz-Echevarría, L.; de Las Heras, J.; Couce, M.L.; Alcalde, C.; Vitoria, I.; Bueno, M.; Blasco-Alonso, J.; Concepción García, M.; Ruiz, M.; Suárez, R.; et al. Non-Alcoholic fatty liver in hereditary fructose intolerance. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Odièvre, M.; Gautier, M.; Rieu, D. Hereditary fructose intolerance in young infants: Development of histologic hepatic lesions under prolonged dietetic therapy (8 cases). Arch. Fr. Pediatr. 1969, 26, 433–443. [Google Scholar]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzicky, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, G.; Nastasio, S.; Sciveres, M. Is liver steatosis diagnostic of non-alcoholic fatty liver disease in patients with hereditary fructose intolerance? Clin. Nutr. 2019, 38, 1960–1961. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; D’Ambrosi, M.; Marcellini, M.; Barbera, C.; Maggiore, G.; Zancan, L.; Giacchino, R.; Vajro, P.; Marazzi, M.G.; Francavilla, R.; et al. Persistence of elevated aminotransferases in Wilson’s disease despite adequate therapy. Hepatology 2004, 39, 1173–1174. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, B.; Gitzelmann, R.; Van Den Berghe, G. Disorders of Fructose Metabolism, 8th ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 1489–1520. [Google Scholar]
- Esposito, G.; Vitagliano, L.; Santamaria, R.; Viola, A.; Zagari, A.; Salvatore, F. Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance. FEBS Lett. 2002, 531, 152–156. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Sex | Age at Diagnosis (year) | Observation Period (year) | Symptoms at Onset | Liver Signs at Onset | Aldo B Mutation | Fructose Intake mg/day |
|---|---|---|---|---|---|---|---|
| 1 * | M | 0.3 | 4.9 | - | B | p.A175D/p.A175D | 0 |
| 2 | M | 0.9 | 7.6 | 1 | A-B | p.A175D/p.A175D | 0 |
| 3 | F | 0.6 | 21.1 | 2-4 | A-B | p.A175D/p.Y204 * | 165 |
| 4 | F | 1.1 | 14.9 | 1 | A-B | p.A175D/p.A175D | 223 |
| 5 | M | 0.8 | 9.7 | - | A-B | p.Y204 */p.Y204 * | 354 |
| 6* | M | 0.8 | 13.9 | 2-3-4 | A-B | p.A150P/p.A175D | 223 |
| 7 | M | 2 | 14.6 | 4 | A-B | p.A150P/p.A175D | 223 |
| 8 | F | 0.6 | 19.2 | 1 | A-B | p.A175DP/p.A175D | 137 |
| 9 | M | 1 | 14.2 | 2 | B | p.A150P/p.A150P | 67 |
| 10 | M | 0.9 | 6.6 | 2-4 | - | p.A150P/p.A175D | 35 |
| 11 | M | 0.9 | 10.5 | 1 | A-B | p.A150P/p.A175D | 723 |
| 12 | F | 1.5 | 11.9 | 2-4 | A | p.A150P/p.A150P | 223 |
| 13 | F | 2.2 | 11.7 | 1 | A-B | p.A150P/p.A150P | 223 |
| 14 * | F | 0 | 7.2 | - | A-B | p.A150P/p.A150P | 91 |
| 15 | M | 2.2 | 19.8 | 1 | A-B | p.N120Kfs */p.N120Kfs * | 91 |
| 16 | M | 1 | 4.3 | 1 | A-B | p.A175D/p.L289Ffs * | 45 |
| 17 | F | 3 | 16.5 | 1 | A-B | p.L257P/p.N335K | 40 |
| 18 | F | 18.5 | 4.5 | 1-4 | B | p.A150P/p.A150P | 104 |
| 19 * | M | 0 | 17 | 1-2-3 | B | p.A150P/p.A150P | 253 |
| 20 | F | 4 | 18.2 | 1 | - | p.A175D/-? | 270 |
| 21 | M | 1 | 6.6 | 1 | A-B | p.A150P/p.A150P | 160 |
| 22 | F | 3 | 13.2 | 1 | B | p.A175D/p.A175D | 163 |
| 23 * | F | 0 | 9.8 | - | B | p.Y204 */p.Y204 * | 154 |
| 24 | F | 0.5 | 16.3 | 1 | B | p.A175D/p.Y204 * | 109 |
| 25 * | M | 0 | 12.1 | - | - | p.A150P/p.A175D | 223 |
| 26 | M | 2.1 | 4.3 | 1 | A-B | p.A175D/p.A175D | 46 |
| 27 | F | 0.6 | 6.9 | 1-3 | A-B | p.A150P/p.A150P | 91 |
| 28 | M | 0.7 | 6.3 | 4 | A-B | p.A150P/p.Y204 * | 325 |
| 29 | F | 10.8 | 15.2 | - | B | p.A150P/p.Y204 * | 198 |
| 30 | F | 1 | 11.4 | 1 | B | p.A150P/p.A175D | 247 |
| 31 | F | 4.7 | 5.6 | 1-2 | - | p.A150P/p.A175D | 91 |
| 32 * | M | 0.2 | 8.6 | - | - | p.A150P/p.A175D | 91 |
| 33 | F | 0.5 | 4.2 | 2-4 | - | p.A150P/p.A175D | 0 |
| 34 | F | 0.3 | 6.7 | 2-4 | - | p.Y204 */p.N335K | 46 |
| 35 | F | 0.3 | 3.2 | 2-4 | B | p.A150P/p.A150P | 182 |
| 36 | M | 0.5 | 24.5 | 1-2-3 | - | p.A150P/p.A150P | 650 |
| 37 | F | 12 | 3.5 | 2 | - | p.Y204 */p.Y204 * | 110 |
| 38 | F | 7.5 | 4 | 2 | - | p.A150P/p.A150P | 91 |
| 39 | M | 5 | 4.1 | 1 | A-B | p.A150P/p.A175D | 6 |
| 40 | M | 1.3 | 5 | 4 | A-B | p.A150P/c.1_624del | 181 |
| 41 | M | 4 | 1.8 | 1 | B | p.A150P/p.A150P | 46 |
| 42 | M | 0.7 | 16.9 | - | A-B | p.A150P/p.A150P | 137 |
| 43 | M | 0.7 | 14.8 | - | B | p.A150P/p.A150P | 199 |
| 44 | F | 8 | 4 | 2 | - | p.A175D/p.L289Ffs * | 235 |
| 45 | F | 2 | 10.2 | 1 | A-B | p.A150P/p.A175D | 434 |
| 46 | M | 0.7 | 11.4 | 1-2-4 | A-B | p.A175D/p.L229P | 91 |
| 47 | F | 3 | 10.2 | 1 | - | p.A150P/p.N120Kfs * | 110 |
| 48 | F | 5.5 | 6.2 | 1 | A-B | p.A175D/p.N335K | 206 |
| Time of Evaluation | ALT (U/L) Mean ± DS | p-Value | |
|---|---|---|---|
| H-Group | nH-Group | ||
| T0 | 117 ± 115 | 61 ± 60 | 0.02 |
| T1 | 91 ± 54 | 35 ± 26 | <0.0001 |
| T2 | 83 ± 44 | 28 ± 19 | <0.0001 |
| T3 | 70 ± 30 | 27 ± 16 | <0.0001 |
| T4 | 74 ± 38 | 24 ± 11 | <0.0001 |
| T5 | 52 ± 33 | 21 ± 8 | <0.0001 |
| T6 | 47 ± 26 | 21 ± 8 | <0.0001 |
| T7 | 85 ± 34 | 22 ± 8 | <0.0001 |
| Time of Evaluation | H-Group | nH-Group | p-Value | |
|---|---|---|---|---|
| GGT IU/L | T0 | 26.1 ± 16.5 | 18.3 ± 12.2 | 0.06 |
| T7 | 22 ± 11.2 | 17.6 ± 8.5 | 0.12 | |
| p-value | 0.38 | 0.79 | ||
| Total Protein g/dL | T0 | 6.97 ± 0.79 | 6.89 ± 0.75 | 0.74 |
| T7 | 6.92 ± 0.62 | 6.98 ± 0.49 | 0.72 | |
| p-value | 0.83 | 0.58 | ||
| Total Bilirubin mg/dL | T0 | 0.37 ± 0.18 | 0.41 ± 0.19 | 0.5 |
| T7 | 0.35 ± 0.18 | 0.4 ± 0.21 | 0.4 | |
| p-value | 0.74 | 0.84 | ||
| Glucose mg/dL | T0 | 77.6 ± 9.5 | 73.2 ± 15.4 | 0.28 |
| T7 | 74.6 ± 10.7 | 78.7 ± 9 | 0.15 | |
| p-value | 0.38 | 0.09 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Dato, F.; Spadarella, S.; Puoti, M.G.; Caprio, M.G.; Pagliardini, S.; Zuppaldi, C.; Vallone, G.; Fecarotta, S.; Esposito, G.; Iorio, R.; et al. Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients 2019, 11, 2397. https://doi.org/10.3390/nu11102397
Di Dato F, Spadarella S, Puoti MG, Caprio MG, Pagliardini S, Zuppaldi C, Vallone G, Fecarotta S, Esposito G, Iorio R, et al. Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients. 2019; 11(10):2397. https://doi.org/10.3390/nu11102397
Chicago/Turabian StyleDi Dato, Fabiola, Simona Spadarella, Maria Giovanna Puoti, Maria Grazia Caprio, Severo Pagliardini, Claudia Zuppaldi, Gianfranco Vallone, Simona Fecarotta, Gabriella Esposito, Raffaele Iorio, and et al. 2019. "Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance" Nutrients 11, no. 10: 2397. https://doi.org/10.3390/nu11102397
APA StyleDi Dato, F., Spadarella, S., Puoti, M. G., Caprio, M. G., Pagliardini, S., Zuppaldi, C., Vallone, G., Fecarotta, S., Esposito, G., Iorio, R., Parenti, G., & Spagnuolo, M. I. (2019). Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients, 11(10), 2397. https://doi.org/10.3390/nu11102397