Abstract
Background: Fast excitatory transmission in the central nervous system is carried out by AMPA-type glutamate receptors. Neuronal hyperexcitability and epilepsy have been associated with the dysregulation of AMPA receptor function. Modulation of the gating kinetics of AMPA receptor function has been proposed to be a desirable target for therapy, especially when the modulation is transmembrane AMPA receptor regulatory protein (TARP)-dependent and AMPA receptor subunit composition-dependent. Methods: Eight dibenzobarrelene-based heterocycles were characterized for their effects on the human embryonic kidney cells expressing homomeric GluA1 and heteromeric GluA1/2 AMPA receptors, either alone or co-expressed with the TARPγ8 auxiliary subunit, using whole-cell patch-clamp electrophysiological recordings, and the current amplitude and kinetics of desensitization and deactivation were measured after rapid glutamate application. Results: Each chemical evaluated suppressed glutamate-induced currents via AMPA receptors and augmented both desensitization and deactivation, indicating a negative allosteric modulatory effect. The co-expression of TARPγ8 diminished, but did not eradicate, the inhibition and acceleration induced by the compounds. The observations indicate that the chemicals diminish agonist-bound open states and facilitate transitions to non-conducting states while maintaining effectiveness. Conclusions: The present study describes a specific kinetic mechanism by which dibenzobarrelene derivatives impair the function of the AMPA receptor and its dependence on auxiliary proteins. The present study provides a mechanistic understanding of AMPA receptor gating modulation and establishes a pharmacological framework for future investigations in more physiologically relevant systems.
1. Introduction
Epilepsy is a chronic neurological disease of the brain that is estimated to affect more than 50 million people worldwide [1,2]. This disease is characterized by recurrent epileptic seizures that are unprovoked [3]. The cause of epileptic seizures is the abnormal excitation of the neuronal network within the hippocampus, resulting in a temporary strong imbalance between excitatory and inhibitory inputs [4]. The primary excitatory neurotransmitter in the nervous system is glutamate, which is critical in the brain as it mediates fast excitatory synaptic transmission through ionotropic receptors and modulates neuronal excitability via metabotropic glutamate receptors [5].
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor is a predominant ionotropic glutamate receptor that mediates excitatory neurotransmission [6]. AMPA receptors can assemble as dimers of dimers due to the different combinations of GluA1, GluA2, GluA3, and GluA4 subunits [7]. Structurally, AMPA receptors are tetrameric ion channels organized as two subunit dimers that associate to form a functional receptor, with distinct dimer interfaces at the amino-terminal and ligand-binding domains (LBD) that are critical for receptor gating and desensitization [8]. The receptor opens upon glutamate binding, allowing the postsynaptic membrane to depolarize due to the influx of cations [9,10]. Calcium-permeable AMPA receptors, which lack the GluA2 subunit or contain an unedited GluA2 at the Q/R site and therefore permit Ca2+ influx, have been most strongly associated with epilepsy [11,12]. Ca2+ influx through these receptors increases neuronal excitability and can amplify excitatory signaling. In epilepsy, seizure activity has been shown to promote the upregulation of GluA2-lacking AMPA receptors, further enhancing Ca2+-dependent excitotoxic mechanisms [13]. Overexcitation mediated by AMPA receptors is influenced by changes in receptor-mediated whole-cell currents and in kinetic properties, which govern channel closure and responsiveness. The two main kinetic processes are the deactivation and desensitization. Deactivation is the decay of the receptor-mediated current after the removal of the agonist. This process typically operates on a sub-millisecond to millisecond timescale. Desensitization describes the decline in current observed during sustained exposure to an agonist. It generally develops over several milliseconds to tens of milliseconds under most experimental conditions [14,15].
Most AMPA receptors are also associated with auxiliary proteins that can affect receptor kinetics and trafficking [16]. Among these, the transmembrane AMPA receptor regulatory proteins (TARPs) are the best characterized [17]. TARPγ8 is predominantly expressed in the hippocampus and is crucial in regulating AMPA receptor trafficking, surface expression, and synaptic plasticity [17,18,19]. The absence of TARPγ8 decreases AMPA receptor expression, especially in the hippocampus CA1 region [19]. Most importantly, TARPγ8 has been extensively investigated as a pharmacological target in treating epilepsy, thereby supporting its role in physiological and pathological AMPA receptor function [17,20].
Despite extensive research into how AMPA receptors are allosterically inhibited and the mechanisms of allosteric competition, there remains a major gap in understanding the exact binding sites for many modulators and the specific interactions between chemical groups, the receptor, and its auxiliary proteins [21,22]. Some compounds have been localized to particular binding sites, but these sites must be fully characterized by their structure–activity relationships [23,24]. Most research has been on homomeric subunits, even though both homomeric and heteromeric subunits represent the full spectrum of receptor function in the brain [25]. Given that allosteric binding sites are not fully characterized and that we do not fully understand how auxiliary proteins influence AMPA receptor pharmacology, further research is needed using structurally diverse ligands across defined receptor assemblies. This research investigates the structure–activity relationship (SAR) of a series of complex molecules featuring rigid polycyclic aromatic scaffolds based on a dibenzobarrelene core and substituted with nitrogen-based heterocyclic substituents, including imidazole and imidazoline-derived moieties, as well as fused heteroaromatic systems (ND-1-ND-8) (Figure 1) [26]. This series of compounds has not been previously investigated against any biological targets. Moreover, the chemical scaffolds appear to be unique and structurally distinct. Notably, the presence of rigid polycyclic frameworks and heteroaromatic moieties shows partial similarity to the reported modulators of AMPA receptors (GluA subtypes), supporting their potential as promising candidates for further exploration in AMPA receptor-related pharmacology [27]. Different classes of AMPA receptor modulators, particularly negative allosteric modulators (NAMs) (non-competitive antagonists), share structural features such as molecular rigidity, heteroaromatic nitrogen atoms, and multiple hydrogen bond donors and acceptors. Representative examples include the clinically approved antiepileptic drug perampanel, as well as 2,3-benzodiazepine derivatives such as GYKI53655 and quinozalin-4-one derivatives such as CP465022 [28,29]. These compounds bind at non-orthosteric sites located in the linker region between the LBD and transmembrane domains, where they act as “wedges” that prevent M3 helix displacement and thereby inhibit ion channel opening [28]. This study will examine whole-cell responses and changes in gating kinetics elicited by a rapid glutamate application in whole-cell patches from human embryonic kidney cells expressing homomeric (GluA1) or heteromeric (GluA1/2) AMPA receptor subunits, in the absence and presence of TARPγ8. The study is designed as a receptor-level pharmacological investigation in a heterologous expression system, primarily to define compound–receptor interactions.
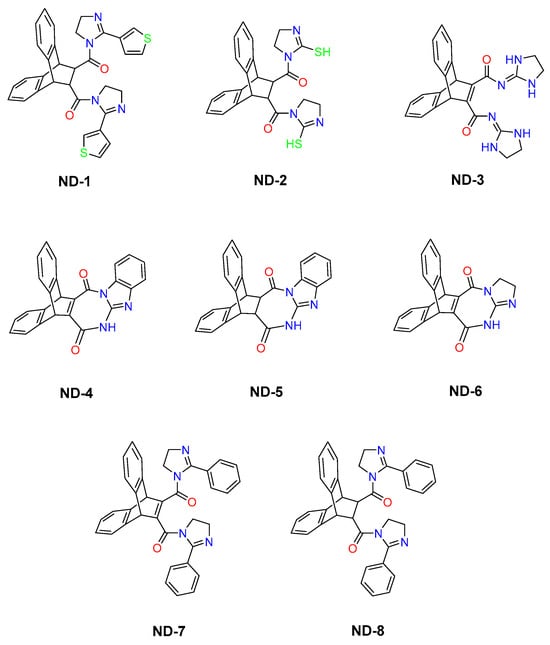
Figure 1.
The molecular structures of 8 dibenzobarrelene derivatives, labeled ND-1 through ND-8.
2. Materials and Methods
2.1. cDNA Construction
For wild-type AMPA receptor subunit expression, GluA1 and GluA2 (Q-form, flip version) cDNAs were employed. To co-express GluA1 and GluA2 in the human embryonic kidney (HEK293T) cells (catalog number 85120602, Sigma-Aldrich, Darmstadt, Germany) for heteromeric AMPA receptor expression, GluA1 and GluA2 cDNAs were co-transfected. The AMPA receptor cDNAs were first obtained from S.F. Heinemann (Salk Institute, La Jolla, CA, USA) and were cloned into mammalian expression plasmid pRK5, which was provided by P.H. Seeburg (Max Planck Institute for Medical Research, Heidelberg, Germany).
All plasmids were maintained in E. coli and confirmed by restriction analysis before being used in this study. The high-copy plasmid DNA was isolated using the QIAGEN Plasmid Mini Kit (Catalog No. 12123, QIAGEN, Hilden, Germany) according to the manufacturer’s protocol, with some standard modifications to maximize yield and purity. In brief, the plasmids were transformed into chemically competent E. coli, plated on LB agar containing the required selective antibiotic, and incubated overnight at 37 °C. Then, a single bacterial colony was picked and grown in selective LB medium to create the starter culture, which was incubated for about 8 h at 37 °C under agitation. The starter culture was then diluted into fresh selective LB medium and incubated for an additional 12–16 h at 37 °C to allow the high-copy plasmids to amplify. Bacterial cultures were obtained by centrifugation, and cell pellets were resuspended and lysed in alkaline conditions using buffers supplied in the kit. After neutralization of the lysate, it was then centrifuged to remove cellular debris. Cell debris was separated from plasmid DNA in a silica-based purification column pre-equilibrated with binding buffer to enable the binding of plasmid DNA by gravity flow. The column was then washed to remove impurities and eluted with the elution buffer supplied in the kit. Finally, DNA was precipitated by isopropanol, washed with ethanol, air-dried, and then resuspended in nuclease-free buffer.
The concentration and purity of plasmid DNA were determined by ultraviolet spectrophotometry at 260 nm, while confirmation of plasmid integrity was done through agarose gel electrophoresis. Only those plasmid DNA preparations that gave high purity ratios were used for further transfection studies. The yield of plasmid DNA per preparation was estimated to be up to 20 µg. For cell identification after transfection, enhanced green fluorescent protein (EGFP; Clontech, Palo Alto, CA, USA) was co-expressed using a separate plasmid as an indicator marker during electrophysiological recordings. The plasmid DNA prepared through this method was used for the transient transfection of HEK293T cells for functional expression of AMPA receptor subunits.
The complete coding region of the TARPγ8 (CACNG8) gene was cloned for heterologous expression in HEK293T cells. The TARPγ8 gene was PCR-amplified with high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA, USA) to introduce a minimum number of point mutations and cloned downstream of a cytomegalovirus (CMV) promoter in the mammalian expression vector pRK5 for high-level transcription in mammalian cells. Cloning was performed via restriction enzyme digestion and ligation (New England Biolabs, Ipswich, MA, USA), and the insert orientation and integrity were confirmed via complete Sanger sequencing of the open reading frame. Additionally, to visualize HEK293T cells transiently expressing the construct for the qualitative assessment of expression, an EGFP tag was added to the TARPγ8 construct. This added the EGFP protein to the N-terminus of the TARPγ8 protein, which was separated from the TARPγ8 protein by a linker. This method allows easy detection of HEK293T cells expressing the construct in whole-cell recordings without changing the biophysical or modulation properties of the TARPγ8 protein.
Additionally, to study specific interactions between the AMPA receptor and the TARPγ8 protein, as well as to reduce variability due to independent expression levels of auxiliary proteins, tandem AMPA receptor–TARPγ8 constructs were created. These constructs joined the open reading frame of the TARPγ8 protein sequence to the C-terminus of the open reading frame sequence of the specific AMPA receptor subunit. Both proteins were joined via a short flexible linker composed of uncharged amino acids. This allows independent protein folding and functional interactions between the receptor and auxiliary protein subunits, while fixing their stoichiometry. All tandem constructs were cloned downstream of the CMV promoter in the pRK5 expression vector. All constructs were confirmed by complete open-reading-frame sequencing.
2.2. Human Embryonic Kidney Cell Transfection
The highly transfectable HEK293T cells that express the SV40 large T antigen were used to express and examine AMPA receptor subunits. HEK293T cells were grown in Dulbecco Modified Eagle Medium (DMEM) (Sigma-Aldrich, Darmstadt, Germany) containing 10% fetal bovine serum (Sigma-Aldrich, Darmstadt, Germany) and 0.1 mg/mL penicillin–streptomycin (Biological Industries, Beit-Haemek, Israel). HEK293T cells were incubated at 37 °C and 5% CO2, and the medium was supplemented. Cells were subcultured twice a week and used for experiments at passages ≤ 20 to preserve transfection efficiency, maintain stable physiology, and reduce variability in electrophysiology. The transfection reagent was the jetPRIME (Polyplus: New York, NY, USA). Cells were co-transfected at a DNA mass ratio of 1:9 (pEGFP-C1 to the GluA subunit plasmid), a design optimized to balance the marker’s visibility with the receptor’s expression fidelity. This prevents overexpression of the fluorescent marker while minimizing nonspecific cellular stresses, thereby minimizing possible interference with the folding and trafficking of AMPA receptors by prioritizing subunit stoichiometry and functional assembly, which are critical for robust electrophysiological analyses [30]. Cells were kept in 12-well plates for 36 h. after transfection. They were then replated on coverslips coated with laminin (1 µg/mL; Sigma-Aldrich, Darmstadt, Germany) for electrophysiology recordings.
2.3. Electrophysiological Recordings
Whole-cell patch-clamp recordings were performed using integrated patch-clamp amplifiers, a data-acquisition system (IPA, Sutter Instruments, Novato, CA, USA), and a rapid solution-exchange system driven by a piezoelectric actuator (Automate Scientific, Berkeley, CA, USA). This system features a two-barrel theta glass pipette operated by a piezoelectric translator. One barrel contained an external solution composed of 150 mM NaCl, 2.8 mM KCl, 0.5 mM MgCl2, 2 mM CaCl2, and 10 mM HEPES, adjusted to pH 7.4 with NaOH, while the other barrel held the chemical compound solution. The internal pipette solution consisted of 110 mM CsF, 30 mM CsCl, 4 mM NaCl, 0.5 mM CaCl2, 10 mM EGTA, and 10 mM HEPES, and 100 μM spermine, with the pH adjusted to 7.2 using CsOH. Patch electrodes made from borosilicate glass with a resistance range of 2–4 MΩ were employed. The performance of the rapid solution-exchange system was assessed by measuring junction potentials at the open tip of the patch pipette, yielding typical 10–90% rise times of 200–300 μs, reflecting the solution exchange rate during rapid agonist application. Cells with peak currents <20 pA were excluded due to low signal-to-noise ratio.
For AMPA receptor-current measurements, deactivation (τw deact) and desensitization (τw des) were determined by applying 10 mM glutamate (Sigma-Aldrich, Darmstadt, Germany) for 500 ms to induce desensitization and for 1 ms to induce deactivation. The weighted tau (τw) was calculated using the formula τw = (τf × af) + (τs × as), where af and as represent the relative amplitudes of the fast (τf) and slow (τs) exponential components, respectively. These measurements were obtained after fitting the desensitization and deactivation currents with two exponential functions, applied from 95% of the peak to the baseline current. All experiments were conducted using cells derived from four independent transfections for homomeric GluA1 receptors and six independent transfections for heteromeric GluA1/2 receptors, performed on different experimental days under conditions of −60 mV potential, pH 7.4, and room temperature (20–23 °C). For each receptor construct, n = 10 individual cells were recorded and analyzed. Pharmacological comparisons were performed using a within-cell paired design.
Compounds were dissolved in DMSO and diluted into an external solution, with the final DMSO concentration not exceeding 0.1%. The eight compounds were tested at a final concentration of 14 µM. The selection of 14 µM as the final concentration for electrophysiological recordings was determined based on full concentration–response experiments. A progressive increase in inhibition was observed across increasing concentrations, beginning at 2 µM, with effects approaching a plateau at 14 µM under the recording conditions. This concentration was therefore selected as the maximal effective concentration to ensure robust and reproducible modulation while avoiding compromised cell viability observed at higher concentrations. Pharmacological effects were assessed within the same recorded cell by first applying glutamate under control conditions, followed by glutamate in the presence of the ND compound, thereby enabling paired within-cell comparisons.
In addition to kinetic analyses, current–voltage (I–V) relationships were assessed to verify receptor expression and TARPγ8 association functionally. Whole-cell patch-clamp recordings used a 1 s voltage ramp (−70 mV to +40 mV) applied during the sustained component of the glutamate-evoked response, after peak activation. The I-V relationship recorded before and after glutamate application was subtracted from those recorded in its presence to isolate the receptor-mediated activity. The resulting I-V relationship was then normalized to the current at −70 mV.
Because GluA2(Q) was used in these experiments, both homomeric GluA1 and heteromeric GluA1/2(Q) receptors are expected to retain calcium permeability and exhibit inward rectification in the presence of intracellular polyamines. Spermine (100 μM) was included in the internal solution to provide a defined polyamine condition and enable the assessment of rectification behavior. The co-expression of TARPγ8 is known to reduce intracellular polyamine block and partially linearize the I–V relationship [31]. In the present study, I–V analysis was used as supportive electrophysiological profiling rather than as a primary determinant of calcium permeability.
2.4. Data Analysis
For each recorded cell, the peak AMPA receptor-mediated currents evoked by glutamate alone (A) and by glutamate in the presence of compound (AI) were measured, and compound effects were quantified as fold inhibition (A/AI), with larger values reflecting a greater fold reduction in current amplitude. Comparisons between glutamate alone and glutamate + ND compounds were performed using a within-cell paired design, as measurements were obtained sequentially from the same recorded cell. Accordingly, paired two-tailed Student’s t-tests were used for two-condition comparisons. When three conditions were compared within the same cell, repeated-measures one-way ANOVA followed by Tukey’s post hoc test was applied. Values were reported as the mean ± SD, and the data were analyzed statistically using GraphPad Prism 9.5.0.730 (x64). The criterion for differences to be statistically significant was set at p < 0.05 and is presented in the form: * p < 0.05; ** p < 0.01; *** p < 0.001; otherwise, it is not significant (ns). The sample size (n) represents the number of individual recorded cells per receptor construct (n = 10). Experiments were performed across four independent transfections for homomeric receptors and six independent transfections for heteromeric receptors, representing biological replicates.
2.5. Ethical Consideration
All experiments were conducted using HEK293T cell lines. No human subjects, patient data, or animals were used. Therefore, no ethical approval was necessary under the institutional and national research regulations.
3. Results
3.1. Dibenzobarrelene Derivatives Inhibited AMPA Receptor-Mediated Peak Currents in the Absence and Presence of TARPγ8
The synthesized compounds ND-1 to ND-8 are dibenzobarrelene-based aza-heterocyclic derivatives that share a rigid polycyclic aromatic scaffold while differing in heterocycle composition, molecular rigidity, and hydrogen-bonding capacity. These compounds can be classified into symmetrical bis-imidazoline amides (ND-1, ND-2, ND-7, ND-8), a di-imidazolidine dicarboxamide (ND-3), fused diazepinedione derivatives (ND-4, ND-5), and a fused imidazo-diazepinedione derivative (ND-6). All compounds were previously synthesized and structurally characterized [26], with full spectroscopic data reported in the original publications and reproduced in the attached Supplementary Materials. Although these compounds have been well characterized chemically, this work is the first to examine their functional effects on AMPA receptor activity. In HEK293T cells expressing either homomeric GluA1 or heteromeric GluA1/2 receptors, glutamate reliably evoked strong inward currents. To illustrate this effect on the receptor activity, representative whole-cell current responses to a 500 ms glutamate pulse are shown in Figure 2. This response was consistent regardless of whether TARPγ8 was co-expressed.
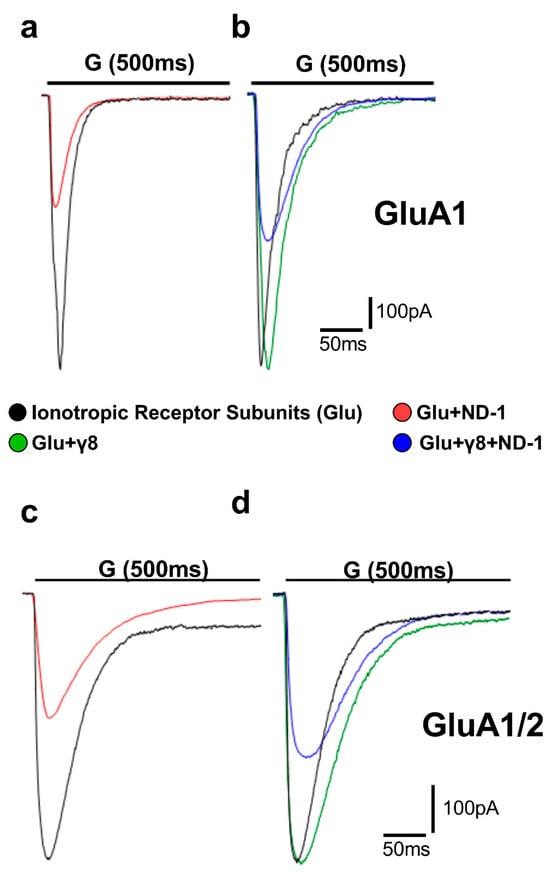
Figure 2.
Representative whole-cell AMPA receptor currents recorded in HEK293T cells. Superimposed representative whole-cell current traces (pA) recorded from AMPA receptor subunits subjected to 500 ms applications of 10 mM glutamate (“G” indicated above the traces) in HEK293T cells expressing homomeric GluA1 (a,b) and heteromeric GluA1/2 (c,d). Traces include responses to glutamate alone (black), glutamate with ND-1 compound (red), glutamate on the subunit co-expressed with TARPγ8 (green), and glutamate with ND-1 on the subunit co-expressed with TARPγ8 (blue). All recordings were obtained under whole-cell patch-clamp conditions at −60 mV, pH 7.4, and 22 °C. Quantitative analysis was performed from n = 10 cells per receptor construct derived from four (GluA1) or six (GluA1/2) independent transfections.
Pre-exposure to the ND compounds significantly reduced the peak amplitude of glutamate-evoked currents across all receptor assemblies tested (Figure 3; Tables S1–S8), consistent with the inhibition of AMPA receptor function. When the compounds were ranked by potency, ND-1 emerged as the most effective inhibitor of peak AMPA receptor-mediated currents. ND-1 produced a pronounced reduction in current amplitude in both GluA1 and GluA1/2 receptors. Co-expression with TARPγ8 partially attenuated this inhibitory effect, but did not abolish it (Figure 3a). ND-2 and ND-3 showed slightly lower, yet comparable, inhibitory efficacy, followed by the fused diazepinedione derivatives ND-4 and ND-5. ND-6 produced moderate inhibition, whereas ND-7 and ND-8 consistently exhibited the weakest effects among the compounds tested (Figure 3b–h). Across all compounds tested, adding TARPγ8 consistently reduced the apparent inhibitory efficacy. This was reflected in reduced fold inhibition compared with receptors expressed alone (Figure S1). We noticed the same trend in both homomeric GluA1 and heteromeric GluA1/2 receptors, suggesting that TARPγ8 dampens the overall extent of compound-driven inhibition of peak currents without disrupting the relative potency ranking among compounds. In other words, the inhibition is weaker, but the hierarchy stays the same.
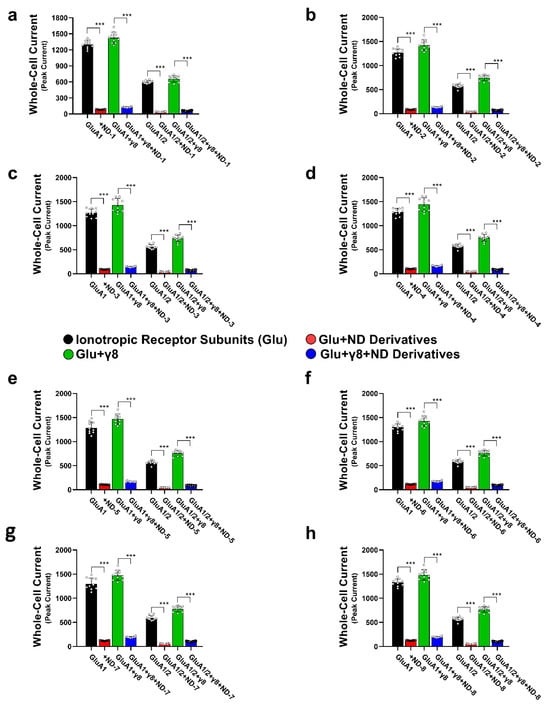
Figure 3.
Effects of dibenzobarrelene derivatives on AMPA receptor-mediated whole-cell currents. This figure demonstrates the inhibitory actions of the eight derivatives on the whole-cell currents mediated by GluA1 and GluA1/2 AMPA receptor subtypes. Panels (a–h) represent data for the individual ND compounds. Whole-cell patch-clamp recordings were done on HEK293T cells expressing GluA1, GluA1/2, GluA1 + γ8, and GluA1/2 + γ8 subunits. TARPless AMPA currents were induced by 10 mM glutamate using a 500 ms application protocol (black bars), and these currents are then compared to currents recorded in the presence of 14 µM ND compounds (red bars). Currents from GluA1 and GluA1/2 co-expressed with TARPγ8 (green bars) were compared to currents recorded in the presence of 14 µM ND compounds (blue bars). Statistical comparisons were performed using paired two-tailed Student’s t-tests and ANOVA, as appropriate, reflecting the within-cell experimental design. *** p < 0.001, reflecting a significant reduction in currents. All the recordings were done in whole-cell patch-clamp mode at −60 mV, pH 7.4, and 22 °C. Data represent mean ± SD from n = 10 cells (each dot above the bar represents an individual cell recorded) per receptor construct obtained from 4 (GluA1) or 6 (GluA1/2) independent transfections.
To quantify these effects, we performed concentration–response experiments and calculated IC50 values (half-maximal inhibitory concentration, defined as the concentration of the compound required to reduce the peak current amplitude by 50%) using nonlinear regression with a four-parameter logistic model (Figure 4; Tables S9 and S10). The data mirrored the inhibition results. Across homomeric GluA1 receptors, IC50 values ranged from 4.21 to 8.75 µM, whereas for heteromeric GluA1/2 receptors they ranged from 4.57 to 5.60 µM. Co-expression with TARPγ8 shifted several concentration–response curves to the right, with IC50 values ranging from 5.63 to 11.15 µM for GluA1 + γ8 and from 5.07 to 8.60 µM for GluA1/2 + γ8 receptors. This TARPγ8-associated shift was observed for the majority of ND derivatives examined (Figure 4). Hill slope (nH) values and corresponding 95% confidence intervals for all fits are provided in Table S9, and goodness-of-fit (R2) values are summarized in Table S10.
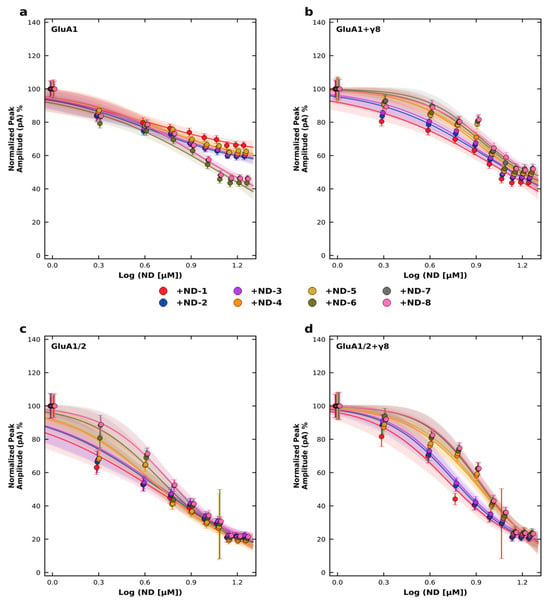
Figure 4.
Concentration–response inhibition of AMPA receptor subtypes by ND compounds. Normalized peak current amplitudes were recorded at −60 mV in HEK293T cells expressing (a) GluA1, (b) GluA1 co-expressed with TARP γ8, (c) GluA1/2, or (d) GluA1/2 co-expressed with TARP γ8. Peak currents were normalized to the control response obtained in the absence of the compound (0 µM), which was set to 100%. ND compounds (ND-1 to ND-8) were applied at concentrations ranging from 0 to 14 µM, and responses are plotted as a function of log[ND] (µM). Data points represent mean ± SD from n = 6–7 cells per concentration. Concentration–response relationships were fitted using a four-parameter logistic model with the maximal response (Top) constrained to 100%. Hill slope (nH), IC50 values, and corresponding 95% confidence intervals were derived from nonlinear regression analysis (GraphPad Prism). Recordings were performed at pH 7.4 and 22 °C.
3.2. Dibenzobarrelene Derivatives Accelerate AMPA Receptor Desensitization
Desensitization kinetics were determined from the weighted time constant (τw des) during a 500 ms glutamate application, in accordance with the well-established function of auxiliary proteins in AMPA receptor gating. The co-expression of TARPγ8 caused a significant retardation of receptor desensitization compared to the desensitization of receptors expressed without the auxiliary protein subunit (Figure 5). This was observed for both homomeric GluA1 and heteromeric GluA1/2 receptors, thus confirming the functional link between TARPγ8 and AMPA receptors under the given experimental conditions. The application of ND compounds resulted in a strong reduction in τw des for all receptor complexes examined, indicating a kinetic acceleration of desensitization (Figure 5). This was found for GluA1 and GluA1/2 receptors, expressed alone and together with TARPγ8. However, the extent to which the compounds could accelerate desensitization was reduced in the presence of the auxiliary subunit. For example, ND-1 induced a significant reduction in τw des in GluA1 receptors, whereas the same compound induced significantly less reduction in τw des when co-expressed with the auxiliary subunit TARPγ8. This is true for all the ND derivatives tested and for both homomeric and heteromeric receptors (Figure 5).
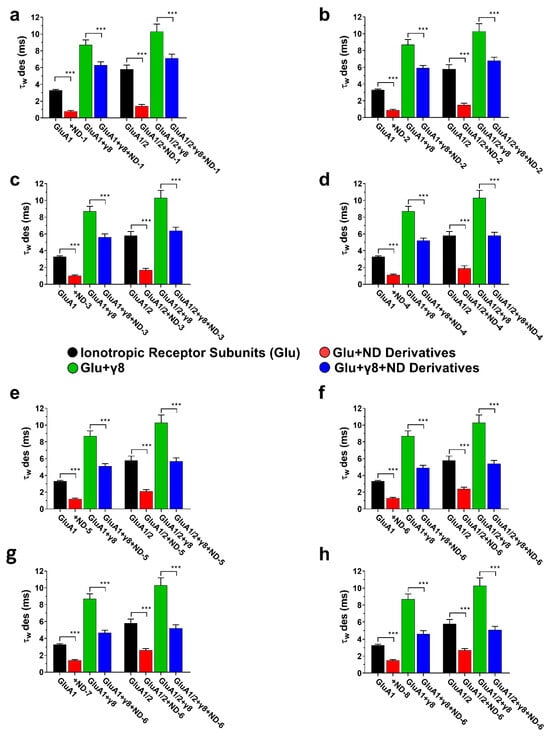
Figure 5.
Desensitization kinetics (τw des) of AMPA receptor subunits in the presence of ND compounds. The bar graphs (a–h) illustrate the weighted desensitization time constants (τw des) for GluA1, GluA1 + γ8, GluA1/2, and GluA1/2 + γ8, demonstrating the impact of ND compounds on controlled desensitization kinetics. Differences in desensitization rates relative to control are significant at *** p < 0.001 (paired two-tailed Student’s t-tests and ANOVA where applicable, reflecting the within-cell experimental design). All recordings were obtained under whole-cell patch-clamp conditions at −60 mV, pH 7.4, and 22 °C. The control currents were induced by 10 mM glutamate using a 500 ms application protocol. Data represent mean ± SD from n = 10 cells per receptor construct obtained from four (GluA1) or six (GluA1/2) independent transfections.
In GluA1 receptors, ND-1 reduced the amplitude and accelerated the desensitization rate (Figure 2a). Co-expression with TARPγ8 slowed the rate of desensitization compared with GluA1 alone, and under these circumstances, the effect of ND-1 on desensitization was reduced (Figure 2b). This was also true for the GluA1/2 receptors. ND-1 reduced the amplitude and accelerated the rate of desensitization for GluA1/2 receptors expressed alone (Figure 2c). Co-expression with TARPγ8 slowed the rate of desensitization and reduced the effect of ND-1 on the amplitude and rate of desensitization (Figure 2d).
3.3. Dibenzobarrelene Derivatives Accelerate AMPA Receptor Deactivation
Deactivation kinetics were determined using brief glutamate pulses (1 ms) to mimic the pattern of receptor activation observed in synapses. In the normal state, the addition of TARPγ8 resulted in a large extension of the weighted deactivation time constant (τw deact) for both GluA1- and GluA1/2-containing receptors, indicating a delay in the process of channel closing following agonist removal (Figure 6). In GluA1 receptors, ND-1 suppressed the amplitude of the maximal current and induced a faster decay of the current after glutamate removal (Figure 6a). Co-expression with TARPγ8 attenuated the decay, and the inclusion of ND-1 restored it to an intermediate state (Figure 6b). This also occurred for the GluA1/2 receptors: the current decay was faster with ND-1 than with the control, and the presence of TARPγ8 attenuated the drug effect (Figure 6c,d). Across all receptor types, the rank order of the drug effect on the decay kinetics was maintained, with ND-1 producing the strongest effect.
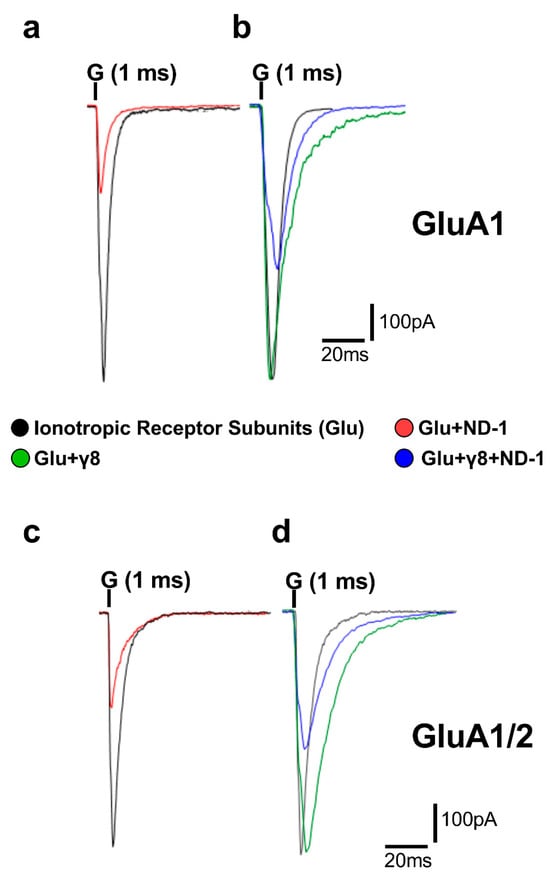
Figure 6.
Whole-cell AMPA receptor currents illustrating deactivation behavior. Superimposed representative whole-cell current traces (pA) recorded from AMPA receptor subunits subjected to 1 ms applications of 10 mM glutamate (“G” indicated above the traces) in HEK293T cells expressing homomeric GluA1 (a,b) and heteromeric GluA1/2 (c,d). Traces include responses to glutamate alone (black), glutamate with ND-1 compound (red), glutamate on the subunit co-expressed with TARPγ8 (green), and glutamate with ND-1 on the subunit co-expressed with TARPγ8 (blue). All recordings were obtained under whole-cell patch-clamp conditions at −60 mV, pH 7.4, and 22 °C. Quantitative analysis was performed from n = 10 cells per receptor construct derived from four (GluA1) or six (GluA1/2) independent transfections.
This finding is consistent with the known role of TARPs in supporting the open or pre-open conformation of the AMPA receptor channels. The addition of ND compounds consistently accelerated deactivation kinetics across all assemblies, as evidenced by a reduction in the weighted deactivation time constant (τw deact) (Figure 7). This occurred in both homomeric and heteromeric assemblies. In assemblies containing TARPγ8, the ND compounds further accelerated deactivation, with the extent of this acceleration moderately reduced compared with assemblies lacking the auxiliary subunit, but significantly less than the reduction observed for the rate of desensitization.
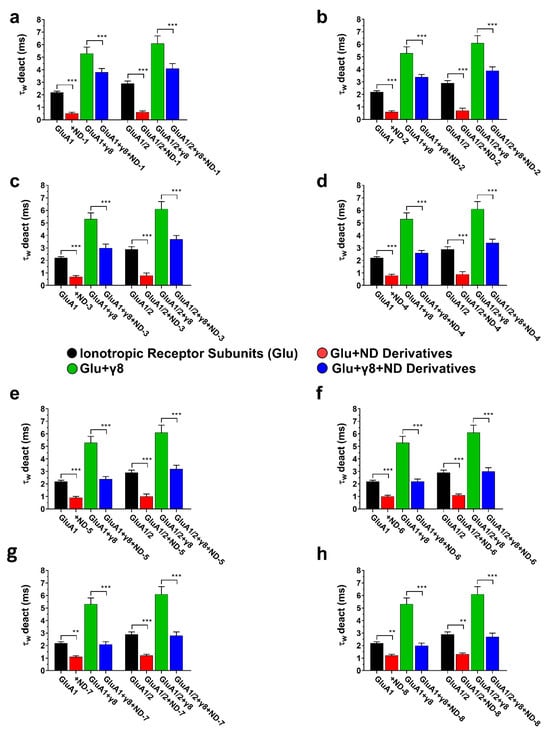
Figure 7.
Deactivation kinetics (τw deact) of AMPA receptor subunits in the presence of ND compounds. The bar graphs (a–h) illustrate the weighted deactivation time constants (τw deact) for GluA1, GluA1 + γ8, GluA1/2, and GluA1/2 + γ8, demonstrating the impact of ND compounds on controlled deactivation kinetics. Differences regarding deactivation rates significant from control are indicated by *** p < 0.001, and ** p < 0.01 (paired two-tailed Student’s t-tests and ANOVA where applicable, reflecting the within-cell experimental design). All recordings were obtained under whole-cell patch-clamp conditions at −60 mV, pH 7.4, and 22 °C. The control currents were induced by 10 mM glutamate using a 1 ms application protocol. Data represent mean ± SD from n = 10 cells per receptor construct obtained from four (GluA1) or six (GluA1/2) independent transfections.
4. Discussion
The goal of this research is to understand the nature of the modulation of AMPA receptor function by a group of structurally similar dibenzobarrelene-containing heterocycles on homomeric GluA1 receptors and heteromeric GluA1/2 receptors in the presence and absence of TARPγ8. The modulation of AMPA receptors is of particular importance because they account for the largest part of fast excitatory synaptic transmission in the central nervous system and because the kinetics of their activation, deactivation, and desensitization underlie the strength of synaptic transmission and the integration of excitatory synaptic inputs in the central nervous system [32,33]. Excessive AMPA receptor activation contributes to neuronal hyperexcitability, particularly when receptor-mediated calcium influx is enhanced [34,35]. While GluA1 homomers are often used experimentally as models of calcium-permeable AMPA receptors, the present study employed the unedited GluA2(Q) variant; therefore, both GluA1 and GluA1/2(Q) receptors retain calcium permeability under the experimental conditions used here. Accordingly, the comparison between subunit assemblies in this work reflects differences in subunit composition and auxiliary protein modulation.
By simultaneously assessing changes in the amplitude of peak whole-cell currents, along with deactivation and desensitization kinetics, the present study provides a mechanistic interpretation of how pharmacological modulation alters the magnitude and timescales of excitatory transmission. Moreover, the direct comparison of receptor assemblies differing in subunit composition and auxiliary subunit association allows a more refined assessment of AMPA receptor functional heterogeneity [11]. Functionally, all tested ND compounds inhibited glutamate-triggered AMPA receptor currents for both GluA1- and GluA1/2-containing receptors, accelerating receptor desensitization and deactivation, while maintaining a consistent rank order of potency across receptor subtypes. Co-expression of TARPγ8 reduced, but did not prevent, the acceleration of AMPA receptor kinetics caused by the compounds, suggesting a modulatory rather than a blocking role for the auxiliary protein modulation of AMPA receptor kinetics. This work sets the stage for evaluating the impact of the TARPγ8 modulation of receptor kinetics on allosteric modulation.
Based on the observation that TARPγ8 reduces, rather than completely prevents, compound-induced inhibition and kinetic acceleration, these actions can be understood within the framework of TARPγ8′s known role as a stabilizer of transitions in the AMPA receptor gates. TARPγ8 is highly concentrated in hippocampal excitatory neurons and represents a major modulatory subunit of AMPA receptors, particularly GluA1/2-containing receptors, which are predominant at forebrain synapses [36,37]. Current electrophysiological and structural studies have also revealed that TARPγ8 retards deactivation and desensitization by stabilizing the pre-active and open-channel conformations [17,38]. At the molecular level, the results of cryo-electron microscopy and molecular dynamics studies show the following interactions of TARPγ8 with the AMPA receptor: There are multiple sites of interaction of TARPγ8 with the AMPA receptor, which involve the extracellular loops and the LBD lobes, as well as interactions of the transmembrane domains of TARPγ8 with the helices of the pore [38]. More specifically, interactions between the extended extracellular loops of γ8 and LBD dimers regulate the balance between activation and desensitization, and interactions between the γ8 helices and the pore region of AMPA receptors are involved in reduced polyamine block and linearization of current–voltage relationships [38,39]. In line with these mechanisms, the co-expression of TARPγ8 in this study resulted in a significant prolongation of the weighted deactivation and desensitization time constants for both GluA1 and GluA1/2 receptors, thus supporting the formation of functional receptor–TARPγ8 complexes under these experimental conditions. Notably, this kinetic slowing provides a molecular mechanism for buffering the effect size of the ND compound, as TARPγ8 suppresses the ability of these compounds to facilitate these transitions by modulating the energy landscape for activation and desensitization. This result places TARPγ8 as a kinetic modulator that determines receptor responsiveness rather than a structural scaffold.
Based on the kinetic stabilization already conferred by TARPγ8, the ND compounds consistently reduced the amplitude of glutamate-evoked peak currents and accelerated deactivation and desensitization in the GluA1 and GluA1/2 assemblies, suggesting a common modulatory profile across subunit compositions. The reduction in peak current amplitude and the acceleration of gating transitions are most consistent with a negative allosteric model, in which the compound’s binding favors a non-conducting conformation [17]. In particular, the partial attenuation of these actions by the co-expression of TARPγ8 suggests a role for γ8 in stabilizing ligand–receptor conformations, thereby reducing the receptor’s sensitivity to the acceleration of the transition to sustained gating. This differential attenuation supports the idea that auxiliary subunits play a stronger modulatory role in the receptor’s long-lasting activity rather than in the rapid receptor closure following ligand unbinding [17]. The coexistence of compound effects even in the presence of TARPγ8 suggests a binding to allosteric sites that are functionally as well as geometrically distinct from the binding interface of the primary TARPγ8-AMPA receptor interaction. This conclusion is consistent with the structural data, which show distinct transmembrane binding pockets at the AMPA receptor–TARP interface. From a pharmacological point of view, the partial inhibition of compound effects by TARPγ8 could serve as a biologically valuable filter, selectively blocking the overinhibition of synapses with auxiliary proteins while leaving the modulation of hyperexcitable receptor populations unaffected by full auxiliary stabilization [40].
Chemically, the modulatory effect of the ND series could be explained by several common structural features associated with allosteric ligands of ionotropic glutamate receptors. All the compounds described are based on the rigid dibenzobarrelene scaffold, which imposes a highly constrained three-dimensional geometry. This is recognized as favorable for affinity to shallow or interfacial sites rather than to deep sites [41,42]. This polycyclic aromatic scaffold provides an extended π surface capable of forming hydrophobic and π-π stacking interactions with aromatic residues, which are often found in the transmembrane and linker regions of AMPA receptors [32]. Superimposed upon the rigid framework, the presence of nitrogen-containing heterocycles (imidazole, imidazoline, diazepinedione, and fused heteroaromatics) and thiol and amide groups introduces the potential for strong hydrogen-bond donor/acceptor interactions. This is an important determinant of ligand binding at the allosteric sites, mediated by polar interactions with backbone carbonyls and side-chain heteroatoms at the LBD-TMD interfaces [11,43]. The SAR is clear in the ND series when rigidity and symmetry are analyzed in the molecules. ND-1, which has a rigid dibenzobarrelene core and symmetrical bis-imidazoline substituents, showed the most potent inhibition and the most effective acceleration of receptor kinetics through the possible H-bonding acceptor interactions of amide’s carbonyl and N3 of imidazoline, while the thiophene rings may introduce polar interactions with the binding site and hydrophobic interactions via benzene rings. Symmetry and rigidity are known to facilitate productive binding in interfacial allosteric sites by providing shape complementarity during ligand–receptor binding [44]. By contrast, ND-7 and ND-8 display a heterocycle orientation that is suboptimal and more flexible, and the presence of a bulky phenyl ring may sterically block H-bonding, resulting in a significantly reduced functional response and weaker binding interactions. This is consistent with a less stable interaction with the receptor’s gating apparatus. The dependence on subtle heterocycle orientation is a clear indication that precise spatial alignment of hydrogen-bond motifs with respect to the rigid aromatic backbone is a critical principle well established for non-competitive AMPA receptor inhibitors such as GYKI and perampanel analogs [45,46].
Mechanistically, the chemical properties of ND ligands match those involving interactions with allosteric sites that favor the stability of active conformations rather than direct glutamate antagonism. Based on structural and modeling studies, negative allosteric AMPA receptor ligands typically target sites associated with the closure of the ligand-binding domain clamshell and the opening of the transmembrane gate [15,43]. The data reported can be interpreted as a chemical destabilization of LBD dimer interfaces, with a high sensitivity to ligands that can alter hydrogen-bonding patterns and hydrophobic packing in these interfaces [11]. Accordingly, the presence of the rigid polycyclic backbone and heteroaromatic nitrogen atoms in the ND compounds is likely to favor the conformational equilibrium of the receptors away from the conducting states, providing a chemically sound explanation for the negative allosteric modulatory effects of the ND compounds.
The pharmacological and biochemical profile of the ND derivatives is very similar to that of AMPA receptors’ NAMs, which reduce peak current and preferentially modulate receptors towards fast non-conducting states [21,45]. The fact that the compounds retain their effectiveness across both GluA1 and GluA1/2 subunits suggests that their modulatory mechanism is not strongly dependent on subunit composition under the conditions tested, but rather reflects a common allosteric influence on channel gating. By promoting both desensitization and deactivation, these compounds are predicted to reduce receptor-mediated charge transfer during sustained glutamatergic stimulation. Notably, a comparable magnitude of peak current suppression together with enhancing receptor kinetics was previously reported for our laboratory’s 2,3-benzodiazepine and benzodioxole NAMs under the same HEK293T/GluA experimental platform. In those studies, molecular docking confirmed engagement of the conserved GluA2 O-site allosteric pocket (PDB 5L1G), with interaction patterns overlapping those of the reference NAM GYKI-Br, thereby providing structural validation for an allosteric gating mechanism. This mechanistic concordance supports the interpretation that the ND series likewise operates through a conserved NAM-like pathway [47,48]. While these prior electrophysiological and structural data strengthen the internal validity of the experimental platform and support a plausible allosteric interpretation, they do not directly confirm that the ND derivatives engage the identical binding site or molecular interactions.
Despite the extensive and coherent functional data in the present work, there are some inherent limitations. Firstly, despite the strongly suggested negative allosteric modulation of the ND compounds based upon their shared kinetic and inhibitory behavior, the lack of any direct structural information, such as cryo-electron microscopy, X-ray crystallography, molecular docking studies, or site-directed mutagenesis analysis, prevents any definitive identification of their binding site and orientation within the receptor complex. Secondly, the functional analysis in the present work has examined only the behavior of two assemblies of AMPA receptors (GluA1 and GluA1/2), together with a single auxiliary receptor (TARPγ8), and thus has not explored the full molecular complexity of native AMPA receptor assemblies. Thirdly, the present study was conducted exclusively in a heterologous HEK293T expression system, which allows pharmacological characterization but does not fully recapitulate the complexity of native neuronal AMPA receptor assemblies and network activity. Although clinically approved AMPA receptor NAMs such as perampanel exist, a direct side-by-side pharmacological comparison under identical experimental conditions was beyond the scope of the present mechanistic investigation and will be valuable for future studies.
5. Conclusions
This study systematically investigated the modulatory effects of eight structurally analogous dibenzobarrelene derivatives on AMPA receptor activity, using specific receptor subunit combinations and auxiliary protein configurations. Using whole-cell patch-clamp electrophysiology in heterologous expression systems, we have demonstrated negative allosteric modulation of these compounds at homomeric GluA1 and heteromeric GluA1/2 receptors, with partial antagonism in the presence of the auxiliary receptor subunit TARPγ8. The data elucidate the kinetic mechanism of action of these compounds, which impede synaptic charge transfer while preserving receptor activation, and highlight the involvement of auxiliary subunits in modifying, but not abolishing, their effect. These observations establish a mechanistic and pharmacological foundation for future structural studies and functional validation in physiologically relevant models of excitatory neurotransmission.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jox16020050/s1, Table S1: Whole-Cell Recordings for Compound ND-1; Table S2: Whole-Cell Recordings for Compound ND-2; Table S3: Whole-Cell Recordings for Compound ND-3; Table S4: Whole-Cell Recordings for Compound ND-4; Table S5 Whole-Cell Recordings for Compound ND-5; Table S6: Whole-Cell Recordings for Compound ND-6; Table S7: Whole-Cell Recordings for Compound ND-7; Table S8: Whole-Cell Recordings for Compound ND-8; Table S9: Hill slope (nH) and IC50 values for ND compounds across AMPA receptor sub-types; Table S10: Goodness-of-fit (R2) values for nonlinear regression of ND compound concentration–response curves; Figure S1: Inhibitory effect of ND derivatives on glutamate-induced currents; ND derivatives’ full IUPAC names.
Author Contributions
Conceptualization, S.B. and M.Q.; methodology, İ.Ç., M.H., S.S. and M.Q.; software, M.Q.; validation, S.B., M.H. and M.Q.; formal analysis, M.Q.; investigation, S.B. and M.Q.; resources, İ.Ç., M.H., S.S. and M.Q.; data curation, S.B. and M.Q.; writing—original draft preparation, S.B. and M.Q.; writing—review and editing, S.B., İ.Ç., M.H., S.S., and M.Q.; supervision, M.Q. and M.H.; project administration, M.H. and M.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors are grateful to An-Najah National University (www.najah.edu, accessed on 2 February 2026) for its support in this research.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Beghi, E. The epidemiology of epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A.; Brigo, F.; Lattanzi, S.; Blumcke, I. Adult epilepsy. Lancet 2023, 402, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Milligan, T.A. Epilepsy: A Clinical Overview. Am. J. Med. 2021, 134, 840–847. [Google Scholar] [CrossRef]
- Bonansco, C.; Fuenzalida, M. Plasticity of hippocampal excitatory-inhibitory balance: Missing the synaptic control in the epileptic brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef]
- Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The role of glutamate receptors in epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Sears, S.M.S.; Hewett, S.J. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp. Biol. Med. 2021, 246, 1069–1083. [Google Scholar] [CrossRef]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E.J.N. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 2009, 462, 745–756. [Google Scholar] [CrossRef]
- Nakagawa, T.; Wang, X.-t.; Miguez-Cabello, F.J.; Bowie, D. The open gate of the AMPA receptor forms a Ca2+ binding site critical in regulating ion transport. Nat. Struct. Mol. Biol. 2024, 31, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, S.S. A model for predicting cation selectivity and permeability in AMPA and NMDA receptors based on receptor subunit composition. Front. Synaptic Neurosci. 2021, 13, 779759. [Google Scholar] [CrossRef]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef]
- Hanada, T. The AMPA Receptor as a Therapeutic Target in Epilepsy: Preclinical and Clinical Evidence; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 39–50. [Google Scholar]
- Guo, C.; Ma, Y.-Y. Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Trussell, L.O.; Fischbach, G.D. Glutamate receptor desensitization and its role in synaptic transmission. Neuron 1989, 3, 209–218. [Google Scholar] [CrossRef]
- Sun, Y.; Olson, R.; Horning, M.; Armstrong, N.; Mayer, M.; Gouaux, E. Mechanism of glutamate receptor desensitization. Nature 2002, 417, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Bissen, D.; Foss, F.; Acker-Palmer, A. AMPA receptors and their minions: Auxiliary proteins in AMPA receptor trafficking. Cell. Mol. Life Sci. 2019, 76, 2133–2169. [Google Scholar] [CrossRef]
- Dohrke, J.N.; Watson, J.F.; Birchall, K.; Greger, I.H. Characterizing the binding and function of TARP γ8-selective AMPA receptor modulators. J. Biol. Chem. 2020, 295, 14565–14577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.-y.; Chang, K.; Suh, Y.H.; Roche, K.W. TARP γ-8 glycosylation regulates the surface expression of AMPA receptors. Biochem. J. 2015, 465, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Tsujita, M.; Yamazaki, M.; Kushiya, E.; Abe, M.; Akashi, K.; Natsume, R.; Kano, M.; Kamiya, H.; Watanabe, M.; et al. Abundant distribution of TARP γ-8 in synaptic and extrasynaptic surface of hippocampal neurons and its major role in AMPA receptor expression on spines and dendrites. Eur. J. Neurosci. 2006, 24, 2177–2190. [Google Scholar] [CrossRef]
- Zhang, D.; Lape, R.; Shaikh, S.A.; Kohegyi, B.K.; Watson, J.F.; Cais, O.; Nakagawa, T.; Greger, I.H. Modulatory mechanisms of TARP γ8-selective AMPA receptor therapeutics. Nat. Commun. 2023, 14, 1659. [Google Scholar] [CrossRef]
- Hale, W.D.; Romero, A.M.; Gonzalez, C.U.; Jayaraman, V.; Lau, A.Y.; Huganir, R.L.; Twomey, E.C. Allosteric Competition and Inhibition in AMPA Receptors. bioRxiv 2023. [Google Scholar] [CrossRef]
- Qneibi, M.; Hawash, M.; Gümüş, M.; Çapan, İ.; Sert, Y.; Bdir, S.; Koca, İ.; Bdair, M. Deciphering the Biophysical Properties of Ion Channel Gating Pores by Coumarin–Benzodiazepine Hybrid Derivatives: Selective AMPA Receptor Antagonists. Mol. Neurobiol. 2023, 61, 4565–4576. [Google Scholar] [CrossRef] [PubMed]
- Stenum-Berg, C.; Musgaard, M.; Chavez-Abiega, S.; Thisted, C.L.; Barrella, L.; Biggin, P.C.; Kristensen, A.S. Mutational analysis and modeling of negative allosteric modulator binding sites in AMPA receptors. Mol. Pharmacol. 2019, 96, 835–850. [Google Scholar] [CrossRef]
- Stenum-Berg, C.; Abiega, S.C.; Thisted, C.L.; Kristensen, A.S. Identification and characterization of the binding pocket for negative allosteric modulators in AMPA receptors. Biophys. J. 2017, 112, 418a. [Google Scholar] [CrossRef][Green Version]
- Ge, Y.; Wang, Y.T. GluA1-homomeric AMPA receptor in synaptic plasticity and neurological diseases. Neuropharmacology 2021, 197, 108708. [Google Scholar] [CrossRef]
- Çapan, İ.; Servi, S. Synthesis of novel aza-heterocyclic derivatives from diester and diacid chlorides having the dibenzobarrelene skeleton. Synth. Commun. 2018, 48, 1164–1171. [Google Scholar] [CrossRef]
- Qneibi, M.; Bdir, S.; Bdair, M.; Aldwaik, S.A.; Sandouka, D.; Heeh, M.; Idais, T.I. AMPA receptor neurotransmission and therapeutic applications: A comprehensive review of their multifaceted modulation. Eur. J. Med. Chem. 2024, 266, 116151. [Google Scholar] [CrossRef]
- Golubeva, E.A.; Lavrov, M.I.; Radchenko, E.V.; Palyulin, V.A. Diversity of AMPA Receptor Ligands: Chemotypes, Binding Modes, Mechanisms of Action, and Therapeutic Effects. Biomolecules 2023, 13, 56. [Google Scholar] [CrossRef]
- Yelshanskaya, M.V.; Singh, A.K.; Sampson, J.M.; Narangoda, C.; Kurnikova, M.; Sobolevsky, A.I. Structural Bases of Noncompetitive Inhibition of AMPA-Subtype Ionotropic Glutamate Receptors by Antiepileptic Drugs. Neuron 2016, 91, 1305–1315. [Google Scholar] [CrossRef]
- Salussolia, C.L.; Corrales, A.; Talukder, I.; Kazi, R.; Akgul, G.; Bowen, M.; Wollmuth, L.P. Interaction of the M4 segment with other transmembrane segments is required for surface expression of mammalian α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 2011, 286, 40205–40218. [Google Scholar] [CrossRef]
- Pierce, V.D.; Niu, L. Stargazin and γ4 slow the channel opening and closing rates of GluA4 AMPA receptors. Sci. Rep. 2019, 9, 9570. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Jonas, P.; Spruston, N. Mechanisms shaping glutamate-mediated excitatory postsynaptic currents in the CNS. Curr. Opin. Neurobiol. 1994, 4, 366–372. [Google Scholar] [CrossRef]
- Rogawski, M.A. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol. Scand. 2013, 127, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Schmidt, D. Modern antiepileptic drug development has failed to deliver: Ways out of the current dilemma. Epilepsia 2011, 52, 657–678. [Google Scholar] [CrossRef]
- Rouach, N.; Byrd, K.; Petralia, R.S.; Elias, G.M.; Adesnik, H.; Tomita, S.; Karimzadegan, S.; Kealey, C.; Bredt, D.S.; Nicoll, R.A. TARP γ-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat. Neurosci. 2005, 8, 1525–1533. [Google Scholar] [CrossRef]
- Schwenk, J.; Harmel, N.; Brechet, A.; Zolles, G.; Berkefeld, H.; Müller, C.S.; Bildl, W.; Baehrens, D.; Hüber, B.; Kulik, A.; et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 2012, 74, 621–633. [Google Scholar] [CrossRef]
- Herguedas, B.; Kohegyi, B.K.; Dohrke, J.-N.; Watson, J.F.; Zhang, D.; Ho, H.; Shaikh, S.A.; Lape, R.; Krieger, J.M.; Greger, I.H. Mechanisms underlying TARP modulation of the GluA1/2-γ8 AMPA receptor. Nat. Commun. 2022, 13, 734. [Google Scholar] [CrossRef]
- Herguedas, B.; Watson, J.F.; Ho, H.; Cais, O.; García-Nafría, J.; Greger, I.H. Architecture of the heteromeric GluA1/2 AMPA receptor in complex with the auxiliary subunit TARP γ8. Science 2019, 364, aav9011. [Google Scholar] [CrossRef]
- Szénási, G.; Hársing, L.G., Jr. Pharmacology and prospective therapeutic usefulness of negative allosteric modulators of AMPA receptors. Drug Discov. Today Ther. Strat. 2004, 1, 69–76. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Twomey, E.C.; Yelshanskaya, M.V.; Grassucci, R.A.; Frank, J.; Sobolevsky, A.I. Structural bases of desensitization in AMPA receptor-auxiliary subunit complexes. Neuron 2017, 94, 569–580. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Hashizume, Y.; Tokuhara, N.; Takenaka, O.; Kohmura, N.; Ogasawara, A.; Hatakeyama, S.; Ohgoh, M.; Ueno, M.; Nishizawa, Y. Perampanel: A novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 2011, 52, 1331–1340. [Google Scholar] [CrossRef]
- Yelshanskaya, M.V.; Singh, A.K.; Narangoda, C.; Williams, R.S.B.; Kurnikova, M.G.; Sobolevsky, A.I. Structural basis of AMPA receptor inhibition by trans-4-butylcyclohexane carboxylic acid. Br. J. Pharmacol. 2022, 179, 3628–3644. [Google Scholar] [CrossRef] [PubMed]
- Qneibi, M.; Jaradat, N.; Hawash, M.; Olgac, A.; Emwas, N. Ortho versus Meta Chlorophenyl-2,3-Benzodiazepine Analogues: Synthesis, Molecular Modeling, and Biological Activity as AMPAR Antagonists. ACS Omega 2020, 5, 3588–3595. [Google Scholar] [CrossRef]
- Hawash, M.; Qneibi, M.; Natsheh, H.; Mohammed, N.H.; Hamda, L.A.; Kumar, A.; Olech, B.; Dominiak, P.M.; Bdir, S.; Bdair, M. Evaluating the Neuroprotective Potential of Novel Benzodioxole Derivatives in Parkinson’s Disease via AMPA Receptor Modulation. ACS Chem. Neurosci. 2024, 15, 2334–2349. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.