Abstract
Background and Clinical Significance: Inherited thrombocytopenia (IT) is a heterogeneous group of disorders caused by mutations in over 45 genes. Among these, ANKRD26-related thrombocytopenia (ANKRD26-RT) accounts for a notable subset and is associated with variable bleeding tendencies and an increased risk of myeloid malignancies. However, the extent of this oncogenic risk appears to vary between specific gene variants. Understanding the genotype–phenotype relationship is essential for patient counseling and management. This report presents a multigenerational family carrying the rare c.−118C > G variant in the 5′ untranslated region of ANKRD26, contributing to the discussion on variant-specific cancer predisposition. Case Presentation: Two sisters aged 57 and 60 presented with lifelong bleeding diathesis and moderate thrombocytopenia. Their symptoms included easy bruising, menorrhagia, and excessive postoperative bleeding. Genetic testing confirmed heterozygosity for the ANKRD26 c.−118C > G variant. Bone marrow analysis revealed abnormal megakaryopoiesis without evidence of dysplasia or somatic mutations. One sister underwent major surgery without complications when managed with prophylactic hemostatic therapy. Their family history included multiple female relatives with similar symptoms, although formal testing was limited. Notably, none of the affected individuals developed hematologic malignancy, and only one developed esophageal cancer, with no current evidence linking this variant to solid tumors. Conclusions: This case underscores the importance of distinguishing between ANKRD26 variants when assessing malignancy risk. While ANKRD26-RT is associated with myeloid neoplasms, the c.−118C > G variant may confer a lower oncogenic potential. Variant-specific risk stratification and genetic counseling are crucial for optimizing surveillance and avoiding unnecessary interventions in low-risk individuals.
1. Introduction
Inherited thrombocytopenia (IT) is part of a growing group of disorders caused by molecular defects involving more than 45 different genes [1,2,3]. The prevalence of IT is estimated at 2–3 per 100,000 individuals, although this is likely an underestimate. Milder cases often go unrecognized, and more severe cases may be misdiagnosed as acquired thrombocytopenia [4].
Inherited thrombocytopenias often present diagnostic challenges due to their clinical overlap with acquired forms, such as immune thrombocytopenia (ITP), myelodysplastic syndromes, or drug-induced thrombocytopenia. In the absence of syndromic features or a family history, these inherited forms may remain undetected for years. Misdiagnosis may lead to inappropriate treatment, including corticosteroids, immunoglobulins, or even splenectomy. Consequently, greater awareness and access to genetic testing are essential for accurate diagnosis and management of IT.
ANKRD26 (Ankyrin Repeat Domain-Containing 26)-related thrombocytopenia (ANKRD26-RT) accounted for 10% of IT cases in a large Italian cohort, making it one of the most common inherited thrombocytopenias [5]. However, the true prevalence for ANKRD26-RT remains unknown. All reported individuals with ANKRD26-RT have an affected parent, but there is significant phenotypic variability within families, which is consistent with variable expressivity. More than 70 families, encompassing over 200 affected individuals, have been described in the literature [4].
ANKRD26-RT is classified as a non-syndromic IT with a predisposition to myeloid neoplasms [6]. The majority of the causative variants are located in a 19-nucleotide region of the 5′ untranslated region (5′UTR; c.-116 to c.−134), which serves as a critical binding site for the transcription factors RUNX1 (runt-related transcription factor 1) and FLI1 (friend leukemia integration 1, also known as transcription factor ERGB), which are essential regulators of thrombopoiesis. The lifetime risk of myeloid transformation remains uncertain, although an 8% risk has been estimated [7]. Importantly, not all sequence variants within ANKRD26-RT have been associated with malignant transformation [8]. It is also worth noting that not only single-nucleotide substitutions have been reported, but also larger truncated mutations in the N-terminal [9,10].
We report a multigenerational family affected by ANKRD26-RT due to the heterozygous c.−118C > G variant. At least four individuals were affected, and none developed a myeloid malignancy. This variant has only been described in two individuals previously: a 32-year-old woman [11] and one other person of unspecified age and gender [12]. In the only previously detailed case of the c.−118C > G variant, the affected woman exhibited moderate thrombocytopenia and mild bleeding symptoms, with no signs of hematologic malignancy during follow-up. These findings are consistent with our current report and suggest that this variant may carry a lower oncogenic potential compared to more frequently reported variants such as c.−128G > A or c.−127C > T. However, due to the rarity of this variant, definitive conclusions cannot be drawn yet [13].
2. Materials/Case
Two sisters, aged 57 and 60, were referred for an evaluation of chronic thrombocytopenia and lifelong bleeding tendencies. Laboratory investigations confirmed moderate thrombocytopenia (see Table 1). Both reported easy bruising from early childhood, and both experienced menorrhagia and iron-deficiency anemia after menarche. Proband I (IV-5) had prolonged bleeding following dental extraction and suffered major hemorrhage after cesarean section; she was advised against further pregnancies. Proband II (IV-7) underwent hysterectomy at age 38 due to menorrhagia and experienced severe postoperative bleeding (see Figure 1 for family tree).

Table 1.
Blood cell counts at diagnosis and last follow-up of the probands.
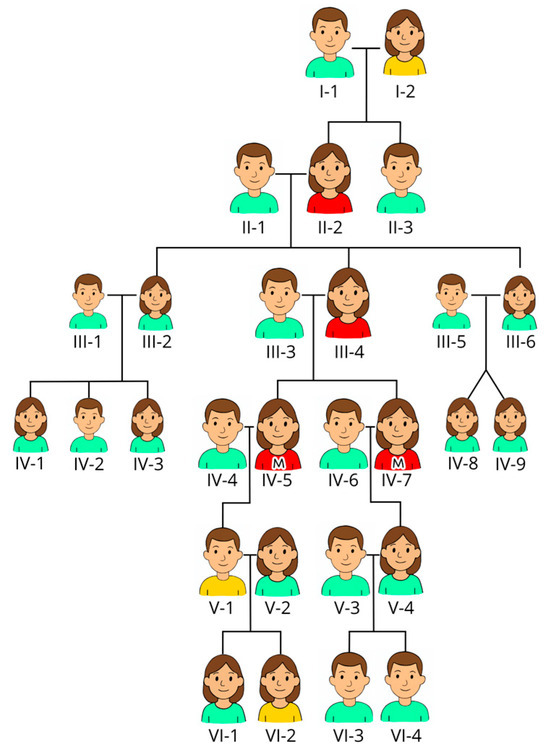
Figure 1.
Pedigree of the family. The males have short hair, and the females have long hair. We have used the color of their shirts as an indicator of affections: red indicates confirmed thrombocytopenia (but mutation is only confirmed in the two sisters marked with M on their shirts, also known as probands); yellow indicates suspicion of thrombocytopenia (likely but not confirmed); and green indicates unaffected persons. Proband I (IV-5. Proband II (IV-7).
Their maternal history revealed similar symptoms. Their mother (III-4) and grandmother (II-2) had documented thrombocytopenia (but the platelet count is not available), easy bruising, and menorrhagia. Their great-grandmother (I-2) died from postpartum hemorrhage, but detailed blood counts were not available at that time, while their great-grandfather (I-1) lived to an advanced age with no signs of bleeding disorder. V-1 has bleeding diathesis along with his daughter (VI-2), but he has refused assessment and has not been tested in terms of either blood counts or mutation status.
Although IT was suspected clinically, the diagnosis was confirmed in 2020 when molecular analysis identified the c.−118C > G variant in ANKRD26 (NM_014915.3).
After IT was suspected, IV-5 underwent major dental surgery and IV-7 received a hip arthroplasty without bleeding complications when treated prophylactically with desmopressin, tranexamic acid, and platelet transfusion. IV-7 died at age 74 from esophageal cancer. Bone marrow analysis performed in 2020 revealed a normal female karyotype (46, XX) and no somatic mutations on the Illumina TruSight Myeloid Panel. The trephine biopsy was slightly hypercellular. The majority (50–60%) of the megakaryocytes were abnormal with two hypolobular nuclei (mono- and bilobular). CD34 + cells accounted for 3–4% of the bone marrow cellularity, and they had a normal phenotype as assessed by flow cytometry. IV-5 remains in good health at 78. Her son (V-1) and granddaughter (VI-2) experience mild bleeding symptoms but have declined further investigation.
3. Methods and Results/Conclusions
3.1. Methods
Whole-exome sequencing was performed using Twist Exome 2.0 (Twist Bioscience, San Francisco, CA, USA) and NextSeq500 (Illumina, San Diego, CA, USA). We applied a standard bioinformatic approach for base calling (RTA, Dubai, United Arab Emirates), read alignment (BWA), and variant detection (best practices variant calling using GATK).
The Illumina’s TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA, USA) was used to identify somatic mutations in the bone marrow from proband IV-7. The panel covers 15 full genes (exons only) and 39 additional genes that are oncogenic hotspots. Alignment and variant calling were performed against GRCh37/hg19 with MiSeq Reporter Software v.2.6.3.2 (Illumina). SNVs and indels <60 bp were annotated by VariantStudio v.2.2. Pathogenicity was determined by using relevant databases. Sanger sequencing was not carried out.
3.2. Conclusions
Although patients with ANKRD26-RT generally have only mild bleeding symptoms, invasive procedures still pose a risk. There are no consensus guidelines for periprocedural hemostasis, but most case series recommend prophylactic measures such as desmopressin, antifibrinolytics, and platelet transfusion when platelet counts fall below 50 × 109/L or in the setting of major surgery. The effective use of this regimen in IV-5 and IV-7 underscores the importance of individualized planning. Long-term management may include iron supplementation for chronic blood loss and gynecologic interventions for menorrhagia.
Thrombocytopenia in ANKRD26-RT is fully penetrant, but is typically mild to moderate with platelet counts ranging from 30 to 150 × 109/L (mean ~48 × 109/L). Counts below 10 × 109/L are rare. Bleeding symptoms are often mild and mucocutaneous in nature, with easy bruising, epistaxis, and menorrhagia being common. Given the subtle phenotype and the reliance on molecular diagnostics, which may not be widely available, the diagnosis is often missed or delayed.
Although pathogenic variants in ANKRD26 have been associated with an increased risk of myeloid malignancies, this risk appears to vary by specific sequence variant. A case series of 118 individuals with confirmed or probable ANKRD26-RT reported an 8% incidence of myeloid malignancy. One report suggested a 24-fold increased risk [8,12], although this may reflect ascertainment bias. Notably, some variants, including c.−118C > G, have not been associated with malignancy to date.
While myeloid malignancies are the primary concern in ANKRD26-RT, there have been isolated reports of other cancers, such as solid tumors, in affected individuals [13]. However, the causal relationship between ANKRD26 variants and these malignancies remains elusive. In our case, IV-7 developed esophageal cancer at age 74, but there is currently no evidence linking the c.−118C > G variant with increased solid tumor risk. Larger cohort studies are needed to clarify whether any ANKRD26 variants predispose individuals to non-hematologic malignancies.
This family contributes to the growing body of evidence suggesting that the malignant potential of ANKRD26-RT may depend on the specific variant. Continued reporting of genotype–phenotype correlations is essential for refining prognostic estimates and guiding individualized management and surveillance strategies.
From a clinical perspective, identifying the specific ANKRD26 variant in affected families allows for more precise counseling regarding bleeding risk and cancer surveillance [14,15,16]. Cascade genetic testing can help identify at-risk relatives who may benefit from tailored monitoring or prophylaxis before surgical interventions. As variant-specific data accumulate, it may become possible to stratify patients by risk and refine surveillance protocols, potentially avoiding unnecessary anxiety or overtesting in low-risk individuals.
In summary, we present a multigenerational case of ANKRD26-RT caused by the c.−118C > G variant, with no cases of myeloid malignancy observed. Two other nucleotide changes have been reported in at least six other families (c.−118C > A and c.−188CT) [5,6,15,16,17]. Our findings support the hypothesis that not all ANKRD26 variants confer an equal risk of malignant transformation. This underscores the need for variant-specific risk stratification and the continued accumulation of genotype–phenotype data to improve patient care.
Author Contributions
E.B.T. and G.E.T. came up with the idea for the study and wrote the manuscript; S.S. and K.T. were responsible for the methods and data for genetic testing, and they also participated in correcting and proofreading the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
There is no need for ethical approval in this case report since there are no human experiments. The ethics committee leader at Oslo University Hospital is Tor Åsmund Martinsen, who was contacted and indicated that no approval was needed.
Informed Consent Statement
The sisters have given their written and oral consent to publish their story.
Data Availability Statement
All data regarding this publication can be found in the medical journal of the two probands, at Oslo University Hospital, Rikshospitalet.
Acknowledgments
We would like to acknowledge Ai Phi Thuy Ho at Kalnes Hospital Trust for her help with the original drawing of the pedigree of the family, making it clear and informative.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Noris, P.; Pecci, A. Hereditary thrombocytopenias: A growing list of disorders. Hematology Am. Soc. Hematol. Educ. Program 2017, 2017, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.T.; Di Paola, J. Genetics of inherited thrombocytopenias. Blood 2022, 139, 3264–3277. [Google Scholar] [CrossRef] [PubMed]
- Lassandro, G.; Palladino, V.; Faleschini, M.; Barone, A.; Boscarol, G.; Cesaro, S.; Chiocca, E.; Farruggia, P.; Giona, F.; Gorio, C.; et al. “Children with Inherited Platelet disorders Surveillance” (CHIPS) retrospective and prospective observational cohort study by Italian Association of Pediatric Hematology and Oncology (AIEOP). Front. Genet. 2022, 10, 967417. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Palmer, E.L.; Botero, J.P. ANKRD26-Related Thrombocytopenia and Predisposition to Myeloid Neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5′UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant for of inherited thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M. ANKRD26-realted thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Vyas, H.; Alcheikh, A.; Lowe, G.; Stevenson, W.S.; Morgan, N.V.; Rabbolini, D.J. Prevalence and natural history of variants in the ANKRD26 gene: A short review and update of reported cases. Platelets 2022, 33, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Marconi, C.; Canobbio, I.; Bozzi, V.; Pippucci, T.; Simonetti, G.; Melazzini, F.; Angori, S.; Martinelli, G.; Saglio, G.; Torti, M.; et al. 5′UTR point substitution and N-terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Wahlster, L.; Verboon, J.M.; Ludwig, L.S.; Black, S.C.; Luo, W.; Garg, K.; Voit, R.A.; Collins, R.L.; Garimella, K.; Costello, M.; et al. Familial thrombocytopenia due to complex structural resulting in a WAC-ANKRD26 fusion transcript. J. Exp. Med. 2021, 218, e20210444. [Google Scholar] [CrossRef] [PubMed]
- Diep, R.T.; Corey, K.; Arcasoy, M.O. A novel nucleotide substitution in the 5′ untranslated region of ANKRD26 gene is associated with inherited thrombocytopenia: A report of two new families. Ann. Hematol. 2019, 98, 1789–1791. [Google Scholar] [CrossRef] [PubMed]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high throughput sequencing of 2396 patients with bleeding, trombotic and plateles disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Expression of ANKRD26 in Cancer—Summary—The Human Protein Atlas. Proteinatlas.org. Available online: https://www.proteinatlas.org/ENSG00000107890-ANKRD26/cancer (accessed on 23 July 2025).
- Hofbrucker-MacKenzie, S.A.; Englisch, A.S.; Izadi, M.; Metzner, K.; Kessels, M.M.; Qualmann, B. Ankrd26 is critical for cell differentiation and cancer-linked mutations affect its key properties. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zidan, N.I.; AbdElmonem, D.M.; Elsheikh, H.M.; Metwally, E.A.; Mokhtar, W.A.; Osman, G.M. Relation between mutations in the 5′ UTR of ANKRD26 gene and inherited thrombocytopenia with predisposition to myeloid malignancies. An Egyptian study. Platelets 2021, 32, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.; Hantel, A.; Lorenz, R.; Neistadt, B.; Wong, J.; Churpek, J.E.; Al Mardini, N.; Shaukat, I.; Gurbuxani, S.; Miller, J.L.; et al. A new family with a germline ANKRD26 mutation and predisposition to myeloid malignancies. Leuk. Lymphoma 2014, 55, 2945–2946. [Google Scholar] [CrossRef] [PubMed]
- Boutroux, H.; Petit, A.; Auvrignon, A.; Lapillonne, H.; Ballerini, P.; Favier, R.; Leverger, G. Childhood diagnosis of genetic thrombocytopenia with mutation in the ankyrine repeat domain 26 gene. Eur. J. Pediatr. 2015, 174, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).