
ANKRD26 Gene Mutation and Thrombocytopenia—Is the Risk of Malignancy Dependent on the Mutation Variant?


Abstract
1. Introduction
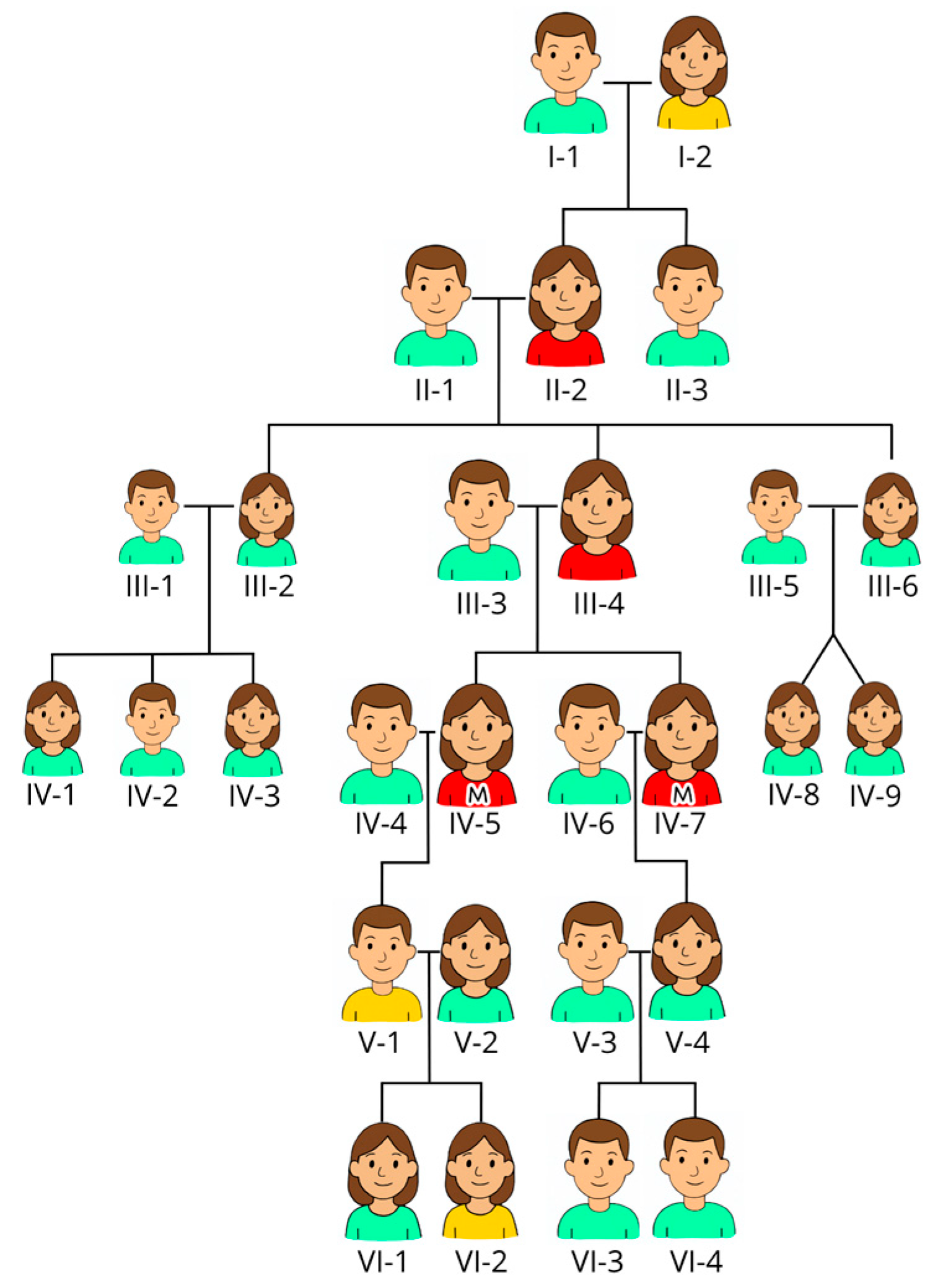
2. Materials/Case
3. Methods and Results/Conclusions
3.1. Methods
3.2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noris, P.; Pecci, A. Hereditary thrombocytopenias: A growing list of disorders. Hematology Am. Soc. Hematol. Educ. Program 2017, 2017, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.T.; Di Paola, J. Genetics of inherited thrombocytopenias. Blood 2022, 139, 3264–3277. [Google Scholar] [CrossRef] [PubMed]
- Lassandro, G.; Palladino, V.; Faleschini, M.; Barone, A.; Boscarol, G.; Cesaro, S.; Chiocca, E.; Farruggia, P.; Giona, F.; Gorio, C.; et al. “Children with Inherited Platelet disorders Surveillance” (CHIPS) retrospective and prospective observational cohort study by Italian Association of Pediatric Hematology and Oncology (AIEOP). Front. Genet. 2022, 10, 967417. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Palmer, E.L.; Botero, J.P. ANKRD26-Related Thrombocytopenia and Predisposition to Myeloid Neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5′UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant for of inherited thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M. ANKRD26-realted thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Vyas, H.; Alcheikh, A.; Lowe, G.; Stevenson, W.S.; Morgan, N.V.; Rabbolini, D.J. Prevalence and natural history of variants in the ANKRD26 gene: A short review and update of reported cases. Platelets 2022, 33, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Marconi, C.; Canobbio, I.; Bozzi, V.; Pippucci, T.; Simonetti, G.; Melazzini, F.; Angori, S.; Martinelli, G.; Saglio, G.; Torti, M.; et al. 5′UTR point substitution and N-terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Wahlster, L.; Verboon, J.M.; Ludwig, L.S.; Black, S.C.; Luo, W.; Garg, K.; Voit, R.A.; Collins, R.L.; Garimella, K.; Costello, M.; et al. Familial thrombocytopenia due to complex structural resulting in a WAC-ANKRD26 fusion transcript. J. Exp. Med. 2021, 218, e20210444. [Google Scholar] [CrossRef] [PubMed]
- Diep, R.T.; Corey, K.; Arcasoy, M.O. A novel nucleotide substitution in the 5′ untranslated region of ANKRD26 gene is associated with inherited thrombocytopenia: A report of two new families. Ann. Hematol. 2019, 98, 1789–1791. [Google Scholar] [CrossRef] [PubMed]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high throughput sequencing of 2396 patients with bleeding, trombotic and plateles disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Expression of ANKRD26 in Cancer—Summary—The Human Protein Atlas. Proteinatlas.org. Available online: https://www.proteinatlas.org/ENSG00000107890-ANKRD26/cancer (accessed on 23 July 2025).
- Hofbrucker-MacKenzie, S.A.; Englisch, A.S.; Izadi, M.; Metzner, K.; Kessels, M.M.; Qualmann, B. Ankrd26 is critical for cell differentiation and cancer-linked mutations affect its key properties. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zidan, N.I.; AbdElmonem, D.M.; Elsheikh, H.M.; Metwally, E.A.; Mokhtar, W.A.; Osman, G.M. Relation between mutations in the 5′ UTR of ANKRD26 gene and inherited thrombocytopenia with predisposition to myeloid malignancies. An Egyptian study. Platelets 2021, 32, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.; Hantel, A.; Lorenz, R.; Neistadt, B.; Wong, J.; Churpek, J.E.; Al Mardini, N.; Shaukat, I.; Gurbuxani, S.; Miller, J.L.; et al. A new family with a germline ANKRD26 mutation and predisposition to myeloid malignancies. Leuk. Lymphoma 2014, 55, 2945–2946. [Google Scholar] [CrossRef] [PubMed]
- Boutroux, H.; Petit, A.; Auvrignon, A.; Lapillonne, H.; Ballerini, P.; Favier, R.; Leverger, G. Childhood diagnosis of genetic thrombocytopenia with mutation in the ankyrine repeat domain 26 gene. Eur. J. Pediatr. 2015, 174, 1399–1403. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| IV-5 14 January 2007 | IV-5 21 November 2022 | IV-7 08 January 2007 | IV-7 01 December 2021 | Reference Value | |
|---|---|---|---|---|---|
| Hemoglobin g/dL | 14.1 | 14.5 | 13.9 | 14.2 | 11.1–15.3 |
| Erythrocytes 1012/L | 4.6 | 4.9 | 4.4 | 3.9–5.2 | |
| MCV fL | 88 | 89 | 94 | 82–98 | |
| Thrombocytes 109/L | 52 | 49 | 51 | 43 | 145–390 |
| Leukocytes 109/L | 13.1 | 13.5 | 14.2 | 12.1 | 3.5–10.0 |
| Granulocytes 109/L | 10.3 | 9.5 | 9.5 | 7.7 | 1.5–7.3 |
| Lymphocytes 109/L | 2.2 | 2.8 | 3.1 | 2.8 | 1.1–3.3 |
| Monocytes 109/L | 0.5 | 1.0 | 1.1 | 0.9 | 0.2–0.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tjønnfjord, E.B.; Tveten, K.; Spetalen, S.; Tjønnfjord, G.E. ANKRD26 Gene Mutation and Thrombocytopenia—Is the Risk of Malignancy Dependent on the Mutation Variant? Hematol. Rep. 2025, 17, 37. https://doi.org/10.3390/hematolrep17040037
Tjønnfjord EB, Tveten K, Spetalen S, Tjønnfjord GE. ANKRD26 Gene Mutation and Thrombocytopenia—Is the Risk of Malignancy Dependent on the Mutation Variant? Hematology Reports. 2025; 17(4):37. https://doi.org/10.3390/hematolrep17040037
Chicago/Turabian StyleTjønnfjord, Eirik B., Kristian Tveten, Signe Spetalen, and Geir E. Tjønnfjord. 2025. "ANKRD26 Gene Mutation and Thrombocytopenia—Is the Risk of Malignancy Dependent on the Mutation Variant?" Hematology Reports 17, no. 4: 37. https://doi.org/10.3390/hematolrep17040037
APA StyleTjønnfjord, E. B., Tveten, K., Spetalen, S., & Tjønnfjord, G. E. (2025). ANKRD26 Gene Mutation and Thrombocytopenia—Is the Risk of Malignancy Dependent on the Mutation Variant? Hematology Reports, 17(4), 37. https://doi.org/10.3390/hematolrep17040037