Abstract
Targeting kinase activity is considered to be an attractive therapeutic strategy to overcome acute myeloid leukemia (AML) since aberrant activation of the kinase pathway plays a pivotal role in leukemogenesis through abnormal cell proliferation and differentiation block. Although clinical trials for kinase modulators as single agents remain scarce, combination therapies are an area of therapeutic interest. In this review, the author summarizes attractive kinase pathways for therapeutic targets and the combination strategies for these pathways. Specifically, the review focuses on combination therapies targeting the FLT3 pathways, as well as PI3K/AKT/mTOR, CDK and CHK1 pathways. From a literature review, combination therapies with the kinase inhibitors appear more promising than monotherapies with individual agents. Therefore, the development of efficient combination therapies with kinase inhibitors may result in effective therapeutic strategies for AML.
1. Introduction
Acute myeloid leukemia (AML) is a clonal disease that is derived from abnormally and, occasionally, poorly differentiated cells of the hematopoietic system [1]. Improvements in AML treatment in younger patients over the past 35 years have largely been due to the dose escalation of chemotherapy and better supportive care [2]. Meanwhile, several attempts to improve antileukemic activity, other than conventional chemotherapy, have emerged. These are epigenetic therapies (e.g., 5-azacitidine), isocitrate dehydrogenase (IDH) inhibitors, and fms-like kinase 3 (FLT3) inhibitors [3,4], which have been employed in clinical practices in these several years. These specific inhibitors were not necessarily developed with the intent of manipulating cellular differentiation; however, they are a central part of the mechanism for these drugs, including cellular differentiation [3].
Accumulated evidences suggests that abnormal activation of signal transduction pathways plays a pivotal role in leukemogenesis, through the blocking of differentiation and abnormal cell proliferation [5]. Kinase inhibitors can serve as differentiation inducers, and the author recently described the application of kinase inhibitors for differentiation therapy in AML [6]. Therefore, targeting kinase activity is considered an attractive therapeutic strategy to overcome AML. Differentiation therapy employing all-trans-retinoic acid for acute promyelocytic leukemia (APL) dramatically improved the clinical outcome in the 1990s. Meanwhile, several attempts to improve antileukemic activity in older patients with diseases other than APL by using hypomethylating agents with low-dose cytarabine showed favorable results [7]. Therefore, the expansion of differentiation therapy for clinical application outside of APL may be attractive for improving clinical outcomes [4]. The author recently described the application of kinase inhibitors for differentiation therapy in AML [6]. Clinical trials for kinase modulators as single agents remain scarce and have shown limited effects [6]. For example, the potential efficacy of epidermal growth factor receptor (EGFR) inhibitors was reported for non-small cell lung cancer, but it was concluded that EGFR inhibitors were not appropriate as single agents for advanced AML [8,9]. Therefore, combination therapy is an area of therapeutic interest that is being pursued.
To follow this issue in the present review, the author summarizes attractive kinase pathways for therapeutic targets and the combination strategies for these pathways. During a search of the literature on PubMed with the keywords “kinase”, “inhibitor”, “combination”, “AML”, “therapy”, and “clinical”, more than 500 papers were found to have been published to date. Based on the search, the FLT3, PI3K/AKT/mTOR, MAPK, AXL, CDK, and CHK1 pathways appear to be common pathways targeted by kinase inhibitor combination therapies. In the present review, the author summarizes these attractive kinase pathways for therapeutic targets and the combination strategies for these pathways.
2. Combination Therapy Targeting the FLT3 Signaling Pathway
Mutations of FLT3 comprise one of the most frequently identified types of genetic alterations in AML [10]. As the biological role of FLT3 is important in the pathogenesis of AML, through the activation of downstream kinase signaling pathways [10,11], the clinical development of FLT3 tyrosine kinase inhibitors (TKIs) has been one of the most active fields in precision medicine for AML [12,13].
Several resistance mechanisms for FLT3 inhibitor therapy have been shown, such as additional mutations in the kinase domain in internal tandem duplication (ITD) patients [14], or another kinase pathway activation [15]. To circumvent resistance, as well as to increase efficiency, ongoing efforts are focusing on the development of combinational strategies. We first discovered [16,17], followed by the others [18,19], who showed that FLT3 inhibitor, in combination with arsenic trioxide, acts synergistically on primary AML cells or AML cell lines with FLT3 mutations. Not only for these in vitro models, thus far, reported results of trials combining FLT3 TKIs with induction and consolidation chemo- therapy in the first-line setting have been encouraging [13,20]. Stone et al. [21] previously reported the result of a phase 1b trial. They investigated several schedules and doses of midostaurin, in combination with cytarabine and daunorubicin induction and post-remission therapy of high dose cytarabine in newly diagnosed AML patients. They revealed that midostaurin, in combination with standard chemotherapy, demonstrated high complete response and overall survival (OS) rates in newly diagnosed younger adults with AML. Subsequently, based on the findings of the RATIFY trial, the US Food and Drug Administration (FDA) approved midostaurin in 2017. The FDA approved midostaurin to be used in combination with standard induction therapy with cytarabine and daunorubicin and consolidation therapy with cytarabine in FLT3-mutated newly diagnosed young (18–59 years) patients with AML [22]. Ofran et al. [23] investigated the roles of midostaurin in patients’ survival who were initially treated with intensive chemotherapy plus midostaurin and then proceed to allo-stem-cell transplantation (SCT) in the first complete remission (CR) [23]. In a multivariate analysis, midostaurin use and allo-SCT in CR1 were the most significant factors affecting overall survival (OS). Midostaurin incorporation into chemotherapy regimens significantly improved CR + CR, with incomplete hematologic recovery rates (p = 0.002) and reduced relapse rates (p = 0.02); it was also remarkably advantageous for FLT3-ITD high-allelic ratio patients (2-year OS of 82%) [23].
FLT3 inhibitors are tyrosine kinase inhibitors and are classified into first- and second-generation inhibitors based on their kinase specificity and potency [24]. First-generation inhibitors include midostaurin and sorafenib. Second-generation inhibitors include quizartinib and gilteritinib. First-generation inhibitors lack specificity to FLT3 and are therefore not as potent as second-generation FLT3 inhibitors, which have been designed to only target FLT3. However, first-generation FLT3 inhibitors can target downstream of FLT3 and may also be effective in parallel signaling and in other targets in AML at diagnosis, which is characterized by the coexistence of multiple leukemic clones [25,26]. Meanwhile, a dominant clone with FLT3 mutations tends to emerge at relapse [26], and it may be better targeted by the second-generation FLT3 inhibitors.
As there are many excellent reviews on FLT3 inhibitors and their combinations [13,27,28,29], in this section, the author introduces essential clinical trials other than FLT3 inhibition plus chemotherapy, such as the combination of the first-generation FLT3 inhibitor sorafenib with the hypomethylating agent 5-azacytidine.
This combination was shown to be well tolerated in older patients with untreated FLT3-ITD AML [30], or underwent a median of 2 prior regimens for treatment (range 0–7) [31]. The majority (53%) of patients experienced grade < 3 adverse effects attributable to sorafenib, and the most common grade ≥ 3 adverse events were thrombocytopenia, neutropenia, anemia, and neutropenia with fever or infection [31]. Another promising combination with FLT3 inhibitor may be venetoclax with decitabine. Maiti et al. [32] reported that the outcomes of newly diagnosed patients with a 2-year OS of 80% compare favorably with prior reports [30] of sorafenib with low-intensity therapy, yielding overall response rates of 78% and a median OS of 5.3–9.2 months.
3. Combination Therapy Targeting the PI3K/AKT/mTOR Signaling Pathway
The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway is one of the key aberrant intracellular axes in AML [33]. Chemotherapeutic combinations such as cytarabine with AKT inhibitor (MK-2206) have been reported [34]. The PI3K/AKT/mTOR signaling pathway, related pathways, and molecule-specific inhibitor combinations are depicted in Figure 1. Sandhöfer et al. [35] demonstrated a broad range of cytotoxic activities for the inhibition of PI3K/mTORC1/2 (BEZ-235), MK-2206, and TORC1 (rapamycin), with high efficacies for cells carrying a lysine methyltransferase (KMT) 2A rearrangement [35]. The pharmacologic inhibition of lysine-specific demethylase 1 (LSD1) promoted a differentiation blockade especially in AML cells with MLL chromosomal translocations [36]. Deb et al. [37] found that mTORC1 signaling was a target of LSD1. From a dropout screen of a genome-wide CRISPR-Cas9, they revealed multiple components of mTORC1 signaling by LSD1 inhibition (LSDi). They also demonstrated that mTORC1 pharmacologic inhibition with LSDi enhanced differentiation in both the cell line and primary cell settings [37]. Abdel-Aziz et al. [38] found that mTOR was involved in mediating the resistance of leukemic cells to LSDi. Of note, the inhibition of mTOR unlocked the resistance of AML cell lines and primary patient-derived blasts to LSDi both in vitro and in vivo [38].
Bertacchini et al. [39] investigated 80 samples of primary cells from AML patients and found that inhibition of Akt and mTOR resulted in paradoxical activation of growth factor receptor tyrosine kinases (RTKs). Accordingly, dual inhibition of RTKs and AKT displayed synergistic potent cytotoxic effects in a pre-clinical model [39]. However, in a phase II study on 23 AML patients with RAS mutations, combined MEK and AKT inhibition had no clinical activity [40]. This may be explained by the fact that the maximum tolerated clinical dose might not reflect the dosing necessary to produce the desired biological effect of this combination [40].
Another kinase pathway related to AKT/mTOR signaling involves PIM kinases and p38a (MAPK14) [41]. Signaling from Akt and PIM kinase converges to control output from the mTOR signaling axis via regulation of upstream and downstream effectors [42]. PIM kinases (PIM1, 2, and 3) are involved in cell proliferation and survival signaling and are emerging as therapeutic targets for various malignancies. Dual inhibition with PIM kinase and AKT inhibitors was reported to show synergistic cytotoxicity in AML [43].
Meja et al. [43] found that a significant portion of primary AML samples showed PIM1 and PIM2 expression, and thus examined the effect of pan-PIM inhibitor AZD1897 on AML cell growth and survival. In their study, PIM inhibition had limited single-agent activity in AML cell lines and primary AML cells, but significant synergy was seen when AZD1897 was combined with Akt inhibitor AZD5363 [43]. PIM kinases are often overexpressed in AML and other hematological malignancies, but the effect of single-agent PIM inhibitor treatment is marginal. Brunen et al. [41] demonstrated that PIM inhibition induced reactive oxygen species production, leading to activation of p38α and downstream AKT/mTOR signaling. Accordingly, inhibition of p38α, combined with PIM kinase inhibition by AZD1208, had a profound effect on AML cells [41].
Recently, metformin, a classic hypoglycemic drug for diabetes, was reported to synergistically sensitize AML cells to Ara-C through mTORC1/P70S6K pathway inhibition [44].
However, concerns about combinations with PI3K/AKT inhibitors have been raised. Liang et al. [45] revealed that GLI1 overexpression in AML cells led to increased AKT phosphorylation and decreased Ara-C sensitivity, which was attenuated by GLI1 inhibition. PI3K inhibition profoundly affected GLI1 expression and co-inhibition of GLI1- and PI3K-induced apoptosis of hematopoietic stem/progenitor cells, raising the possibility for serious side effects of this treatment.
Clinical trials on mTOR inhibitors have been conducted. In the early 2010s, the results of the phase Ib GOELAMS study of the mTOR inhibitor RAD001 [46] and the phase II GIMEMA study (AML-1107) of temsirolimus [47] were published. Overall, these studies revealed that the inhibitors had acceptable toxicity and led to improved outcomes after treatment. The latter study examined the combination of an mTOR inhibitor, temsirolimus, and low dose of clofarabine in older patients with AML as salvage therapy. The overall remission rate (ORR) was 21% (8% complete remission (CR), and 13% CR without full blood count recovery) in 53 evaluable patients. The median disease-free survival was 3.5 months, and the median overall survival was 4 months (9.1 months for responders). In 2018, a clinical trial combining an mTORC1 inhibitor (sirolimus) and MEC (mitoxantrone, etoposide, and cytarabine) was performed in relapsed, refractory, or untreated high-risk AML patients [48]. The ORR among patients with mTORC1 inhibition and baseline target activation during therapy was 71% (12/17), compared with 20% (2/10) in patients without target inhibition. These data provide clinical confirmation that activation of mTORC1 mediates chemotherapy resistance in AML patients.
4. Combination Therapy Targeting the MAPK Signaling Pathway
One of the most aberrantly activated oncogenic pathways in AML is the RAS-RAF-MEK-ERK (MAPK) pathway [49]. However, in clinical trials, the targeting of this pathway by MEK inhibitors was not proven to be effective. Jain et al. [50] previously demonstrated the effect of selumetinib, an oral small-molecule inhibitor of MEK, as a modest single antileukemic agent in advanced AML. In parallel, common drug-related toxicities were mild, such as grade 1–2 diarrhea, fatigue, nausea, vomiting, and skin rash. Together with these, a combination with drugs that target other signaling pathways than MEK should be considered in AML.
MAPK-interacting kinases 1 and 2 (MNK1/2) are downstream effectors of this pathway that control the activation of eukaryotic translation factor 4E (eIF4E) [51] (Figure 1). eIF4E was reported to be overexpressed in AML and to play a role in AML pathogenesis [52,53]. Saurez et al. [54] demonstrated an effect of tomivosertib, the highly selective MNK1/2 inhibitor, on AML cells. The inhibition of Mnk was also reported to enhance the apoptotic activity of cytarabine in AML cells [55]. Furthermore, eIF4E inhibition was shown to enhance the effect of FLT3 inhibitors on both internal tandem duplication and tyrosine kinase domain mutants [56]. Altman et al. [57] examined whether cercosporamide, an antifungal agent that acts as a unique Mnk inhibitor, exhibits antileukemic properties. They found that treatment of AML cells with cercosporamide resulted in dose-dependent suppression of eIF4E phosphorylation and that the combination of cercosporamide with cytarabine resulted in enhanced antileukemic responses in a xenograft mouse model in vivo [57].
Antiapoptotic Bcl-2 family members are critical for the survival of AML cells. The combination of venetoclax and tomivosertib showed synergistic anti-leukemic responses in AML cells [54]. In a similar context, Tambe et al. [58] reported that pan-RAF inhibition, but not MEK inhibition, caused cell death in 29% of AML samples. Pan-RAF inhibition was not toxic to normal bone marrow cells. Furthermore, pan-RAF inhibition induced apoptosis in AML cells and synergized with BCL2 inhibition [58]. Cremer et al. [59] conducted a genome-scale open-reading-frame resistance screen and identified RAS-MAPK-ERK pathway activation as a major mechanism of resistance to SYK inhibitors. They further demonstrated that an MEK and SYK inhibitor combination was synergistic in vitro and in vivo [59]. Gefitinib, an EGFR inhibitor, was reported to induce differentiation [60] through an off-target effect of Syk family kinase inhibition [61]. Recently, it was reported that phosphorylated EGFR and EGFR ligand were expressed in 19% and 29%, respectively, of blast cells from APL patients, but not in those from healthy controls [62]. The same study further showed that the combination of gefitinib with ATRA and ATO promoted myeloid cell differentiation in ATRA- and ATO-resistant APL cells [62].
5. Combination Therapy Targeting AXL
AXL, named after the Greek word “anexelekto”, meaning uncontrolled, is a member of the TAM family of receptor tyrosine kinases. AXL potentially drives cell proliferation through effector molecules in the PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, JAK/STAT, and NF-kB signaling pathways [63]. Inhibition of AXL sensitized AML stem/progenitor cells to venetoclax treatment, with strong synergistic effects in vitro and in xenotransplantation models [64]. It was reported that combined treatment with the DNA methyltransferase inhibitor decitabine and histone deacetylase inhibitor vorinostat synergistically inhibited AML cell viability and induced AXL expression [65]. Triple combination treatment with AXL-specific inhibitor BGB324 further increased the sensitivity compared with the decitabine–vorinostat combination treatment [65].
6. Combination Therapy Targeting the CDK Signaling Pathway
C Cyclin-dependent kinases (CDKs) 1, 2, 4, and 6 are mainly involved in regulation of the cell cycle, while CDK7, 8, and 9 play roles in regulating transcription to further influence survival and cell proliferation by driving the target gene expressions [66]. Among the CDKs, CDK9 is probably the most attractive target, in combination with other inhibitors or chemotherapy, for hematological malignancies [67,68,69,70,71,72]. Note that Zeidner et al. [72] demonstrated that Alvocidib, a potent and nonselective CDK9 inhibitor, can be safely administrated prior to 7 + 3 (cytarabine + daunorubicin) induction with encouraging clinical activity. There was one dose-limiting toxicity of cytokine release syndrome. The most common grade ≥ 3 nonhematologic toxicities were diarrhea (44%) and tumor lysis syndrome (34%) [72].
CDK9 is a transcriptional regulator of myeloid cell leukemia-1 (MCL-1) that can influence apoptosis induction [73,74]. CDK9 is also a global transcriptional regulator that forms part of the super-elongation complex controlling RNA polymerase II phosphorylation and elongation [73,75] (Figure 2). Several studies have reported effects of other CDKs, such as CDK2 [76] and CDK6 [77,78]. CDK2 suppression was reported to synergize with all-trans-retinoic acid to overcome the myeloid differentiation blockade of AML cells [76].
Recently, histone methyltransferase EZH2 loss and the subsequent reduction in trimethylation of histone H3K27 were reported to result in the de-repression of HOX genes as a novel pathway for acquired resistance to tyrosine kinase inhibitors and cytotoxic drugs in AML [79]. Specifically, CDK1 inhibition prevented the degradation of EZH2, thereby restored drug sensitivity, suggesting the importance of CDK inhibition in leukemia therapy [79].
7. Combination Therapy Targeting the CHK1 Signaling Pathway
The DNA damage checkpoint is regulated by two signaling pathways, ATM-CHK2-p53 and ATR-CHK1-cdc25A, of which the ATM-CHK2-p53 pathway is impaired in many cancers [80] (Figure 3). Consequently, agents that inhibit ATR-CHK1-cdc25A, especially CHK1, are very attractive for the development of efficient therapies [81] (Figure 3). CHK1 is a protein kinase that regulates cell cycle progression in response to checkpoint activation. It was reported that cytarabine, a gold-standard chemotherapeutic agent for AML, activates the replication checkpoint kinases CHK1 and ATR. In turn, cytarabine regulates a series of cellular responses that aid survival during replication stress [82]. Indeed, a selective CHK1 inhibitor was shown to enhance the cytotoxicity of cytarabine [82]. CPX-351 is a liposomal formulation encapsulating cytarabine and daunorubicin that has received approval for treatment of AML. Recently, the addition of a CHK1 inhibitor, MK-8776 or CHK1 knockdown, was found to enhance CPX-351-induced apoptosis in multiple AML cell lines and primary samples [83]. Although there are many promising results in pre-clinical models, clinical success has not been achieved to date [84]. For example, a randomized phase II trial of combination therapy with Ara-C and CHK1 inhibitor MK-8776 produced somewhat disappointing results [85]. Thirty-two patients with relapsed or primary refractory AML were randomized 1:1 to receive either AraC with MK-8776 (Arm A, 14 patients); or AraC alone (Arm B, 18 patients). Response rates and survival were similar between the two groups in spite of evidence that MK-8776 augmented DNA damage in circulating leukemic blasts. There was an increase in asymptomatic grade III prolongation of the QTcF interval among patients receiving MK-8776, Arm B, which was also reported in the phase I trial and is likely attributable to MK-8776 [86]. Di Tullio et al. [87] shed light on another combination strategy involving granulocyte-colony-stimulating factor (G-CSF). They reported that the CHK inhibitor GDC-0575 enhanced the cytotoxicity of Ara-C in different AML cell lines and had effects on AML-cell-line-injected NOD/Scid gamma IL2Rγ null mice. They further revealed that persistent residual leukemic cells became responsive after treatment involving G-CSF administration [87].


Figure 1.
Depicted is a schematic presentation of the pathways described in this review. Effects of inhibitors are shown in pink [22,32,35,36,37,38,39,41,43,48,50,51,54,55,57,58,64,67,68,69,70,73,74,82,83,84]. Yellow circles show the molecules frequently targeted by the kinase inhibitors, while gray circles are less frequently targeted, described in the review. MK2, MAPK-activated protein kinases; IRS1, insulin receptor substrate 1; Mnk1/2, MAPK-interacting kinase 1/2; FOXO, Forkhead box O; mTORC1, mTOR complex 1, TCP: tranylcypromine.
Figure 2.
Inhibition of CDK9 reduces MCL-1 expression. The depicted figure is a modification from Tibes et al. [75] that was created with BioRender. Blue arrows and red lines are showing effects and inhibitory effects, respectively. Brd4, bromodomain-containing protein 4; CDK, cyclin-dependent kinase; MCL-1, myeloid leukemia-1; RNA pol II, RNA polymerase II.
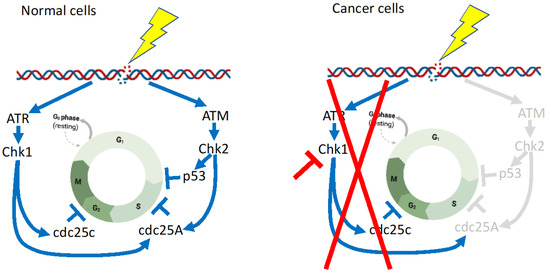
Figure 3.
Differences in the DNA damage checkpoint between normal cells and cancer cells. The ATM-CHK2-p53 pathway is impaired in many cancers [80], and thus CHK1 inhibition is very effective for cancers. The depicted figures are modifications from Smith et al. [80] and Goto et al. [81], with minor modifications created with BioRender. Regulatory pathways in normal cells are shown in blue lines and arrows, whereas these are abrogated in cancer cells, shown in red lines. Pathways shown in gray, are inactivated in cancer cells.
A major cause of treatment failure is resistance to chemotherapeutic agents, and one strategy to overcome such chemoresistance is to target the antiapoptotic Bcl-2 protein. The Bcl-2-selective inhibitor ABT-199 showed encouraging preclinical results [88,89]. Mcl-1, a member of the antiapoptotic BCL-1 protein family, is a key regulator of mitochondrial homeostasis [90]. Mcl-1 was demonstrated to contribute to ABT-199 resistance [91]. Zhao et al. [92] found that CHK1 inhibitor LY2603618 decreased Mcl-1. Simultaneous treatment with LY2603618 and ABT-199 resulted in the synergistic induction of apoptosis in both AML cell lines and primary patient samples [92].
8. Concluding Remarks
Table 1 shows a summary of the agents described in this review. Combinations with FLT3 have been the most attractive form of targeted therapy in AML [30,31,32]. One of the most aberrantly activated oncogenic pathways in AML is the RAS-RAF-MEK-ERK (MAPK) pathway, but in clinical trials, the targeting of this pathway by MEK inhibitors was not proven to be effective [50]. Although there is no clinical activity for MEK and AKT inhibition [40] or Ara-C and CHK1 inhibition [85], several combination therapies have shown clinical efficacy for AML treatment [30,31,32,46,47,48,72]. Among the kinase pathways described in this review (Figure 1), in my opinion, combination therapies that target the FLT3 are the most promising therapies [30,31,32], and also the PI3K/AKT/mTOR signaling pathway shows some efficiency, even in clinical settings [46,47,48]. In addition, at the pre-clinical level, various combination therapies targeting PI3K/AKT/mTOR, MAPK, CDK, and CHK1 pathways seem attractive [35,37,38,41,43,44,67,68,78,82,83]. In the short term, these preclinical and clinical data continue to be rapidly generated—not only FLT3 but others for successful targeted therapy for AML. Determining the optimal combinations and clarifying the mechanisms of inhibitor effects may lead to the development of efficient integrated therapies. The next stage is to decide in which phase of the treatment it should be used, as a part of first-line induction therapy, as consolidation, in a relapse, or in refractory setting. Based on these findings, early recognition of the genomic and prognostic subtype of these gene aberrations, followed by individualized remission–induction or maintenance therapy with kinase inhibitors, is a highly awaited next-generation therapy for AML.

Table 1.
Combinations of the kinase inhibitors mainly described in this review.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research (21K07346) from the Ministry of Education, Science and Culture, Japan, Kyowa-Kirin Research Support (Kyowa-Kirin Co. Ltd.), and Daiichi-Sankyo Research Support (Daiichi-Sankyo Inc.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
I thank my students and staffs in my lab, especially Souma Suzuki, Yuri Sato, and Susumu Suzuki for related experiments to inspire ideas for mechanisms of the action of the kinases.
Conflicts of Interest
The author declares no competing interest.
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 16010. [Google Scholar] [CrossRef]
- Stubbins, R.J.; Karsan, A. Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 2021, 11, 193. [Google Scholar] [CrossRef]
- Takahashi, S. Current Understandings of Myeloid Differentiation Inducers in Leukemia Therapy. Acta Haematol. 2021, 144, 380–388. [Google Scholar] [CrossRef]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef]
- Takahashi, S. Kinase Inhibitors and Interferons as Other Myeloid Differentiation Inducers in Leukemia Therapy. Acta Haematol. 2022, 145, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Vey, N. Low-intensity regimens versus standard-intensity induction strategies in acute myeloid leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720913010. [Google Scholar] [CrossRef] [PubMed]
- Deangelo, D.J.; Neuberg, D.; Amrein, P.C.; Berchuck, J.; Wadleigh, M.; Sirulnik, L.A.; Galinsky, I.; Golub, T.; Stegmaier, K.; Stone, R.M. A phase II study of the EGFR inhibitor gefitinib in patients with acute myeloid leukemia. Leuk. Res. 2014, 38, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Abou Dalle, I.; Cortes, J.E.; Pinnamaneni, P.; Lamothe, B.; Diaz Duque, A.; Randhawa, J.; Pemmaraju, N.; Jabbour, E.; Ferrajoli, A.; Wierda, W.G.; et al. A Pilot Phase II Study of Erlotinib for the Treatment of Patients with Relapsed/Refractory Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 30–39. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Mutations of FLT3 receptor affect its surface glycosylation, intracellular localization, and downstream signaling. Leuk. Res. Rep. 2020, 13, 100187. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Lowenberg, B. Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef]
- Smith, C.C. The growing landscape of FLT3 inhibition in AML. Hematology Am. Soc. Hematol. Educ. Program. 2019, 2019, 539–547. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Harigae, H.; Yokoyama, H.; Ishikawa, I.; Abe, S.; Imaizumi, M.; Sasaki, T.; Kaku, M. Synergistic effect of arsenic trioxide and flt3 inhibition on cells with flt3 internal tandem duplication. Int. J. Hematol. 2006, 84, 256–261. [Google Scholar] [CrossRef]
- Takahashi, S. Combination therapy with arsenic trioxide for hematological malignancies. Anticancer. Agents Med. Chem. 2010, 10, 504–510. [Google Scholar] [CrossRef]
- Nagai, K.; Hou, L.; Li, L.; Nguyen, B.; Seale, T.; Shirley, C.; Ma, H.; Levis, M.; Ghiaur, G.; Duffield, A.; et al. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget 2018, 9, 32885–32899. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Gong, P.; Gabrilove, J.; Waxman, S.; Jing, Y. Arsenic Trioxide and Sorafenib Induce Synthetic Lethality of FLT3-ITD Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Tecik, M.; Adan, A. Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches. Onco Targets Ther. 2022, 15, 1449–1478. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Leiba, R.; Frisch, A.; Horesh, N.; Henig, I.; Yehudai-Ofir, D.; Moshe, Y.; Neaman, M.; Ganzel, C.; Gal-Rabinovich, K.; et al. Midostaurin in combination with chemotherapy is most effective in patients with acute myeloid leukemia presenting with high FLT3-ITD allelic ratio who proceed to allogeneic stem cell transplantation while in first complete remission. Eur. J. Haematol. 2021, 106, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Majothi, S.; Adams, D.; Loke, J.; Stevens, S.P.; Wheatley, K.; Wilson, J.S. FLT3 inhibitors in acute myeloid leukaemia: Assessment of clinical effectiveness, adverse events and future research-a systematic review and meta-analysis. Syst. Rev. 2020, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Pratz, K.W.; Sato, T.; Murphy, K.M.; Stine, A.; Rajkhowa, T.; Levis, M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010, 115, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cai, J.; Cheng, J.; Yang, W.; Zhu, Y.; Li, H.; Lu, T.; Chen, Y.; Lu, S. FLT3 Inhibitors in Acute Myeloid Leukemia: Challenges and Recent Developments in Overcoming Resistance. J. Med. Chem. 2021, 64, 2878–2900. [Google Scholar] [CrossRef]
- Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib Combined with 5-azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef]
- Tabe, Y.; Tafuri, A.; Sekihara, K.; Yang, H.; Konopleva, M. Inhibition of mTOR kinase as a therapeutic target for acute myeloid leukemia. Expert. Opin. Ther. Targets 2017, 21, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Lin, Y.M.; Lai, Y.L.; Chen, C.Y.; Hu, C.Y.; Tien, H.F.; Ou, D.L.; Lin, L.I. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med. Oncol. 2015, 32, 206. [Google Scholar] [CrossRef]
- Sandhofer, N.; Metzeler, K.H.; Rothenberg, M.; Herold, T.; Tiedt, S.; Groiss, V.; Carlet, M.; Walter, G.; Hinrichsen, T.; Wachter, O.; et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 2015, 29, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Deb, G.; Wingelhofer, B.; Amaral, F.M.R.; Maiques-Diaz, A.; Chadwick, J.A.; Spencer, G.J.; Williams, E.L.; Leong, H.S.; Maes, T.; Somervaille, T.C.P. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 2020, 34, 1266–1277. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.K.; Pallavicini, I.; Ceccacci, E.; Meroni, G.; Saadeldin, M.K.; Varasi, M.; Minucci, S. Tuning mTORC1 activity dictates the response of acute myeloid leukemia to LSD1 inhibition. Haematologica 2020, 105, 2105–2117. [Google Scholar] [CrossRef]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E., 3rd; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in Patients with Acute Myeloid Leukemia with RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e413. [Google Scholar] [CrossRef]
- Brunen, D.; Garcia-Barchino, M.J.; Malani, D.; Jagalur Basheer, N.; Lieftink, C.; Beijersbergen, R.L.; Murumagi, A.; Porkka, K.; Wolf, M.; Zwaan, C.M.; et al. Intrinsic resistance to PIM kinase inhibition in AML through p38alpha-mediated feedback activation of mTOR signaling. Oncotarget 2016, 7, 37407–37419. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.; Stengel, C.; Sellar, R.; Huszar, D.; Davies, B.R.; Gale, R.E.; Linch, D.C.; Khwaja, A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br. J. Haematol. 2014, 167, 69–79. [Google Scholar] [CrossRef]
- Yuan, F.; Cheng, C.; Xiao, F.; Liu, H.; Cao, S.; Zhou, G. Inhibition of mTORC1/P70S6K pathway by Metformin synergistically sensitizes Acute Myeloid Leukemia to Ara-C. Life Sci. 2020, 243, 117276. [Google Scholar] [CrossRef]
- Liang, H.; Zheng, Q.L.; Fang, P.; Zhang, J.; Zhang, T.; Liu, W.; Guo, M.; Robinson, C.L.; Chen, S.B.; Chen, X.P.; et al. Targeting the PI3K/AKT pathway via GLI1 inhibition enhanced the drug sensitivity of acute myeloid leukemia cells. Sci. Rep. 2017, 7, 40361. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Saint Marcoux, F.; Recher, C.; Prebet, T.; Chevallier, P.; Cahn, J.Y.; Leguay, T.; Bories, P.; Witz, F.; et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013, 27, 1479–1486. [Google Scholar] [CrossRef]
- Amadori, S.; Stasi, R.; Martelli, A.M.; Venditti, A.; Meloni, G.; Pane, F.; Martinelli, G.; Lunghi, M.; Pagano, L.; Cilloni, D.; et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: Results of a phase II GIMEMA study (AML-1107). Br. J. Haematol. 2012, 156, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kasner, M.T.; Mick, R.; Jeschke, G.R.; Carabasi, M.; Filicko-O’Hara, J.; Flomenberg, N.; Frey, N.V.; Hexner, E.O.; Luger, S.M.; Loren, A.W.; et al. Sirolimus enhances remission induction in patients with high-risk acute myeloid leukemia and mTORC1 target inhibition. Investig. New. Drugs 2018, 36, 657–666. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; McQueen, T.; Chism, D.; Milella, M.; Estey, E.; Kaldjian, E.; Sebolt-Leopold, J.; Konopleva, M.; Andreeff, M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 2005, 19, 1543–1549. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.; Culjkovic, B.; Cocolakis, E.; Rousseau, C.; Beslu, N.; Amri, A.; Caplan, S.; Leber, B.; Roy, D.C.; Miller, W.H., Jr.; et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood 2009, 114, 257–260. [Google Scholar] [CrossRef]
- Topisirovic, I.; Guzman, M.L.; McConnell, M.J.; Licht, J.D.; Culjkovic, B.; Neering, S.J.; Jordan, C.T.; Borden, K.L. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol. Cell. Biol. 2003, 23, 8992–9002. [Google Scholar] [CrossRef] [PubMed]
- Suarez, M.; Blyth, G.T.; Mina, A.A.; Kosciuczuk, E.M.; Dolniak, B.; Dinner, S.; Altman, J.K.; Eklund, E.A.; Saleiro, D.; Beauchamp, E.M.; et al. Inhibitory effects of Tomivosertib in acute myeloid leukemia. Oncotarget 2021, 12, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Diab, S.; Yu, M.; Adams, J.; Islam, S.; Basnet, S.K.; Albrecht, H.; Milne, R.; Wang, S. Inhibition of Mnk enhances apoptotic activity of cytarabine in acute myeloid leukemia cells. Oncotarget 2016, 7, 56811–56825. [Google Scholar] [CrossRef]
- Nishida, Y.; Zhao, R.; Heese, L.E.; Akiyama, H.; Patel, S.; Jaeger, A.M.; Jacamo, R.O.; Kojima, K.; Ma, M.C.J.; Ruvolo, V.R.; et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia 2021, 35, 2469–2481. [Google Scholar] [CrossRef]
- Altman, J.K.; Szilard, A.; Konicek, B.W.; Iversen, P.W.; Kroczynska, B.; Glaser, H.; Sassano, A.; Vakana, E.; Graff, J.R.; Platanias, L.C. Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood 2013, 121, 3675–3681. [Google Scholar] [CrossRef]
- Tambe, M.; Karjalainen, E.; Vaha-Koskela, M.; Bulanova, D.; Gjertsen, B.T.; Kontro, M.; Porkka, K.; Heckman, C.A.; Wennerberg, K. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia 2020, 34, 3186–3196. [Google Scholar] [CrossRef]
- Cremer, A.; Ellegast, J.M.; Alexe, G.; Frank, E.S.; Ross, L.; Chu, S.H.; Pikman, Y.; Robichaud, A.; Goodale, A.; Haupl, B.; et al. Resistance Mechanisms to SYK Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 214–231. [Google Scholar] [CrossRef]
- Stegmaier, K.; Corsello, S.M.; Ross, K.N.; Wong, J.S.; Deangelo, D.J.; Golub, T.R. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood 2005, 106, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.K.; Berchuck, J.E.; Ross, K.N.; Kakoza, R.M.; Clauser, K.; Schinzel, A.C.; Ross, L.; Galinsky, I.; Davis, T.N.; Silver, S.J.; et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell. 2009, 16, 281–294. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.Y.; Pereira-Martins, D.A.; Weinhauser, I.; Ortiz, C.; Candido, L.A.; Lange, A.P.; De Abreu, N.F.; Mendonza, S.E.S.; de Deus Wagatsuma, V.M.; Do Nascimento, M.C.; et al. The Combination of Gefitinib with ATRA and ATO Induces Myeloid Differentiation in Acute Promyelocytic Leukemia Resistant Cells. Front. Oncol. 2021, 11, 686445. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Rothe, K.; Chen, M.; Grasedieck, S.; Li, R.; Nam, S.E.; Zhang, X.; Novakovskiy, G.E.; Ahn, Y.H.; Maksakova, I.; et al. Targeting AXL kinase sensitizes leukemic stem and progenitor cells to venetoclax treatment in acute myeloid leukemia. Blood 2021, 137, 3641–3655. [Google Scholar] [CrossRef]
- Young, C.S.; Clarke, K.M.; Kettyle, L.M.; Thompson, A.; Mills, K.I. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget 2017, 8, 51429–51446. [Google Scholar] [CrossRef]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef]
- Phillips, D.C.; Jin, S.; Gregory, G.P.; Zhang, Q.; Xue, J.; Zhao, X.; Chen, J.; Tong, Y.; Zhang, H.; Smith, M.; et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia 2020, 34, 1646–1657. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Bogenberger, J.; Whatcott, C.; Hansen, N.; Delman, D.; Shi, C.X.; Kim, W.; Haws, H.; Soh, K.; Lee, Y.S.; Peterson, P.; et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017, 8, 107206–107222. [Google Scholar] [CrossRef]
- Luedtke, D.A.; Su, Y.; Ma, J.; Li, X.; Buck, S.A.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. Inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in preclinical models of acute myeloid leukemia. Signal. Transduct. Target. Ther. 2020, 5, 17. [Google Scholar] [CrossRef]
- McCalmont, H.; Li, K.L.; Jones, L.; Toubia, J.; Bray, S.C.; Casolari, D.A.; Mayoh, C.; Samaraweera, S.E.; Lewis, I.D.; Prinjha, R.K.; et al. Efficacy of combined CDK9/BET inhibition in preclinical models of MLL-rearranged acute leukemia. Blood Adv. 2020, 4, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Lee, D.J.; Frattini, M.; Fine, G.D.; Costas, J.; Kolibaba, K.; Anthony, S.P.; Bearss, D.; Smith, B.D. Phase I Study of Alvocidib Followed by 7+3 (Cytarabine + Daunorubicin) in Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Schoenwaelder, N.; Sender, S.; Junghanss, C.; Maletzki, C. Cyclin-Dependent Kinase Inhibitors in Hematological Malignancies-Current Understanding, (Pre-)Clinical Application and Promising Approaches. Cancers 2021, 13, 2497. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Deckwerth, T.L.; Kerwin, W.S.; Casalini, J.R.; Merrell, A.J.; Grenley, M.O.; Burns, C.; Ditzler, S.H.; Dixon, C.P.; Beirne, E.; et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci. Rep. 2017, 7, 18007. [Google Scholar] [CrossRef]
- Tibes, R.; Bogenberger, J.M. Transcriptional Silencing of MCL-1 Through Cyclin-Dependent Kinase Inhibition in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1205. [Google Scholar] [CrossRef]
- Shao, X.; Xiang, S.; Fu, H.; Chen, Y.; Xu, A.; Liu, Y.; Qi, X.; Cao, J.; Zhu, H.; Yang, B.; et al. CDK2 suppression synergizes with all-trans-retinoic acid to overcome the myeloid differentiation blockade of AML cells. Pharmacol. Res. 2020, 151, 104545. [Google Scholar] [CrossRef]
- Uras, I.Z.; Maurer, B.; Nebenfuehr, S.; Zojer, M.; Valent, P.; Sexl, V. Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib. Int. J. Mol. Sci. 2018, 19, 3987. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert. Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Goto, H.; Inagaki, M. [DNA damage checkpoint and cancer--pros and cons of Chk1 inhibitors]. Seikagaku 2013, 85, 145–151. [Google Scholar]
- Schenk, E.L.; Koh, B.D.; Flatten, K.S.; Peterson, K.L.; Parry, D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Karnitz, L.M.; Kaufmann, S.H. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin. Cancer Res. 2012, 18, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Vincelette, N.D.; Ding, H.; Huehls, A.M.; Flatten, K.S.; Kelly, R.L.; Kohorst, M.A.; Webster, J.; Hess, A.D.; Pratz, K.W.; Karnitz, L.M.; et al. Effect of CHK1 Inhibition on CPX-351 Cytotoxicity in vitro and ex vivo. Sci. Rep. 2019, 9, 3617. [Google Scholar] [CrossRef]
- Jammal, N.; Rausch, C.R.; Kadia, T.M.; Pemmaraju, N. Cell cycle inhibitors for the treatment of acute myeloid leukemia: A review of phase 2 & 3 clinical trials. Expert. Opin. Emerg. Drugs 2020, 25, 491–499. [Google Scholar] [CrossRef]
- Webster, J.A.; Tibes, R.; Morris, L.; Blackford, A.L.; Litzow, M.; Patnaik, M.; Rosner, G.L.; Gojo, I.; Kinders, R.; Wang, L.; et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk. Res. 2017, 61, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef]
- Di Tullio, A.; Rouault-Pierre, K.; Abarrategi, A.; Mian, S.; Grey, W.; Gribben, J.; Stewart, A.; Blackwood, E.; Bonnet, D. The combination of CHK1 inhibitor with G-CSF overrides cytarabine resistance in human acute myeloid leukemia. Nat. Commun. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Xiang, W.; Yang, C.Y.; Bai, L. MCL-1 inhibition in cancer treatment. Onco Targets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Niu, X.; Li, X.; Edwards, H.; Wang, G.; Wang, Y.; Taub, J.W.; Lin, H.; Ge, Y. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget 2016, 7, 34785–34799. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).