

Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment


Abstract
1. Introduction
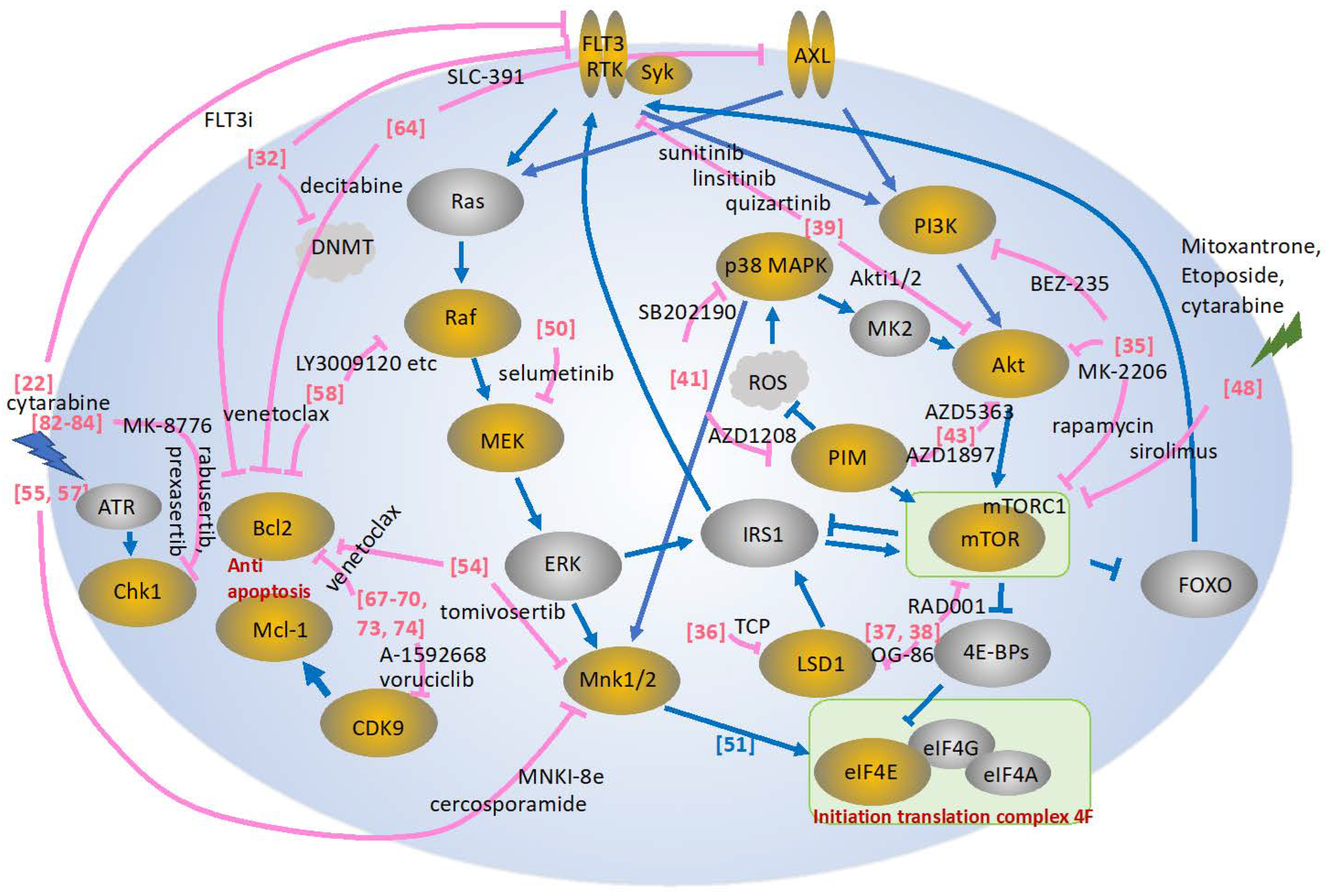
2. Combination Therapy Targeting the FLT3 Signaling Pathway
3. Combination Therapy Targeting the PI3K/AKT/mTOR Signaling Pathway
4. Combination Therapy Targeting the MAPK Signaling Pathway
5. Combination Therapy Targeting AXL
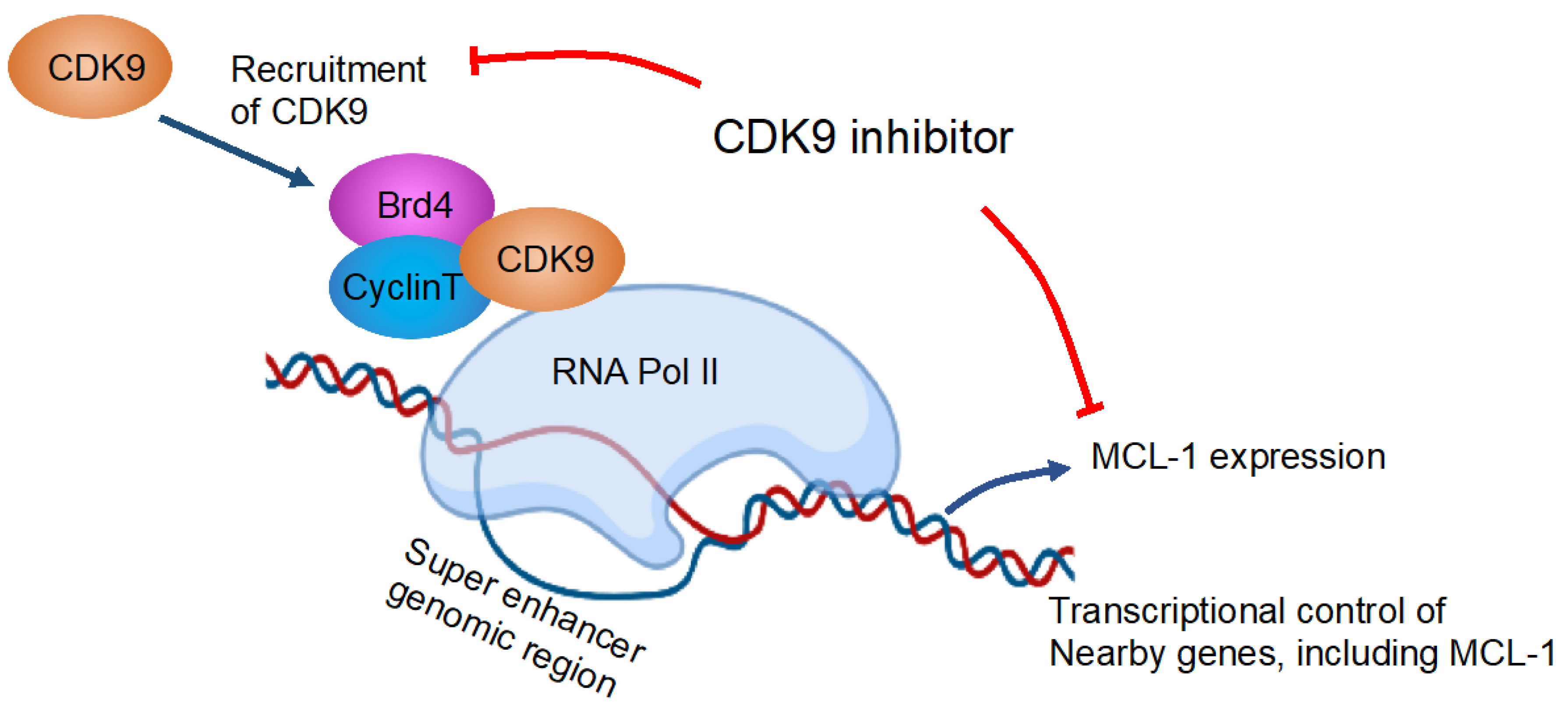
6. Combination Therapy Targeting the CDK Signaling Pathway
7. Combination Therapy Targeting the CHK1 Signaling Pathway


8. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 16010. [Google Scholar] [CrossRef]
- Stubbins, R.J.; Karsan, A. Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 2021, 11, 193. [Google Scholar] [CrossRef]
- Takahashi, S. Current Understandings of Myeloid Differentiation Inducers in Leukemia Therapy. Acta Haematol. 2021, 144, 380–388. [Google Scholar] [CrossRef]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef]
- Takahashi, S. Kinase Inhibitors and Interferons as Other Myeloid Differentiation Inducers in Leukemia Therapy. Acta Haematol. 2022, 145, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Vey, N. Low-intensity regimens versus standard-intensity induction strategies in acute myeloid leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720913010. [Google Scholar] [CrossRef] [PubMed]
- Deangelo, D.J.; Neuberg, D.; Amrein, P.C.; Berchuck, J.; Wadleigh, M.; Sirulnik, L.A.; Galinsky, I.; Golub, T.; Stegmaier, K.; Stone, R.M. A phase II study of the EGFR inhibitor gefitinib in patients with acute myeloid leukemia. Leuk. Res. 2014, 38, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Abou Dalle, I.; Cortes, J.E.; Pinnamaneni, P.; Lamothe, B.; Diaz Duque, A.; Randhawa, J.; Pemmaraju, N.; Jabbour, E.; Ferrajoli, A.; Wierda, W.G.; et al. A Pilot Phase II Study of Erlotinib for the Treatment of Patients with Relapsed/Refractory Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 30–39. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Mutations of FLT3 receptor affect its surface glycosylation, intracellular localization, and downstream signaling. Leuk. Res. Rep. 2020, 13, 100187. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Wei, A.H.; Lowenberg, B. Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef]
- Smith, C.C. The growing landscape of FLT3 inhibition in AML. Hematology Am. Soc. Hematol. Educ. Program. 2019, 2019, 539–547. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Harigae, H.; Yokoyama, H.; Ishikawa, I.; Abe, S.; Imaizumi, M.; Sasaki, T.; Kaku, M. Synergistic effect of arsenic trioxide and flt3 inhibition on cells with flt3 internal tandem duplication. Int. J. Hematol. 2006, 84, 256–261. [Google Scholar] [CrossRef]
- Takahashi, S. Combination therapy with arsenic trioxide for hematological malignancies. Anticancer. Agents Med. Chem. 2010, 10, 504–510. [Google Scholar] [CrossRef]
- Nagai, K.; Hou, L.; Li, L.; Nguyen, B.; Seale, T.; Shirley, C.; Ma, H.; Levis, M.; Ghiaur, G.; Duffield, A.; et al. Combination of ATO with FLT3 TKIs eliminates FLT3/ITD+ leukemia cells through reduced expression of FLT3. Oncotarget 2018, 9, 32885–32899. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Gong, P.; Gabrilove, J.; Waxman, S.; Jing, Y. Arsenic Trioxide and Sorafenib Induce Synthetic Lethality of FLT3-ITD Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Tecik, M.; Adan, A. Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches. Onco Targets Ther. 2022, 15, 1449–1478. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Leiba, R.; Frisch, A.; Horesh, N.; Henig, I.; Yehudai-Ofir, D.; Moshe, Y.; Neaman, M.; Ganzel, C.; Gal-Rabinovich, K.; et al. Midostaurin in combination with chemotherapy is most effective in patients with acute myeloid leukemia presenting with high FLT3-ITD allelic ratio who proceed to allogeneic stem cell transplantation while in first complete remission. Eur. J. Haematol. 2021, 106, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Majothi, S.; Adams, D.; Loke, J.; Stevens, S.P.; Wheatley, K.; Wilson, J.S. FLT3 inhibitors in acute myeloid leukaemia: Assessment of clinical effectiveness, adverse events and future research-a systematic review and meta-analysis. Syst. Rev. 2020, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Pratz, K.W.; Sato, T.; Murphy, K.M.; Stine, A.; Rajkhowa, T.; Levis, M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010, 115, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cai, J.; Cheng, J.; Yang, W.; Zhu, Y.; Li, H.; Lu, T.; Chen, Y.; Lu, S. FLT3 Inhibitors in Acute Myeloid Leukemia: Challenges and Recent Developments in Overcoming Resistance. J. Med. Chem. 2021, 64, 2878–2900. [Google Scholar] [CrossRef]
- Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib Combined with 5-azacytidine in Older Patients with Untreated FLT3-ITD Mutated Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef]
- Tabe, Y.; Tafuri, A.; Sekihara, K.; Yang, H.; Konopleva, M. Inhibition of mTOR kinase as a therapeutic target for acute myeloid leukemia. Expert. Opin. Ther. Targets 2017, 21, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.W.; Lin, Y.M.; Lai, Y.L.; Chen, C.Y.; Hu, C.Y.; Tien, H.F.; Ou, D.L.; Lin, L.I. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med. Oncol. 2015, 32, 206. [Google Scholar] [CrossRef]
- Sandhofer, N.; Metzeler, K.H.; Rothenberg, M.; Herold, T.; Tiedt, S.; Groiss, V.; Carlet, M.; Walter, G.; Hinrichsen, T.; Wachter, O.; et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 2015, 29, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Deb, G.; Wingelhofer, B.; Amaral, F.M.R.; Maiques-Diaz, A.; Chadwick, J.A.; Spencer, G.J.; Williams, E.L.; Leong, H.S.; Maes, T.; Somervaille, T.C.P. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 2020, 34, 1266–1277. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.K.; Pallavicini, I.; Ceccacci, E.; Meroni, G.; Saadeldin, M.K.; Varasi, M.; Minucci, S. Tuning mTORC1 activity dictates the response of acute myeloid leukemia to LSD1 inhibition. Haematologica 2020, 105, 2105–2117. [Google Scholar] [CrossRef]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E., 3rd; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in Patients with Acute Myeloid Leukemia with RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e413. [Google Scholar] [CrossRef]
- Brunen, D.; Garcia-Barchino, M.J.; Malani, D.; Jagalur Basheer, N.; Lieftink, C.; Beijersbergen, R.L.; Murumagi, A.; Porkka, K.; Wolf, M.; Zwaan, C.M.; et al. Intrinsic resistance to PIM kinase inhibition in AML through p38alpha-mediated feedback activation of mTOR signaling. Oncotarget 2016, 7, 37407–37419. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.; Stengel, C.; Sellar, R.; Huszar, D.; Davies, B.R.; Gale, R.E.; Linch, D.C.; Khwaja, A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br. J. Haematol. 2014, 167, 69–79. [Google Scholar] [CrossRef]
- Yuan, F.; Cheng, C.; Xiao, F.; Liu, H.; Cao, S.; Zhou, G. Inhibition of mTORC1/P70S6K pathway by Metformin synergistically sensitizes Acute Myeloid Leukemia to Ara-C. Life Sci. 2020, 243, 117276. [Google Scholar] [CrossRef]
- Liang, H.; Zheng, Q.L.; Fang, P.; Zhang, J.; Zhang, T.; Liu, W.; Guo, M.; Robinson, C.L.; Chen, S.B.; Chen, X.P.; et al. Targeting the PI3K/AKT pathway via GLI1 inhibition enhanced the drug sensitivity of acute myeloid leukemia cells. Sci. Rep. 2017, 7, 40361. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Saint Marcoux, F.; Recher, C.; Prebet, T.; Chevallier, P.; Cahn, J.Y.; Leguay, T.; Bories, P.; Witz, F.; et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013, 27, 1479–1486. [Google Scholar] [CrossRef]
- Amadori, S.; Stasi, R.; Martelli, A.M.; Venditti, A.; Meloni, G.; Pane, F.; Martinelli, G.; Lunghi, M.; Pagano, L.; Cilloni, D.; et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: Results of a phase II GIMEMA study (AML-1107). Br. J. Haematol. 2012, 156, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kasner, M.T.; Mick, R.; Jeschke, G.R.; Carabasi, M.; Filicko-O’Hara, J.; Flomenberg, N.; Frey, N.V.; Hexner, E.O.; Luger, S.M.; Loren, A.W.; et al. Sirolimus enhances remission induction in patients with high-risk acute myeloid leukemia and mTORC1 target inhibition. Investig. New. Drugs 2018, 36, 657–666. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; McQueen, T.; Chism, D.; Milella, M.; Estey, E.; Kaldjian, E.; Sebolt-Leopold, J.; Konopleva, M.; Andreeff, M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 2005, 19, 1543–1549. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.; Culjkovic, B.; Cocolakis, E.; Rousseau, C.; Beslu, N.; Amri, A.; Caplan, S.; Leber, B.; Roy, D.C.; Miller, W.H., Jr.; et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood 2009, 114, 257–260. [Google Scholar] [CrossRef]
- Topisirovic, I.; Guzman, M.L.; McConnell, M.J.; Licht, J.D.; Culjkovic, B.; Neering, S.J.; Jordan, C.T.; Borden, K.L. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol. Cell. Biol. 2003, 23, 8992–9002. [Google Scholar] [CrossRef] [PubMed]
- Suarez, M.; Blyth, G.T.; Mina, A.A.; Kosciuczuk, E.M.; Dolniak, B.; Dinner, S.; Altman, J.K.; Eklund, E.A.; Saleiro, D.; Beauchamp, E.M.; et al. Inhibitory effects of Tomivosertib in acute myeloid leukemia. Oncotarget 2021, 12, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Diab, S.; Yu, M.; Adams, J.; Islam, S.; Basnet, S.K.; Albrecht, H.; Milne, R.; Wang, S. Inhibition of Mnk enhances apoptotic activity of cytarabine in acute myeloid leukemia cells. Oncotarget 2016, 7, 56811–56825. [Google Scholar] [CrossRef]
- Nishida, Y.; Zhao, R.; Heese, L.E.; Akiyama, H.; Patel, S.; Jaeger, A.M.; Jacamo, R.O.; Kojima, K.; Ma, M.C.J.; Ruvolo, V.R.; et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia 2021, 35, 2469–2481. [Google Scholar] [CrossRef]
- Altman, J.K.; Szilard, A.; Konicek, B.W.; Iversen, P.W.; Kroczynska, B.; Glaser, H.; Sassano, A.; Vakana, E.; Graff, J.R.; Platanias, L.C. Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood 2013, 121, 3675–3681. [Google Scholar] [CrossRef]
- Tambe, M.; Karjalainen, E.; Vaha-Koskela, M.; Bulanova, D.; Gjertsen, B.T.; Kontro, M.; Porkka, K.; Heckman, C.A.; Wennerberg, K. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia 2020, 34, 3186–3196. [Google Scholar] [CrossRef]
- Cremer, A.; Ellegast, J.M.; Alexe, G.; Frank, E.S.; Ross, L.; Chu, S.H.; Pikman, Y.; Robichaud, A.; Goodale, A.; Haupl, B.; et al. Resistance Mechanisms to SYK Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 214–231. [Google Scholar] [CrossRef]
- Stegmaier, K.; Corsello, S.M.; Ross, K.N.; Wong, J.S.; Deangelo, D.J.; Golub, T.R. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood 2005, 106, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.K.; Berchuck, J.E.; Ross, K.N.; Kakoza, R.M.; Clauser, K.; Schinzel, A.C.; Ross, L.; Galinsky, I.; Davis, T.N.; Silver, S.J.; et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell. 2009, 16, 281–294. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.Y.; Pereira-Martins, D.A.; Weinhauser, I.; Ortiz, C.; Candido, L.A.; Lange, A.P.; De Abreu, N.F.; Mendonza, S.E.S.; de Deus Wagatsuma, V.M.; Do Nascimento, M.C.; et al. The Combination of Gefitinib with ATRA and ATO Induces Myeloid Differentiation in Acute Promyelocytic Leukemia Resistant Cells. Front. Oncol. 2021, 11, 686445. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Rothe, K.; Chen, M.; Grasedieck, S.; Li, R.; Nam, S.E.; Zhang, X.; Novakovskiy, G.E.; Ahn, Y.H.; Maksakova, I.; et al. Targeting AXL kinase sensitizes leukemic stem and progenitor cells to venetoclax treatment in acute myeloid leukemia. Blood 2021, 137, 3641–3655. [Google Scholar] [CrossRef]
- Young, C.S.; Clarke, K.M.; Kettyle, L.M.; Thompson, A.; Mills, K.I. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget 2017, 8, 51429–51446. [Google Scholar] [CrossRef]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef]
- Phillips, D.C.; Jin, S.; Gregory, G.P.; Zhang, Q.; Xue, J.; Zhao, X.; Chen, J.; Tong, Y.; Zhang, H.; Smith, M.; et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia 2020, 34, 1646–1657. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Bogenberger, J.; Whatcott, C.; Hansen, N.; Delman, D.; Shi, C.X.; Kim, W.; Haws, H.; Soh, K.; Lee, Y.S.; Peterson, P.; et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017, 8, 107206–107222. [Google Scholar] [CrossRef]
- Luedtke, D.A.; Su, Y.; Ma, J.; Li, X.; Buck, S.A.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. Inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in preclinical models of acute myeloid leukemia. Signal. Transduct. Target. Ther. 2020, 5, 17. [Google Scholar] [CrossRef]
- McCalmont, H.; Li, K.L.; Jones, L.; Toubia, J.; Bray, S.C.; Casolari, D.A.; Mayoh, C.; Samaraweera, S.E.; Lewis, I.D.; Prinjha, R.K.; et al. Efficacy of combined CDK9/BET inhibition in preclinical models of MLL-rearranged acute leukemia. Blood Adv. 2020, 4, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Lee, D.J.; Frattini, M.; Fine, G.D.; Costas, J.; Kolibaba, K.; Anthony, S.P.; Bearss, D.; Smith, B.D. Phase I Study of Alvocidib Followed by 7+3 (Cytarabine + Daunorubicin) in Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Schoenwaelder, N.; Sender, S.; Junghanss, C.; Maletzki, C. Cyclin-Dependent Kinase Inhibitors in Hematological Malignancies-Current Understanding, (Pre-)Clinical Application and Promising Approaches. Cancers 2021, 13, 2497. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Deckwerth, T.L.; Kerwin, W.S.; Casalini, J.R.; Merrell, A.J.; Grenley, M.O.; Burns, C.; Ditzler, S.H.; Dixon, C.P.; Beirne, E.; et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci. Rep. 2017, 7, 18007. [Google Scholar] [CrossRef]
- Tibes, R.; Bogenberger, J.M. Transcriptional Silencing of MCL-1 Through Cyclin-Dependent Kinase Inhibition in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1205. [Google Scholar] [CrossRef]
- Shao, X.; Xiang, S.; Fu, H.; Chen, Y.; Xu, A.; Liu, Y.; Qi, X.; Cao, J.; Zhu, H.; Yang, B.; et al. CDK2 suppression synergizes with all-trans-retinoic acid to overcome the myeloid differentiation blockade of AML cells. Pharmacol. Res. 2020, 151, 104545. [Google Scholar] [CrossRef]
- Uras, I.Z.; Maurer, B.; Nebenfuehr, S.; Zojer, M.; Valent, P.; Sexl, V. Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib. Int. J. Mol. Sci. 2018, 19, 3987. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert. Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Goto, H.; Inagaki, M. [DNA damage checkpoint and cancer--pros and cons of Chk1 inhibitors]. Seikagaku 2013, 85, 145–151. [Google Scholar]
- Schenk, E.L.; Koh, B.D.; Flatten, K.S.; Peterson, K.L.; Parry, D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Karnitz, L.M.; Kaufmann, S.H. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin. Cancer Res. 2012, 18, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Vincelette, N.D.; Ding, H.; Huehls, A.M.; Flatten, K.S.; Kelly, R.L.; Kohorst, M.A.; Webster, J.; Hess, A.D.; Pratz, K.W.; Karnitz, L.M.; et al. Effect of CHK1 Inhibition on CPX-351 Cytotoxicity in vitro and ex vivo. Sci. Rep. 2019, 9, 3617. [Google Scholar] [CrossRef]
- Jammal, N.; Rausch, C.R.; Kadia, T.M.; Pemmaraju, N. Cell cycle inhibitors for the treatment of acute myeloid leukemia: A review of phase 2 & 3 clinical trials. Expert. Opin. Emerg. Drugs 2020, 25, 491–499. [Google Scholar] [CrossRef]
- Webster, J.A.; Tibes, R.; Morris, L.; Blackford, A.L.; Litzow, M.; Patnaik, M.; Rosner, G.L.; Gojo, I.; Kinders, R.; Wang, L.; et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk. Res. 2017, 61, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef]
- Di Tullio, A.; Rouault-Pierre, K.; Abarrategi, A.; Mian, S.; Grey, W.; Gribben, J.; Stewart, A.; Blackwood, E.; Bonnet, D. The combination of CHK1 inhibitor with G-CSF overrides cytarabine resistance in human acute myeloid leukemia. Nat. Commun. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Xiang, W.; Yang, C.Y.; Bai, L. MCL-1 inhibition in cancer treatment. Onco Targets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Niu, X.; Li, X.; Edwards, H.; Wang, G.; Wang, Y.; Taub, J.W.; Lin, H.; Ge, Y. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget 2016, 7, 34785–34799. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Design of the Study | Results | Refs | |
|---|---|---|---|
| Combination Therapy Targeting the FLT3 Signaling Pathway | |||
| Phase III study of whether the addition of midostaurin to standard chemotherapy would prolong overall survival in patients with FLT3 mutation. | Clinical | Overall survival was significantly longer in the midostaurin group than in the placebo group (hazard ratio for death, 0.78; one-sided p = 0.009), as was event-free survival (hazard ratio for event or death, 0.78; one-sided p = 0.002). | [22] |
| Phase II study of sorafenib and azacytidine on 27 patients with untreated FLT3 mutated AML | Clinical | The regimen was well tolerated in elderly patients with untreated FLT3 mutated AML with no early deaths. | [30] |
| Phase II study of sorafenib and azacytidine on 43 AML patients (range, 24–87 years; median, 64 years) were enrolled; 37 were evaluable for response | Clinical | The combination of AZA and sorafenib is effective for patients with relapsed AML and FLT-3-ITD. | [31] |
| Phase II study of gilteritinib, or sorafenib, or midostaurin, venetoclax and decitabine on 25 patients with FLT3 mutated, newly diagnosed (ND) with AML > 60 years (n = 12) and relapsed/refractory (R/R) patients > 18 years (n = 13). | Clinical | Triplet therapy with FLT3i, venetoclax, and decitabine is safe and an excellent frontline option for older patients with ND FLT3mut AML, and it is effective for R/R AML. | [32] |
| Combination Therapy Targeting the PI3K/AKT/mTOR Signaling Pathway | |||
| Specific allosteric AKT inhibitor (MK-2206) and cytarabine in AML cells. | Pre-clinical | MK-2206 is an active agent in AML, and its efficacy in combination with cytarabine is implicated. | [34] |
| Inhibition of mTORC1 (rapamycin), AKT (MK-2206), and PI3K/mTORC1/2 (BEZ-235) in primary samples and cell lines. | Pre-clinical | Implicating a possible therapeutic benefit in the MLL-mutated subgroup of PI3K/mTOR inhibition. | [35] |
| mTORC1 pharmacologic inhibition or knockdown of mTORC1 components in combination with LSD1 in both primary cell settings and cell line in vitro and in vivo. | Pre-clinical | Dual LSD1 and mTORC1 inhibition represents a possible combination strategy for enhanced differentiation in AML with MLL-translocation. | [37] |
| Dual inhibition of Akt and RTKs on AML cells | Pre-clinical | Dual inhibition of Akt and RTKs displays strong synergistic cytotoxic effects in AML cells and downmodulates Akt signaling to a much greater extent than either drug alone. | [39] |
| Phase II study of combined MEK and AKT inhibition on 23 AML patients with RAS mutations. | Clinical | Combined MEK and AKT inhibition had no clinical activity in patients with RAS-mutated AML. | [40] |
| p38α inhibitors and PIM kinase inhibitor AZD1208 treatment on hematological tumor cell lines in vitro and in vivo. | Pre-clinical | p38α inhibitors sensitize hematological tumor cell lines to AZD1208 treatment in vitro and in vivo. | [41] |
| Dual inhibition of PIM and AKT kinase inhibitors in AML cell lines and primary AML cells. | Pre-clinical | A significant synergy was seen when AZD1897 was combined with the Akt inhibitor AZD5363 in AML cell lines and primary AML cells. | [43] |
| Metformin, Ara-C, and mTORC1/P70S6K pathway inhibition on AML cells. | Pre-clinical | Metformin could synergistically sensitize AML cells to Ara-C via inhibiting the mTORC1/P70S6K pathway. | [44] |
| Efficacy of RAD001, an mTOR inhibitor, combined with chemotherapy for first-relapsed AML patients. | Clinical | A 70 mg dose of RAD001 at d1 and d7 of an induction chemotherapy regimen for AML has acceptable toxicity and may improve treatment. | [46] |
| Efficacy of mTOR inhibitor temsirolimus and low dose of clofarabine in older patients as salvage therapy in AML. | Clinical | The predictive value of target inhibition and the acceptable safety profile promote further investigation. | [47] |
| Sirolimus, an mTORC1 inhibitor, and MEC (mitoxantrone, etoposide, and cytarabine) in high-risk AML patients with untreated, refractory, or relapsed condition. | Clinical | The ORR was 71% (12/17) among patients with mTORC1 inhibition and baseline target activation during treatment, compared with 20% (2/10) in patients without target inhibition. | [48] |
| Combination Therapy Targeting the MAPK Signaling Pathway | |||
| Combination of MNK1/2 inhibitor, tomivosertib, and Bcl-2 inhibitor venetoclax in AML cell lines. | Pre-clinical | Combination of tomivosertib and venetoclax resulted in synergistic anti-leukemic responses in AML cell lines. | [54] |
| Ara-C with either MNKI-8e, an MNK inhibitor, or knockdown of Mnks by short hairpin RNA in MV4-11 AML cells. | Pre-clinical | In Ara-C-treated MV4-11 cells, the MAPK-Mnk-eIF4E pathway plays a critical role. | [55] |
| Combination of cercosporamide; Mnk inhibitor, with cytarabine in primitive leukemic progenitors (CFU-L) from AML patients; and a xenograft mouse model. | Pre-clinical | The combination of cercosporamide with cytarabine resulted in enhanced antileukemic responses. | [57] |
| Pan-RAF inhibitors and BCL2 inhibitor on AML samples and AML cell lines. | Pre-clinical | Pan-RAF inhibition, alone or combined with BCL2 inhibition, is effective in primary AML samples and AML cell lines. | [58] |
| MEK inhibitor (PD0325901) and SYK inhibitor (entospletinib, PRT062607) in AML cell lines, primary AML samples, and AML model mice. | Pre-clinical | MEK and SYK inhibitor combination was synergistic both in vitro and in vivo. | [59] |
| Combination Therapy Targeting AXL | |||
| Combined treatment with DNA methyltransferase inhibitor decitabine, histone deacetylase inhibitor vorinostat, and AXL-specific inhibitor BGB324 on OCI-AML3 cells and xenograft models. | Pre-clinical | Triple combination increased the sensitivity of OCI-AML3 cells to decitabine and vorinostat, as shown through viability assays, and significantly extended the survival of mice xenograft models. | [65] |
| Combination Therapy Targeting the CDK Signaling Pathway | |||
| CDK9 inhibitor (A-1592668 or the related analog A-1467729) and venetoclax in a number of hematologic cell lines and primary NHL patient samples. | Pre-clinical | CDK9 inhibitor plus venetoclax combination was well tolerated in vivo and demonstrated efficacy superior to either agent alone in both lymphoma and AML mouse models. | [67] |
| BET bromodomain inhibitor BI 894,999 effect on AML and lymphoma cell line, ex vivo treated AML, and MM primary patient samples and AML xenografts. | Pre-clinical | BI 894,999 is active as monotherapy in AML xenografts and, in addition, leads to strongly enhanced antitumor effects in combination with CDK9 inhibitors. | [68] |
| CDK inhibitor alvocidib and BCL2 inhibitor venetoclax (ABT-199) on AML cells, AML patient samples, and AML xenograft model. | Pre-clinical | Alvocidib potentiates venetoclax anti-leukemic activity in AML cells, AML patient samples, and AML xenograft models. | [69] |
| Effect of targeting CDK9 with voruciclib in combination with venetoclax on AML cell lines and primary patient samples. | Pre-clinical | Targeting CDK9 with voruciclib in combination with venetoclax results in synergistic antileukemic activity against AML cell lines and primary patient samples. | [70] |
| Effect of CDK9 inhibitor, CDKI-73, and BET bromodomain inhibitor JQ1 on AML cell lines and patient-derived xenograft (PDX) model. | Pre-clinical | CDK 9, bromodomain, and extraterminal inhibitors are synergistic in MLL-rearranged leukemia. | [71] |
| Phase I dose-escalation study of alvocidib on days 1–3, followed by 7 + 3, was performed in newly diagnosed AML ≤ 65 years. | Clinical | Alvocidib can be safely administered prior to 7 + 3 induction with encouraging clinical activity. | [72] |
| Combination Therapy Targeting the CHK1 Signaling Pathway | |||
| Effects of Chk1 inhibitor SCH 900776 and cytarabine were examined using AML cell lines, clinical AML isolates, and normal myeloid progenitors. | Pre-clinical | CHK1 inhibitor SCH 900776 enhanced cytotoxicity of cytarabine in AML lines, clinical AML isolates, and normal myeloid progenitors. | [82] |
| Effect of Chk1 inhibitor MK-8776 and CPX-351 (a liposomal formulation encapsulating a 5:1 molar ratio of cytarabine and daunorubicin) in AML cell lines and primary AML samples. | Pre-clinical | MK-8776 (CHK1 inhibitor; rabusertib or prexasertib) or CHK1 knockdown enhanced CPX-351 effect and induced apoptosis in multiple AML cell lines and primary samples. | [83] |
| Randomized phase II trial of Ara-C combined with CHK1 inhibitor MK-8776. Patients with relapsed or primary refractory AML were randomized 1:1 to receive either AraC with MK-8776 (Arm A: 14 patients) or AraC alone (Arm B: 18 patients). | Clinical | Response rates and survival were similar between the two groups. | [85] |
| Effect of CHK1 inhibitor GDC-0575, Ara-C, and G-CSF in human AML cell line, primary AML cells, human cord blood cells, and AML cell xenografted mice. | Pre-clinical | Combination of CHK1 inhibitor with G-CSF overcame cytarabine resistance in human AML cell lines and had effects on AML-cell-line-injected NOD/Scid gamma IL2Rγ null mice. | [87] |
| Effect of CHK1 inhibitor LY2603618 and Bcl2 inhibitor ABT-199 in human AML cell line and primary AML cells. | Pre-clinical | Simultaneous treatment with CHK1 inhibitor LY2603618 and ABT-199 resulted in synergistic induction of apoptosis in both AML cell lines and primary patient samples. | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, S. Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment. Hematol. Rep. 2023, 15, 331-346. https://doi.org/10.3390/hematolrep15020035
Takahashi S. Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment. Hematology Reports. 2023; 15(2):331-346. https://doi.org/10.3390/hematolrep15020035
Chicago/Turabian StyleTakahashi, Shinichiro. 2023. "Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment" Hematology Reports 15, no. 2: 331-346. https://doi.org/10.3390/hematolrep15020035
APA StyleTakahashi, S. (2023). Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment. Hematology Reports, 15(2), 331-346. https://doi.org/10.3390/hematolrep15020035