Abstract
Background: Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Modulator therapies have the ability to improve CFTR function in CF patients, but despite the clear evidence of benefits regarding CFTR modulator therapy, including improved lung function, the reduced rate of exacerbations, and an overall improved quality of life, studies focusing on the reduction rates of P. aeruginosa infections during modulator therapy expressed the need for future research on this topic. Objective: This study aimed to evaluate the impact of CFTR modulator therapies on the prevalence, density, and persistence of P. aeruginosa infection in CF patients and to explore the mechanisms involved. Methods: A systematic literature review was performed by searching five major databases (PubMed, Cochrane Library, Scopus, Google Scholar, and Web of Science), and 21 relevant articles investigating the link between CFTR therapy and P. aeruginosa infections were selected following the PRISMA guidelines. Results: The data indicated that Ivacaftor and the combination Elexacaftor/Tezacaftor/Ivacaftor (ETI) can reduce total bacterial load and markers of systemic inflammation. However, clonal lines of P. aeruginosa persist in most cases, and complete eradication is rare, mainly due to biofilm formation and antimicrobial resistance. Conclusions: Although CFTR-modulating therapies help to improve clinical condition and reduce inflammation, they do not consistently lead to the elimination of P. aeruginosa.
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, expressed on the apical surface of epithelial cells, which regulates ion transport through chloride channels [1]. The dysfunction of the CFTR protein leads to ion imbalance, the depletion of airway surface fluid, and pH alteration, thus impairing mucociliary clearance and host immune defenses, ultimately leading to higher susceptibility to chronic airway infections. CFTR dysfunction induces thick mucus accumulation in the airway lumen, alteration in airway microenvironment with airway surface dehydration, and impaired mucociliary clearance [2,3].
A major opportunistic pathogen that causes recurring pulmonary infections in patients diagnosed with CF is Pseudomonas aeruginosa. Its infections are associated with a higher morbidity and mortality rate in many demographic groups, including in patients with pneumonia, chronic obstructive pulmonary disease (COPD), and CF [4]. The World Health Organization (WHO) included it on its priority list of bacterial pathogens, for which the research and development of new strategies are needed [5]. P. aeruginosa is highly capable of causing both acute and chronic infections. Its pathogenic profile comes from a variable and broad depository of virulence factors and antibiotic resistance stored in its genome. Infections with this pathogen are correlated with increased pulmonary exacerbations, accelerated decline in lung function, impaired quality of life, and early death in patients with CF [4,6].
Under normal physiological conditions, CFTR operates as a cAMP-regulated chloride and bicarbonate channel and is upregulated at the apical plasma membrane of various epithelial tissues. It has a key role in maintaining electrolyte and fluid balance, therefore regulating the composition and volume of epithelial secretions [7]. In a mutation in the CFTR gene, CFTR protein expression and/or function is impaired, leading to abnormal ion transport and dehydration of the epithelial surface [7]. This process leads to thick mucus accumulation, chronic inflammation, and recurrent infection, which over time causes tissue damage and structural remodeling [7,8].
Modulator therapies have the ability to improve CFTR function and mutant CFTR protein in CF patients and are classified into the following five main categories according to their respective effects on CFTR mutations: enhancers, correctors, stabilizers, read-through agents, and enhancers [9].
Recent CFTR modulators have revolutionized CF care, correcting defects, improving health, and slowing disease progression. Highly effective CFTR modulator therapy, including Elexacaftor/Tezacaftor/Ivacaftor (ETI), has been found to reduce significantly upper and lower respiratory symptoms and is approved for up to 90% of adults with CF genetic disorders [10,11,12].
Despite the clear evidence of benefits regarding CFTR modulator therapy, including improvement in lung function, a reduced rate of exacerbations, and an overall improved quality of life, observational studies focusing on the interaction between P. aeruginosa and CFTR modulators demonstrate uncertain results [13,14].
This study aimed to investigate the infection rates of P. aeruginosa during CFTR modulator therapy in patients with cystic fibrosis. By conducting a systematic review, we wanted to assess whether CFTR modulators influence the frequency of respiratory infections caused by P. aeruginosa, which is known to be difficult to treat.
2. Materials and Method
The narrative systematic review follows the PRISMA guidelines (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), which ensures methodological transparency, reproducibility, and comprehensive reporting of the search strategy, selection process, data extraction, and findings synthesis.
We conducted a multi-step process to analyze the literature regarding P. aeruginosa infections during CFTR modulator therapy.
2.1. Search Strategy
Our study included a comprehensive current literature search conducted across multiple online databases (PubMed, Cochrane Library, Scopus, Google Scholar, Web of Science) in order to identify potentially relevant studies related to our topic of interest. The search strategy also employed medical subject headings, such as “P. aeruginosa”, “CFTR modulators”, “biofilms”, and “cystic fibrosis”.
2.2. Eligibility Criteria
The inclusion criteria were predefined to include studies that focused on the interaction between CFTR modulator therapy and P. aeruginosa infections in CF patients and were assessed based on this criteria during our initial screening. Once the search results were retrieved, we excluded duplicates, studies older than 10 years, and those that did not provide full-text availability. Titles and abstracts were screened for relevance. Studies that did not meet our inclusion criteria at this stage were excluded. Full-text articles of eligible studies were later retrieved and assessed, while focusing on their study design, population, intervention process, and outcomes (as expressed in Figure 1). Our selection process aimed at ensuring that the systematic review provided a comprehensive and unbiased analysis of available literature.
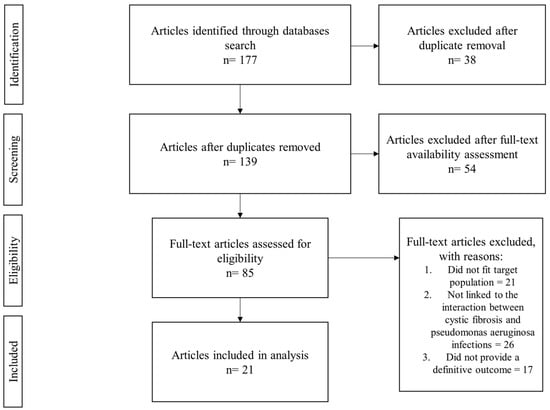
Figure 1.
Flowchart of the selection process.
3. Results
3.1. Article Characteristics
At the end of our selection process, 21 studies were included in this review. Out of the 21 articles, 6 were observational cohort studies (28.57%) [15,16,17,18,19,20], 5 were randomized controlled trials (23.80%) [10,13,14,21,22], 3 were longitudinal cohort studies (14.28%) [23,24,25], 2 were prospective monocentric studies (9.52%) [11,26], prospective observational studies (9.52%) [12,27], and experimental studies (9.52%) [28,29], and 1 was a qualitative analysis study (4.76%) [30].
A diverse range of treatment modalities for culture-positive P. aeruginosa infections and CFTR modulator therapy were assessed, as described in Table 1.

Table 1.
Article protocol and outcome characteristics.
3.2. Protocol
During our assessment of the studies, we identified and outlined the protocols used in the selected articles. The most frequent protocol used was Elexacaftor/Tezacaftor/Ivacaftor (46.61% [10,11,12,13,14,15,16,21,27], followed by Ivacaftor (9.52%) [17,23], with the following protocols: ex vivo analyses of Ki-67 expression in antigen-specific CD154 (+) T cells [25], Ivacaftor, and antibiotics [18]; rRNA gene amplicon sequencing of sinus, throat, and sputum samples [19]; the physiological effects of ETI [30]; antibiotic treatment to combat pseudomonas biofilms [22]; Ivacaftor, Ivacaftor/Lumacaftor, Tezacaftor/Ivacaftor [24]; Symkevi/ETI [26]; Ivacaftor, Lumacaftor, Tezacaftor, Elexacaftor, and ETI combined with antibiotics [28]; phage and ciprofloxacin alone and in combination [29]; and Orkambi [20] being tied at 4.76%.
Assessment of methodological quality and reliability of evidence.
The quality of the selected studies was assessed using the JBI (Joanna Briggs Institute) tool, adapted according to each type of study. Each tool contains between 8 and 13 items, covering internal validity, the selection of participants, the measurement of exposure and outcome, and the management of confounding factors. A score was calculated for each study and was expressed as the ratio between the number of items that met the criteria and the total number of applicable items. The JBI scale has a system of scores that consist of ≥8/10 or ≥10/13, which indicate good quality, 7/10, which expresses moderate quality, and scores ≤ 6/10, which suggest a high risk of bias [31].
The GRADE (Grading of Recommendations, Assessment, Development and Evaluation) scale was used to assess the level of confidence in the included studies. This grading classifies the overall certainty of evidence into the following four main categories: high, moderate, low, and very low, which take the risk of bias, inconsistency of results, indirectness of evidence, imprecision of estimates, and risk of selective publication into account. The GRADE scale has a grading that consists of a high score, which indicates a strong evidence that the study was not affected by significant limitations, while a low or very low score reflects uncertainty about the estimated effect [32].
The evaluation was conducted by two separate authors, and discrepancies were discussed until a consensus was reached.
3.3. Study Group Demographics
Out of the 21 selected studies that employed a study group, there were a total of 4332 patients with CF registered, with a distribution of 48% female and 49% male, with the remaining 3% being the number of patients with unspecified sex. We found that among these patients, 3008 had at least one copy of the F508del CFTR mutation, making up for 69.4% of the total registered patients. This result is in line with other studies regarding the frequency of the F508del, it being the most common mutation [33,34,35].
3.4. Description of Studies
3.4.1. ETI
In a study conducted by Lee T. et al. that involved 468 patients, regarding the annual rate of lung function decline of CF patients on ETI treatment, discovered that on average, pulmonary function was not lost over a two-year period, assessed through a mean annualized rate of change in percent predicted forced expiratory volume in 1 s (ppFEV1), thus demonstrating that CFTR modulator therapy has the potential of stopping lung function decline over an extended period of time in CF patients [15].
Ledger E. et al. conducted a randomized controlled trial, involving 11 patients with CF, to investigate how P. aeruginosa in CF patients may change in an altered lung environment after the initiation of CFTR therapy. They showed that clonal lineages of P. aeruginosa persisted even after CFTR therapy, with no evidence of displacement by alternative strains, sustained mucoid morphology, and continued resistance to antibiotics in isolates [21].
Long D. et al. investigated through a randomized controlled trial involving 15 patients with CF, of which 10 were culture-positive with P. aeruginosa, whether methods for this pathogen whole genome hybridization enrichment could enhance detection from cfDNA. Read counts of P. aeruginosa for the 10 culture-positive patients increased by 3505-fold on average, thus indicating that the sequencing power can potentially be reduced by that same factor without a negative impact on assay performance, but relative levels of normalized P. aeruginosa cfDNA remained unchanged when compared among patients. Their results express that plasma cfDNA sequencing can identify P. aeruginosa respiratory culture positivity in CF patients, even those treated with CFTR modulators [10].
In a prospective monocentric study conducted by Schnell A. et al., 69 patients were evaluated on the effects of ETI treatment on clinical, biochemical data, and P. aeruginosa colonization rate. Marked improvements on biochemical markers of systemic inflammation were observed, as white blood cell count and the level of immunoglobulin A, G, M, and albumin within 24 weeks of therapy were monitored. The authors concluded that ETI treatment was effective in ameliorating lung function and sweat chloride concentration. Colonization status of P. aeruginosa assessment revealed a conversion from a positive to negative detection in 36% of cases after a one-year period of therapy [11].
Similar results were obtained in a prospective observational study ran by Migliorisi G. et al., who enrolled 13 patients with CF, with the aim of defining the clinical and microbiological implications of ETI treatment administration. They reported that airway infection rates decreased, and pulmonary exacerbations were drastically reduced after a one-year period of therapy; however, P. aeruginosa showed continuous colonization rates, although slightly reduced [27].
One study conducted by Aspinall S. et al. discusses the lived experience of 12 CF patients undergoing ETI therapy and the psychological aspects involved to determine the disease burden during CFTR therapy. They concluded that individuals undergoing ETI therapy experience increased anxiety and fear of returning to life pre-ETI treatment [30].
Sutharsan S. et al. employed an observational cohort study that aimed at evaluating the real-world impact of ETI on lung function, pulmonary exacerbations frequency, sweat chloride concentration, and nutritional status on 2645 CF patients. Over the first year of ETI, they observed an increase in ppFEV1 by 11.3%, a decrease of 75.9% in pulmonary exacerbation frequency, and a decrease in mean sweat chloride concentration of 50.9 mmol/L [16].
3.4.2. Ivacaftor
Rowe S. et al. conducted a longitudinal cohort study that involved 151 patients with CF and expressed significant clinical and physiologic improvements on the initiation of Ivacaftor, with an improvement in predicted FEV1% (forced expiratory volume in 1 s) from baseline to 6 months, an improvement in baseline body mass index (BMI), decreased sweat chloride from baseline to 6 months, and a reduction in P. aeruginosa infection [23].
In an observational cohort study by Durfey S. et al., involving 10 patients with CF, the combination of Ivacaftor and an intensive three and a half months of antibiotic course was investigated on the impact on chronic P. aeruginosa clearance. Ivacaftor alone improved CFTR activity and lung function and inflammation within 48 h and reduced P. aeruginosa density by ~10 fold within a week. However, at the end of the study, they concluded that all persistently P. aeruginosa culture-positive CF patients remained infected by their pretreatment strain, suggesting that chronic CF infection with this pathogen resist eradication even after marked and rapid modulator-induced improvements in lung infection and inflammation parameters and aggressive antibiotic treatment [18].
Heltshe S. et al. enrolled 151 CF patients in a longitudinal observational cohort study to examine changes in CF respiratory pathogens with Ivacaftor and the correlation with baseline characteristics and their clinical response. Of the 89 patients that were culture-positive for P. aeruginosa the year prior to Ivacaftor use, 26 were culture-negative the year following treatment, with 52 other culture-negative patients remaining uninfected. They showed a 35% reduction in P. aeruginosa positivity in the year after Ivacaftor treatment, compared to the year prior, also showing reduced odds of mucoid P. aeruginosa and Aspergillus but not S. aureus or other common CF pathogens [17].
Westholter D. et al. conducted a longitudinal cohort study with peripheral blood mononuclear cells and serum samples collected from 108 patients with CF in order to evaluate if CFTR modulator therapy also targets T cells and thereby influences immune cell abnormalities in CF. They concluded that P. aeruginosa impairs regulatory T cells in CF patients [24].
3.4.3. Experimental
Eschenhagen P. et al. performed ex vivo analyses of Ki-67 expression in antigen-specific CD154 (+) T cells against bacterial and fungal respiratory pathogens in CF after the initiation of highly effective CFTR modulator therapy and showed a significant decrease in mean Ki-67 expression in antigen-specific CD154 (+) T cells against P. aeruginosa, Aspergillus fumigatus, Scedosporium apiospermum, and Candida albicans, but not Staphylococcus aureus or mean total serum IgG and IgE, and they showed a significant increase in BMI and FEV1 after the initiation of ETI treatment [25].
The same results were registered in another observational cohort study conducted by Armbruster C. et al. with a cohort of 19 CF patients. They expressed that patients remained infected throughout their upper and lower respiratory tract with the same strain of P. aeruginosa after the initiation of ETI treatment, and that those strains continued to evolve in response to the newly CFTR-corrected airway [19].
3.4.4. Orkambi
In an observational cohort study by Adam D. et al., 22 CF patients were observed and evaluated during Orkambi combination treatment for the effects on the repair of the CF primary airway epithelia in infectious conditions. Their results showed that combined treatment with VX-809 and VX-770 contributed to a greater beneficial impact on airway epithelial repair and a slight improvement in airway epithelial repair and transepithelial resistance, even in the presence of P. aeruginosa exoproducts [20].
3.4.5. Biofilm
A randomized controlled trial by Yau Y. et al. that enrolled 88 patients with CF aimed to determine whether P. aeruginosa’s antimicrobial susceptibility testing grown as a biofilm, instead of planktonically, improves the efficacy of antibiotic treatment on pulmonary exacerbations. Their results show that biofilm antimicrobial susceptibility testing did not improve microbiological or clinical outcomes compared to the conventional methods of treatment of pulmonary exacerbations in CF patients with chronic P. aeruginosa infection [22].
4. Discussion
Impact of CFTR modulators on P. aeruginosa colonization.
Our study aimed to investigate the infection rates of P. aeruginosa during CFTR modulator therapy in patients with cystic fibrosis. P. aeruginosa displays a resistance to a wide variety of antibiotics. Typically, P. aeruginosa has three primary mechanisms used to suppress antibiotics, which can be classified as intrinsic resistance, acquired resistance, and adapted resistance [36]. Intrinsic resistance refers to its low-outer membrane permeability and through antibiotic expulsion out of the cell using efflux pumps, leading to enzymes that inactivate antibiotics [4,37]. Acquired resistance is expressed through multifactorial mutational change or chromosomal mutation and the capability of the horizontal transfer of resistance genes [38,39]. Lastly, its adaptive resistance leads to the formation of sputum-suspended aggregates, also named biofilms, in the patient’s lungs, where it serves as a barrier, thus limiting antibiotic access to bacterial cells [36,40]. Current literature reports a 36% conversion from a positive P. aeruginosa to a negative P. aeruginosa status following 12 months of ETI treatment and also a 35% reduction in P. aeruginosa mucoid detection following Ivacaftor therapy [11,17]. However, P. aeruginosa long-term persistence remains high [29,37,39,41].
Persistence of colonies and biofilm as a therapeutic barrier.
Sputum-suspended aggregates, or biofilms, consist of matrix-associated exopolysaccharides (EPS), extracellular DNA, and proteins [40,41]. This structure produces chemical and nutrient gradients that affect cells differently. It also provides physical protection against antimicrobials and immune host cells [42]. Biofilm-grown P. aeruginosa has a constant and gradual adaptation that bolsters its defensive capabilities and survivability. In cases where a failed attempt at its eradication occurs, the infection becomes chronic and leads to further inflammation and scarring [42]. This often leads to a decrease in lung function, quality of life, and increased mortality in infected cystic fibrosis patients [38,43]. After the initial colonization of the airways, P. aeruginosa transitions from a planktonic to a biofilm-like phenotype, particularly under the influence of a microenvironment with low pH, hypoxia, and osmotic stress [44]. This transition is associated with a profound bacterial transcriptomic reprogramming, including the activation of the las and rhl regulatory systems, as well as the overexpression of the algD gene, responsible for alginate synthesis. Post-transplant histopathologic studies have shown that areas of increased biofilm density correspond topographically with regions of severe bronchiectasis and parietal pulmonary fibrosis, supporting the idea that biofilm is not only an effect of chronic infection but an active factor in tissue progression [45].
This chronic picture perpetuates a vicious circle; the biofilm maintains the inflammation, which in turn, sustains the survival of the biofilm. Therefore, therapeutic strategies combining CFTR modulators with anti-biofilm agents (such as DNase, quorum sensing inhibitors or bacteriophages) are promising but still under-tested in randomized clinical trials [46].
Current research gaps and limitations of included studies.
Until 2012, CF therapies were mainly focused on disease sign and symptom management through inhalation and physical therapy alongside numerous daily medications, including antibiotics, anti-inflammatory agents, and mucolytics, assessed by a multidisciplinary healthcare team [47,48,49]. With the help of fundamental advances in the development of preclinical cell models and the implementation of cell-based high-throughput screening essays, new treatment modalities appeared that target the primary CFTR defect [50]. These CFTR modulators can restore both the folding and cross-linking of the mutant CFTR protein or increase the probability of channel opening when the protein is localized to the plasma membrane [20,21,25].
As expressed in our study assessment process, we outlined the most frequently used CFTR modulator protocols, the main ones being the triple combination of Elexacaftor–Tezacaftor–Ivacaftor, followed by Ivacaftor-only treatment. Due to the relatively recent introduction of CFTR modulators, there is currently a lack of prospective observational and experimental studies that assess the interaction between these modulators and P. aeruginosa.
Ivacaftor was the first CFTR potentiator that expressed clinically significant improvements in lung function and nutritional status in patients with cystic fibrosis [17,23]. It was approved by the FDA for numerous CFTR residual mutations in in vitro studies [51], with its clinical benefits confirmed in several clinical studies [17,52,53]. However, a couple of studies reported that indeed Ivacaftor treatment showed significant clinical and physiological improvements in CF patients, but not only do these patients remain colonized with P. aeruginosa, they also show that the same clonal lineages persist, as opposed to eradicating the preexisting strains [21,23]. Of note, despite reporting an improved airway obstruction, the biomarkers of airway inflammation showed no meaningful improvement with the addition of Ivacaftor [25]. One likely explanation of this infection persistence is the irreversible structural damage that causes defective pathogen clearance, with P. aeruginosa being known for its adaptability to a CF lung environment [27].
The triple combination, or ETI, is the first triple combination of modulator drugs approved for cystic fibrosis patients aged two or higher with at least one F508del mutation. This mutation is the most common among patients with CF, making it accessible for most patients [54]. There continues to be a gap in our understanding of whether or how these CFTR modulators affect the microbiological profile in CF patients.
While Ivacaftor showed an initial reduction in the sputum and isolation of bacterial pathogens, Heltshe et al. showed that following Ivacaftor treatment, P. aeruginosa detection rates were decreased by 35% over the course of one year, with 26 out of 89 culture-positive patients becoming culture-negative [17]. Similar results were expressed by Durfey et al., who observed a ten-fold reduction in P. aeruginosa density following one week of Ivacaftor treatment; however, long-term eradication was not achieved [18]. P. aeruginosa density and strains rebounded and were still present even after intensive antibiotic therapy [17,18,54]. In similar studies examining ETI’s impact on P. aeruginosa infections, several researchers showed a decrease in total bacterial load and a normalization of systemic inflammation markers, reflected by a reduction in P. aeruginosa RNA quantity in the sputum samples of CF patients following ETI treatment [11,19,28]. However, as in the case of Ivacaftor, although a reduction in bacterial load was recorded, some studies found that 100% of involved patients remained colonized with the same strain of P. aeruginosa even after six to twelve months of CFTR therapy [19,21]. Additional research is needed to determine how CF patients continue to change post-ETI treatment.
Therapeutic implications and combined strategies.
Molecular synergy between inhaled antibiotics and CFTR modulators has been suggested in the literature [18,55]. However, current evidence does not fully support this hypothesis. One study that evaluated the bacterial density of P. aeruginosa reported a decrease in CF patients receiving CFTR modulators with concurrent inhaled antibiotics, with very few patients reporting a cleared infection [18]. Currently, there is insufficient data to support the discontinuation or continuation of inhaled antibiotic therapy during CFTR modulator therapy. Additional data is needed to develop clear guidelines.
Translational perspectives and future research directions.
New treatments are emerging with the hope of improving the CFTR function, pulmonary function, and overall quality of life of CF patients. Recent studies involving a new triple therapy of Vanzacaftor–Tezacaftor–Deutivacaftor show promising results in this field [56,57,58].
Vanzacaftor–Tezacaftor–Deutivacaftor therapy has been approved for CF patients aged six and above, demonstrating significant increases in lung function and the correction of the defective CFTR protein, alongside a reduction in sweat chloride levels [59].
Two phase-three studies demonstrated non-inferiority to ETI with significant improvements in both lung function, through a substantial increase in ppFEV1 in the studied population, and the correction of CFTR proteins [60,61]. Whereas Ivacaftor treatment is taken twice a day, the combination of Vanzacaftor–Tezacaftor–Deutivacaftor is recommended once a day, potentially improving the adherence to prescribed treatment in CF patients [62,63]. However, infection rates in the target population were not studied; thus, further research into this topic is needed to determine if this new therapy also reduces infection rates caused by P. aeruginosa in CF patients.
5. Conclusions
P. aeruginosa infections remain a recurrent problem in CF, even in the context of next-generation CFTR modulator therapies. Although reviewed studies show a reduction in bacterial load and improvement in inflammatory markers following treatment with Ivacaftor or ETI, the complete eradication of the bacteria is rarely achieved, and persistent colonies continue to affect patients.
These findings highlight the need for further research into the connection between CFTR modulators and the resistance mechanisms of P. aeruginosa, analyzing both the role of biofilm and chronic inflammation in maintaining colonization rates.
Integrating both anti-biofilm and modulator therapy into a personalized therapeutic program could lead to an improvement in the control of chronic infections and a higher quality of life.
Author Contributions
Conceptualization, A.F.C. and C.C.P.; methodology, C.C.P., V.C. and A.M.; validation, C.O., C.C.P. and M.M.; investigation, A.F.C.; resources, Ș.D.-R. and S.L.; data curation, A.F.C., A.M. and M.M.; writing—original draft preparation, C.C.P. and V.C.; writing—review and editing, A.F.C., C.C.P. and M.M.; visualization, C.O.; supervision, Ș.D.-R., S.L. and C.O.; project administration, A.F.C. and C.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The publication costs of the present article were covered by “Victor Babes” University of Medicine and Pharmacy Timisoara.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CF | Cystic Fibrosis |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| COPD | Chronic Obstructive Pulmonary Disease |
| WHO | World Health Organization |
| ETI | Elexacaftor–Tezacaftor–Ivacaftor |
| EPS | Exopolysaccharides |
| FEV1 | Forced Expiratory Volume in 1 s |
| GRADE | Grading of Recommendations, Assessment, Development, and Evaluation |
| JBI | Joanna Briggs Institute |
| ppFEV1 | Percent Predicted Forced Expiratory Volume in 1 s |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
References
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Bacterial Priority List; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Williams, D.; Fothergill, J.L.; Evans, B.; Caples, J.; Haldenby, S.; Walshaw, M.J.; Brockhurst, M.A.; Winstanley, C.; Paterson, S. Transmission and lineage displacement drive rapid population genomic flux in cystic fibrosis airway infections of a Pseudomonas aeruginosa epidemic strain. Microb. Genom. 2018, 4, e000167. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef]
- Long, D.R.; Holmes, E.A.; Goss, C.H.; Singh, P.K.; Waalkes, A.; Salipante, S.J. Cell-Free DNA Detects Pseudomonas aeruginosa Lung Infection in Modulator-treated People with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2023, 208, 944–947. [Google Scholar] [CrossRef]
- Schnell, A.; Hober, H.; Kaiser, N.; Ruppel, R.; Geppert, A.; Tremel, C.; Sobel, J.; Plattner, E.; Woelfle, J.; Hoerning, A. Elexacaftor—Tezacaftor—Ivacaftor treatment improves systemic infection parameters and Pseudomonas aeruginosa colonization rate in patients with cystic fibrosis a monocentric observational study. Heliyon 2023, 9, e15756. [Google Scholar] [CrossRef]
- Nichols, D.P.; Paynter, A.C.; Heltshe, S.L.; Donaldson, S.H.; Frederick, C.A.; Freedman, S.D.; Gelfond, D.; Hoffman, L.R.; Kelly, A.; Narkewicz, M.R.; et al. Clinical Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis: A Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2020, 395, 1694, Erratum in Lancet 2019, 394, 1940–1948. https://doi.org/10.1016/S0140-6736(19)32597-8. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Sawicki, G.S.; Altenburg, J.; Millar, S.J.; Geiger, J.M.; Jennings, M.T.; Lou, Y.; McGarry, L.J.; Van Brunt, K.; Linnemann, R.W. Effect of elexacaftor/tezacaftor/ivacaftor on annual rate of lung function decline in people with cystic fibrosis. J. Cyst. Fibros. 2023, 22, 402–406. [Google Scholar] [CrossRef]
- Sutharsan, S.; Dillenhoefer, S.; Welsner, M.; Stehling, F.; Brinkmann, F.; Burkhart, M.; Ellemunter, H.; Dittrich, A.M.; Smaczny, C.; Eickmeier, O.; et al. Impact of elexacaftor/tezacaftor/ivacaftor on lung function, nutritional status, pulmonary exacerbation frequency and sweat chloride in people with cystic fibrosis: Real-world evidence from the German CF Registry. Lancet Reg. Health Eur. 2023, 32, 100690. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M.; GOAL (the G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Durfey, S.L.; Pipavath, S.; Li, A.; Vo, A.T.; Ratjen, A.; Carter, S.; Morgan, S.J.; Radey, M.C.; Grogan, B.; Salipante, S.J.; et al. Combining Ivacaftor and Intensive Antibiotics Achieves Limited Clearance of Cystic Fibrosis Infections. mBio 2021, 12, e0314821. [Google Scholar] [CrossRef]
- Armbruster, C.R.; Hilliam, Y.K.; Zemke, A.C.; Atteih, S.; Marshall, C.W.; Moore, J.; Koirala, J.; Krainz, L.; Gaston, J.R.; Lee, S.E.; et al. Persistence and evolution of Pseudomonas aeruginosa following initiation of highly effective modulator therapy in cystic fibrosis. mBio 2024, 15, e0051924. [Google Scholar] [CrossRef]
- Adam, D.; Bilodeau, C.; Sognigbé, L.; Maillé, É.; Ruffin, M.; Brochiero, E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J. Cyst. Fibros. 2018, 17, 705–714. [Google Scholar] [CrossRef]
- Ledger, E.L.; Smith, D.J.; Teh, J.J.; Wood, M.E.; Whibley, P.E.; Morrison, M.; Goldberg, J.B.; Reid, D.W.; Wells, T.J. Impact of CFTR Modulation on Pseudomonas aeruginosa Infection in People With Cystic Fibrosis. J. Infect. Dis. 2024, 230, e536–e547. [Google Scholar] [CrossRef]
- Yau, Y.C.; Ratjen, F.; Tullis, E.; Wilcox, P.; Freitag, A.; Chilvers, M.; Grasemann, H.; Zlosnik, J.; Speert, D.; Corey, M.; et al. Randomized controlled trial of biofilm antimicrobial susceptibility testing in cystic fibrosis patients. J. Cyst. Fibros. 2015, 14, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Westhölter, D.; Beckert, H.; Straßburg, S.; Welsner, M.; Sutharsan, S.; Taube, C.; Reuter, S. Pseudomonas aeruginosa infection, but not mono or dual-combination CFTR modulator therapy affects circulating regulatory T cells in an adult population with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Eschenhagen, P.N.; Bacher, P.; Grehn, C.; Mainz, J.G.; Scheffold, A.; Schwarz, C. Proliferative activity of antigen-specific CD154+ T cells against bacterial and fungal respiratory pathogens in cystic fibrosis decreases after initiation of highly effective CFTR modulator therapy. Front. Pharmacol. 2023, 14, 1180826. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Dayman, N.; Blyth, N.; Madge, J.; Gaillard, E. Impact of CFTR modulators on exercise capacity in adolescents with cystic fibrosis. ERJ Open Res. 2024, 10, 00687-2023. [Google Scholar] [CrossRef] [PubMed]
- Migliorisi, G.; Collura, M.; Ficili, F.; Pensabene, T.; Bongiorno, D.; Collura, A.; Di Bernardo, F.; Stefani, S. Elexacaftor-Tezacaftor-Ivacaftor as a Final Frontier in the Treatment of Cystic Fibrosis: Definition of the Clinical and Microbiological Implications in a Case-Control Study. Pharmaceuticals 2022, 15, 606. [Google Scholar] [CrossRef]
- Cigana, C.; Giannella, R.; Colavolpe, A.; Alcalá-Franco, B.; Mancini, G.; Colombi, F.; Bigogno, C.; Bastrup, U.; Bertoni, G.; Bragonzi, A. Mutual Effects of Single and Combined CFTR Modulators and Bacterial Infection in Cystic Fibrosis. Microbiol. Spectr. 2023, 11, e0408322. [Google Scholar] [CrossRef]
- Luscher, A.; Simonin, J.; Falconnet, L.; Valot, B.; Hocquet, D.; Chanson, M.; Resch, G.; Köhler, T.; van Delden, C. Combined Bacteriophage and Antibiotic Treatment Prevents Pseudomonas aeruginosa Infection of Wild Type and cftr- Epithelial Cells. Front. Microbiol. 2020, 11, 1947. [Google Scholar] [CrossRef]
- Aspinall, S.A.; Mackintosh, K.A.; Hill, D.M.; Cope, B.; McNarry, M.A. Evaluating the Effect of Kaftrio on Perspectives of Health and Wellbeing in Individuals with Cystic Fibrosis. Int. J. Environ. Res. Public Health 2022, 19, 6114. [Google Scholar] [CrossRef]
- Aromataris, E.; Munn, Z. JBI Manual for Evidence Synthesis; Joanna Briggs Institute: Adelaide, Australia, 2020. [Google Scholar]
- Guyatt, G.H.; Oxman, A.D.; Vist, G.E.; Kunz, R.; Falck-Ytter, Y.; Alonso-Coello, P.; Schünemann, H.J.; GRADE Working Group. GRADE: An emerging consensus on rating quality of evidence and strength of Reccomandation. BMJ 2008, 336, 924–926. [Google Scholar] [CrossRef]
- Petrova, N.; Balinova, N.; Marakhonov, A.; Vasilyeva, T.; Kashirskaya, N.; Galkina, V.; Ginter, E.; Kutsev, S.; Zinchenko, R. Ethnic Differences in the Frequency of CFTR Gene Mutations in Populations of the European and North Caucasian Part of the Russian Federation. Front. Genet. 2021, 12, 678374. [Google Scholar] [CrossRef]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef]
- Varkki, S.D.; Aaron, R.; Chapla, A.; Danda, S.; Medhi, P.; Jansi Rani, N.; Paul, G.R. CFTR mutations and phenotypic correlations in people with cystic fibrosis: A retrospective study from a single centre in south India. Lancet Reg. Health Southeast Asia 2024, 27, 100434. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Kapouni, N.; Moustaki, M.; Douros, K.; Loukou, I. Efficacy and Safety of Elexacaftor-Tezacaftor-Ivacaftor in the Treatment of Cystic Fibrosis: A Systematic Review. Children 2023, 10, 554. [Google Scholar] [CrossRef]
- Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410. [Google Scholar] [CrossRef]
- Valladares, K.N.; Jones, L.I.; Barnes, J.W.; Krick, S. Highly Effective Modulator Therapy: Implications for the Microbial Landscape in Cystic Fibrosis. Int. J. Mol. Sci. 2024, 25, 11865. [Google Scholar] [CrossRef]
- Burgel, P.R.; Ballmann, M.; Drevinek, P.; Heijerman, H.; Jung, A.; Mainz, J.G.; Peckham, D.; Plant, B.J.; Schwarz, C.; Taccetti, G.; et al. Considerations for the use of inhaled antibiotics for Pseudomonas aeruginosa in people with cystic fibrosis receiving CFTR modulator therapy. BMJ Open Respir. Res. 2024, 11, e002049. [Google Scholar] [CrossRef]
- Perrem, L.; Ratjen, F. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr. Pulmonol. 2019, 54, S46–S55. [Google Scholar] [CrossRef]
- Chung, J.; Eisha, S.; Park, S.; Morris, A.J.; Martin, I. How Three Self-Secreted Biofilm Exopolysaccharides of Pseudomonas aeruginosa, Psl, Pel, and Alginate, Can Each Be Exploited for Antibiotic Adjuvant Effects in Cystic Fibrosis Lung Infection. Int. J. Mol. Sci. 2023, 24, 8709. [Google Scholar] [CrossRef]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial Extracellular Polysaccharides in Biofilm Formation and Function. Microb. Biofilms 2015, 3, 223–247. [Google Scholar] [CrossRef]
- Santos-Fernandez, E.; Martin-Souto, L.; Antoran, A.; Areitio, M.; Aparicio-Fernandez, L.; Bouchara, J.P.; Schwarz, C.; Rementeria, A.; Buldain, I.; Ramirez-Garcia, A. Microbiota and fungal-bacterial interactions in the cystic fibrosis lung. FEMS Microbiol Rev. 2023, 47, fuad029. [Google Scholar] [CrossRef]
- Burgel, P.R.; Southern, K.W.; Addy, C.; Battezzati, A.; Berry, C.; Bouchara, J.P.; Brokaar, E.; Brown, W.; Azevedo, P.; Durieu, I.; et al. Standards for the care of people with cystic fibrosis (CF); recognising and addressing CF health issues. J. Cyst. Fibros. 2024, 23, 187–202. [Google Scholar] [CrossRef]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H.A. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef]
- Soares, A.; Alexandre, K.; Etienne, M. Tolerance and Persistence of Pseudomonas aeruginosa in Biofilms Exposed to Antibiotics: Molecular Mechanisms, Antibiotic Strategies and Therapeutic Perspectives. Front. Microbiol. 2020, 11, 2057. [Google Scholar] [CrossRef]
- Moser, C.; Jensen, P.Ø.; Thomsen, K.; Kolpen, M.; Rybtke, M.; Lauland, A.S.; Trøstrup, H.; Tolker-Nielsen, T. Immune Responses to Pseudomonas aeruginosa Biofilm Infections. Front. Immunol. 2021, 12, 625597. [Google Scholar] [CrossRef]
- Jarzynka, S.; Makarewicz, O.; Weiss, D.; Minkiewicz-Zochniak, A.; Iwańska, A.; Skorupa, W.; Padzik, M.; Augustynowicz-Kopeć, E.; Olędzka, G. The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study. Pathogens 2023, 12, 1440. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2019, 7, e40, Erratum in Lancet Respir. Med. 2020, 8, 65–124. https://doi.org/10.1016/S2213-2600(19)30337-6. [Google Scholar] [CrossRef]
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision-making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Salvatore, D.; Carnovale, V.; Iacotucci, P.; Braggion, C.; Castellani, C.; Cimino, G.; Colangelo, C.; Francalanci, M.; Leonetti, G.; Lucidi, V.; et al. Effectivenesss of ivacaftor in severe cystic fibrosis patients and non-G551D gating mutations. Pediatr. Pulmonol. 2019, 54, 1398–1403. [Google Scholar] [CrossRef]
- Zhang, M.; Brindle, K.; Robinson, M.; Ingram, D.; Cavany, T.; Morice, A. Chronic cough in cystic fibrosis: The effect of modulator therapy on objective 24-h cough monitoring. ERJ Open Res. 2022, 8, 00031-2022. [Google Scholar] [CrossRef]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M.; GOALe(2) Investigators. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Nichols, D.P.; Odem-Davis, K.; Riekert, K.A.; Sawicki, G.S.; Donaldson, S.H.; Ratjen, F.; Konstan, M.W.; Simon, N.; Rosenbluth, D.B.; et al. Evaluating the Impact of Stopping Chronic Therapies after Modulator Drug Therapy in Cystic Fibrosis: The SIMPLIFY Clinical Trial Study Design. Ann. Am. Thorac. Soc. 2021, 18, 1397–1405. [Google Scholar] [CrossRef]
- Dobiáš, R.; Škríba, A.; Pluháček, T.; Petřík, M.; Palyzová, A.; Káňová, M.; Čubová, E.; Houšť, J.; Novák, J.; Stevens, D.A.; et al. Noninvasive Combined Diagnosis and Monitoring of Aspergillus and Pseudomonas Infections: Proof of Concept. J. Fungi 2021, 7, 730. [Google Scholar] [CrossRef]
- Hoppe, J.E.; Kasi, A.S.; Pittman, J.E.; Jensen, R.; Thia, L.P.; Robinson, P.; Tirakitsoontorn, P.; Ramsey, B.; Mall, M.A.; Taylor-Cousar, J.L.; et al. Vanzacaftor-tezacaftor-deutivacaftor for children aged 6-11 years with cystic fibrosis (RIDGELINE Trial VX21-121-105): An analysis from a single-arm, phase 3 trial. Lancet Respir. Med. 2025, 13, e19, Erratum in Lancet Respir. Med. 2025, 13, 244–255. https://doi.org/10.1016/S2213-2600(24)00407-7. [Google Scholar] [CrossRef]
- Uluer, A.Z.; MacGregor, G.; Azevedo, P.; Indihar, V.; Keating, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Rubenstein, R.C.; et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: Randomised, double-blind, controlled, phase 2 trials. Lancet Respir. Med. 2023, 11, 550–562. [Google Scholar] [CrossRef]
- Keating, C.; Yonker, L.M.; Vermeulen, F.; Prais, D.; Linnemann, R.W.; Trimble, A.; Kotsimbos, T.; Mermis, J.; Braun, A.T.; O’Carroll, M.; et al. Vanzacaftor-tezacaftor-deutivacaftor versus elexacaftor-tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years and older (SKYLINE Trials VX20-121-102 and VX20-121-103): Results from two randomised, active-controlled, phase 3 trials. Lancet Respir. Med. 2025, 13, e19, Erratum in Lancet Respir. Med. 2025, 13, 256–271. https://doi.org/10.1016/S2213-2600(24)00411-9. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).