Virtual Clinical Trials Guided Design of an Age-Appropriate Formulation and Dosing Strategy of Nifedipine for Paediatric Use


 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HPLC Analytical Method Development
2.3. Dose Banding and Selection of Strengths
2.4. Dissolution
2.5. Kinetic Modelling
2.6. Extended-Release Mini Tablet Production
2.7. Hardness
2.8. Friability
2.9. Development of a Nifedipine PBPK Model for Use in Virtual Clinical Trials
2.9.1. Validation in Adults
2.9.2. Formulation Performance in Virtual Adults
2.9.3. Formulation Performance in Virtual Paediatrics
2.9.4. Developing a Dosing Approach in Children
2.9.5. Predictive Performance
2.10. Statistical Analysis
3. Results and Discussion
3.1. Acceptability of Mini-Tablets
3.2. Paediatric Formulation Development Approach for Extended Release Mini-Tablets and Formulation Composition Optimisation
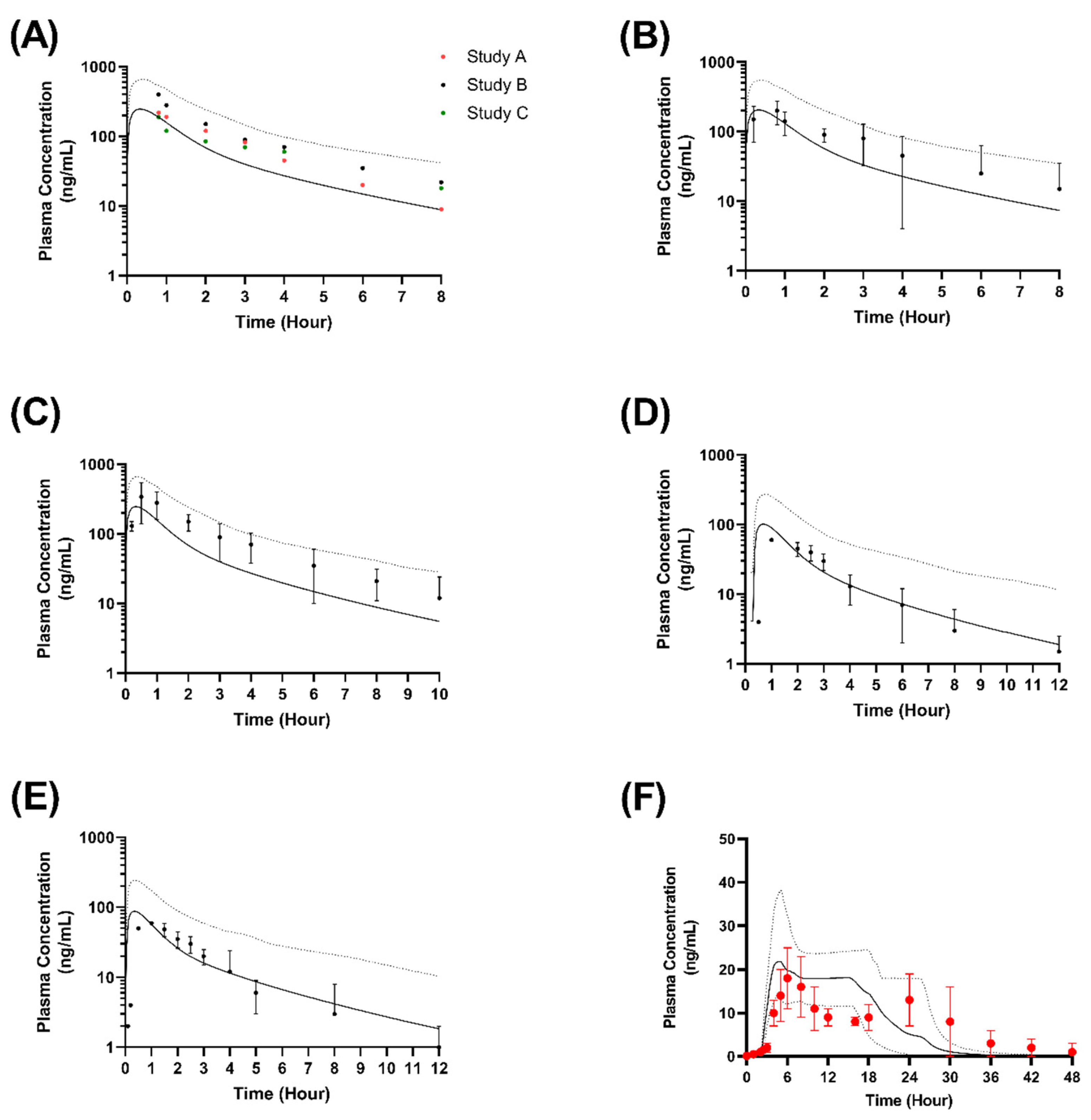
3.3. Virtual Clinical Trials Analysis of Nifedipine Model in Adults and Children
3.4. Formulation Performance in Virtual Adults
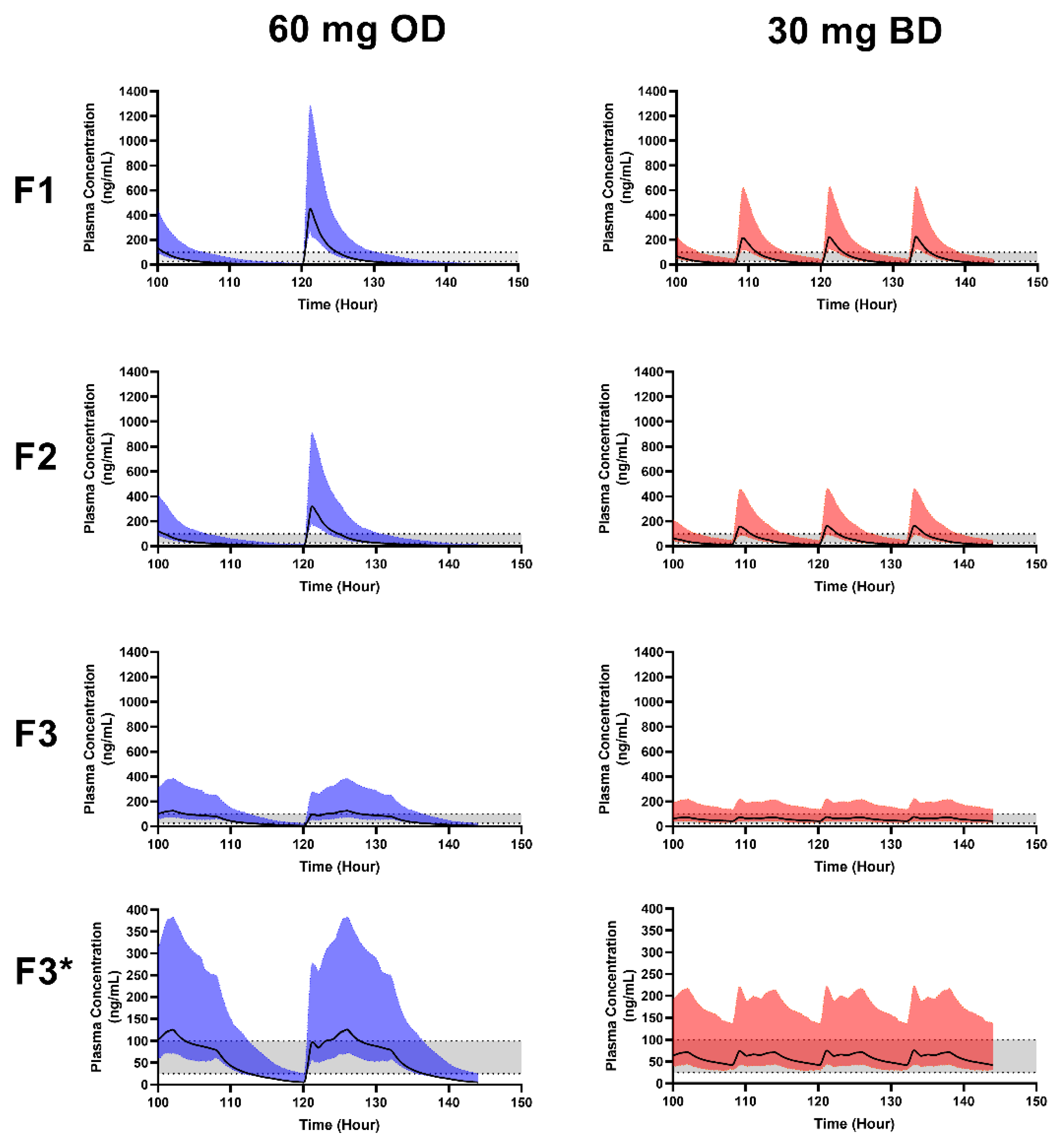
3.5. Formulation Performance in Virtual Paediatrics
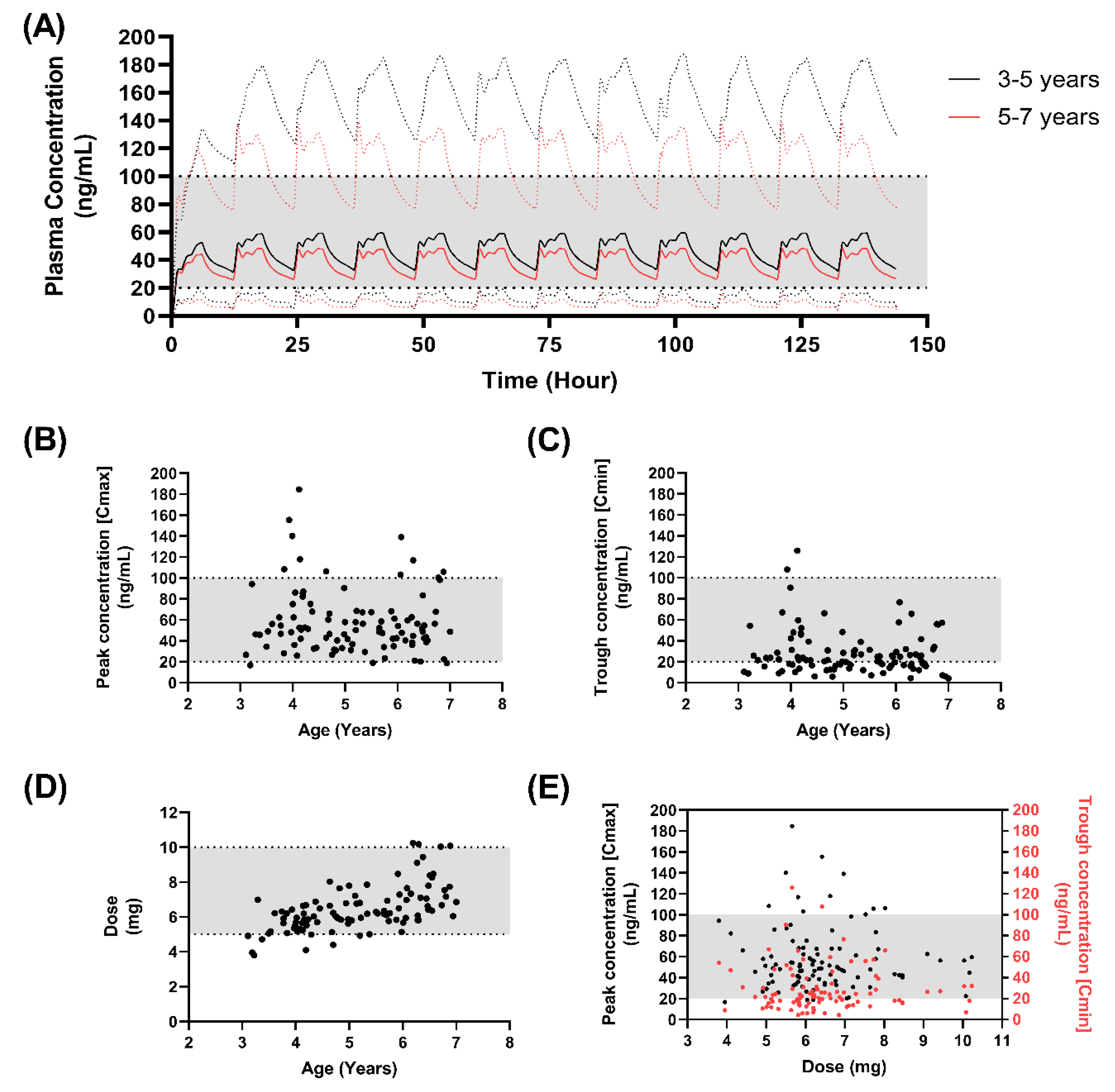
3.6. Developing a Dosing Approach in Children
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, K.M.; Patel, J.B.; Schaefer, T.J. Nifedipine; StatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Royster, R.L.; Butterworth, J.F.; Groban, L.; Slaughter, T.F.; Zvara, D.A. Chapter 8—Cardiovascular Pharmacology. In Essentials of Cardiac Anesthesia; Kaplan, J.A., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2008; pp. 118–163. [Google Scholar]
- Snider, M.E.; Nuzum, D.S.; Veverka, A. Long-acting nifedipine in the management of the hypertensive patient. Vasc. Health Risk Manag. 2008, 4, 1249–1257. [Google Scholar]
- Furberg, C.D.; Psaty, B.M.; Meyer, J.V. Nifedipine. Circulation 1995, 92, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Psaty, B.M.; Heckbert, S.R.; Koepsell, T.D.; Siscovick, D.S.; Raghunathan, T.E.; Weiss, N.S.; Rosendaal, F.R.; Lemaitre, R.N.; Smith, N.L.; Wahl, P.W.; et al. The Risk of Myocardial Infarction Associated With Antihypertensive Drug Therapies. Jama 1995, 274, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, M.J.; Brox, A.; Bestawros, A.N. Calcium channel blockers: An update. Am. J. Med. 2004, 116, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.W.; Deming, D.D.; Hamada, N.; Perkin, R.M.; Sahney, S. Evaluation of the safety of short-acting nifedipine in children with hypertension. Pediatr. Nephrol. 2002, 17, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Blaszak, R.T.; Savage, J.A.; Ellis, E.N. The use of short-acting nifedipine in pediatric patients with hypertension. J. Pediatr. 2001, 139, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Yiu, V.; Orrbine, E.; Rosychuk, R.J.; MacLaine, P.; Goodyer, P.; Girardin, C.; Gowrishankar, M.; Ogborn, M.; Midgley, J.; Filler, G.; et al. The safety and use of short-acting nifedipine in hospitalized hypertensive children. Pediatr. Nephrol. 2004, 19, 644–650. [Google Scholar] [CrossRef]
- Grossman, E.; Messerli, F.H. Chapter 63—Hypertensive Urgencies and Emergencies. In Comprehensive Hypertension; Lip, G.Y.H., Hall, J.E., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 761–774. [Google Scholar]
- Kankali, R.; Kirby, D.; Gerrard, A. Dosing accuracy and stability of enteral nifedipine for paediatric patients. Arch. Dis. Child. 2015, 100, e1. [Google Scholar] [CrossRef]
- Tuleu, C.; Grangé, J.; Seurin, S. The need for pædiatric formulation: Oral administration of nifedipine in children, a proof of concept. J. Drug Deliv. Sci. Technol. 2005, 15, 319–324. [Google Scholar] [CrossRef]
- Sahney, S. A review of calcium channel antagonists in the treatment of pediatric hypertension. Paediatr. Drugs 2006, 8, 357–373. [Google Scholar] [CrossRef]
- Khan, D.; Kirby, D.; Bryson, S.; Shah, M.; Rahman Mohammed, A. Paediatric specific dosage forms: Patient and formulation considerations. Int. J. Pharm. 2022, 616, 121501. [Google Scholar] [CrossRef]
- Upton, R.N.; Foster, D.J.R.; Abuhelwa, A.Y. An introduction to physiologically-based pharmacokinetic models. Pediatr. Anesth. 2016, 26, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, J.E.; Freriksen, J.J.; de Hoop-Sommen, M.A.; van Bussel, L.P.; Driessen, S.H.; Orlebeke, A.E.; Verscheijden, L.F.; Greupink, R.; de Wildt, S.N. Feasibility of a Pragmatic PBPK Modeling Approach: Towards Model-Informed Dosing in Pediatric Clinical Care. Clin. Pharmacokinet. 2022, 61, 1705–1717. [Google Scholar] [CrossRef]
- Cheung, K.W.K.; van Groen, B.D.; Burckart, G.J.; Zhang, L.; de Wildt, S.N.; Huang, S.M. Incorporating ontogeny in physiologically based pharmacokinetic modeling to improve pediatric drug development: What we know about developmental changes in membrane transporters. J. Clin. Pharmacol. 2019, 59, S56–S69. [Google Scholar] [CrossRef]
- Verscheijden, L.F.; Koenderink, J.B.; Johnson, T.N.; de Wildt, S.N.; Russel, F.G. Physiologically-based pharmacokinetic models for children: Starting to reach maturation? Pharmacol. Ther. 2020, 211, 107541. [Google Scholar] [CrossRef]
- Wang, K.; Jiang, K.; Wei, X.; Li, Y.; Wang, T.; Song, Y. Physiologically Based Pharmacokinetic Models Are Effective Support for Pediatric Drug Development. AAPS PharmSciTech 2021, 22, 208. [Google Scholar] [CrossRef] [PubMed]
- Ince, I.; Solodenko, J.; Frechen, S.; Dallmann, A.; Niederalt, C.; Schlender, J.; Burghaus, R.; Lippert, J.; Willmann, S. Predictive pediatric modeling and simulation using ontogeny information. J. Clin. Pharmacol. 2019, 59, S95–S103. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, B.D.; Nicolaï, J.; Kuik, A.C.; Van Cruchten, S.; Van Peer, E.; Smits, A.; Schmidt, S.; De Wildt, S.N.; Allegaert, K.; De Schaepdrijver, L.; et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol. Rev. 2021, 73, 597–678. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.; Mbasu, R.; Nicolaï, J.; Barnouin, K.; Kotian, A.; Mooij, M.G.; Kist, N.; Wijnen, R.M.; Ungell, A.L.; Cutler, P.; et al. Ontogeny of Small Intestinal Drug Transporters and Metabolizing Enzymes Based on Targeted Quantitative Proteomics. Drug Metab. Dispos. 2021, 49, 1038–1046. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Grimstein, M.; Fan, J.; Grillo, J.A.; Huang, S.M.; Zhu, H.; Wang, Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J. Clin. Pharmacol. 2020, 60, S160–S178. [Google Scholar] [CrossRef]
- Grimstein, M.; Yang, Y.; Zhang, X.; Grillo, J.; Huang, S.M.; Zineh, I.; Wang, Y. Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J. Pharm. Sci. 2019, 108, 21–25. [Google Scholar] [CrossRef]
- El-Khateeb, E.; Burkhill, S.; Murby, S.; Amirat, H.; Rostami-Hodjegan, A.; Ahmad, A. Physiological-based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20-years; in-depth analysis of applications, organizations, and platforms. Biopharm. Drug Dispos. 2021, 42, 107–117. [Google Scholar] [CrossRef]
- Zakaria, Z.; Badhan, R.K.S. The impact of CYP2B6 polymorphisms on the interactions of efavirenz with lumefantrine: Implications for paediatric antimalarial therapy. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2018, 119, 90–101. [Google Scholar] [CrossRef]
- Dennison, T.J.; Smith, J.C.; Badhan, R.K.S.; Mohammed, A.R. Formulation and Bioequivalence Testing of Fixed-Dose Combination Orally Disintegrating Tablets for the Treatment of Tuberculosis in the Paediatric Population. J. Pharm. Sci. 2020, 109, 3105–3113. [Google Scholar] [CrossRef]
- Badhan, R.K.S.; Gittins, R. Precision dosing of methadone during pregnancy: A pharmacokinetics virtual clinical trials study. J. Subst. Abus. Treat. 2021, 130, 108521. [Google Scholar] [CrossRef] [PubMed]
- Almurjan, A.; Macfarlane, H.; Badhan, R.K.S. The application of precision dosing in the use of sertraline throughout pregnancy for poor and ultrarapid metabolizer CYP 2C19 subjects: A virtual clinical trial pharmacokinetics study. Biopharm. Drug Dispos. 2021, 42, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Almurjan, A.; Macfarlane, H.; Badhan, R.K.S. Precision dosing-based optimisation of paroxetine during pregnancy for poor and ultrarapid CYP2D6 metabolisers: A virtual clinical trial pharmacokinetics study. J. Pharm. Pharmacol. 2020, 72, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Badhan, R.K.S. The Application of Virtual Therapeutic Drug Monitoring to Assess the Pharmacokinetics of Imatinib in a Chinese Cancer Population Group. J. Pharm. Sci. 2022, 112, 599–609. [Google Scholar] [CrossRef]
- Yu, H.; Singh Badhan, R.K. The Pharmacokinetics of Gefitinib in a Chinese Cancer Population Group: A Virtual Clinical Trials Population Study. J. Pharm. Sci. 2021, 110, 3507–3519. [Google Scholar] [CrossRef]
- Van der Vossen, A.C.; Hanff, L.M.; Vulto, A.G.; Fotaki, N. Potential prediction of formulation performance in paediatric patients using biopharmaceutical tools and simulation of clinically relevant administration scenarios of nifedipine and lorazepam. Br. J. Clin. Pharmacol. 2019, 85, 1728–1739. [Google Scholar] [CrossRef]
- Khan, D.; Kirby, D.; Bryson, S.; Shah, M.; Mohammed, A.R. Development of an Age-Appropriate Mini Orally Disintegrating Carvedilol Tablet with Paediatric Biopharmaceutical Considerations. Pharmaceutics 2021, 13, 831. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Chetty, M.; Rose, R.H.; Abduljalil, K.; Patel, N.; Lu, G.; Cain, T.; Jamei, M.; Rostami-Hodjegan, A. Applications of linking PBPK and PD models to predict the impact of genotypic variability, formulation differences, differences in target binding capacity and target site drug concentrations on drug responses and variability. Front. Pharmacol. 2014, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sudo, T.; Sakamoto, K.; Tateishi, T.; Fujimura, A.; Kumagai, Y.; Ebihara, A. The Influence of Pretreatment Periods with Diltiazem on Nifedipine Kinetics. J. Clin. Pharmacol. 1993, 33, 222–225. [Google Scholar] [CrossRef]
- Tateishi, T.; Ohashi, K.; Sudo, T.; Sakamoto, K.; Toyosaki, N.; Hosoda, S.; Toyo-Oka, T.; Kumagai, Y.; Sugimoto, K.; Fujimara, A.; et al. Dose Dependent Effect of Diltiazem on the Pharmacokinetics of Nifedipine. J. Clin. Pharmacol. 1989, 29, 994–997. [Google Scholar] [CrossRef]
- Ohashi, K.; Tateishi, T.; Sudo, T.; Sakamoto, K.; Toyosaki, N.; Hosoda, S.; Toyo-oka, T.; Sugimoto, K.; Kumagai, Y.; Ebihara, A. Effects of diltiazem on the pharmacokinetics of nifedipine. J. Cardiovasc. Pharmacol. 1990, 15, 96–101. [Google Scholar] [CrossRef]
- Smith, S.R.; Kendall, M.J.; Lobo, J.; Beerahee, A.; Jack, D.B.; Wilkins, M.R. Ranitidine and cimetidine; drug interactions with single dose and steady-state nifedipine administration. Br. J. Clin. Pharmacol. 1987, 23, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Weibull, W. A statistical distribution function of wide applicability. J. Appl. Mech. 1951, 18, 293–297. [Google Scholar] [CrossRef]
- Wonnemann, M.; Schug, B.; Anschütz, M.; Brendel, E.; Nucci, G.D.; Blume, H. Comparison of two marketed nifedipine modified-release formulations: An exploratory clinical food interaction study. Clin. Ther. 2008, 30, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Stever, L.M.; Demitraszek, C.J.; Murray, S.A.; Ali, A.N. Chapter 17—Beta adrenergic antagonists and antianginal drugs. In Side Effects of Drugs Annual. 43; Ray, S.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 219–236. [Google Scholar]
- Johnson, T.N.; Rostami-Hodjegan, A.; Tucker, G.T. Prediction of the Clearance of Eleven Drugs and Associated Variability in Neonates, Infants and Children. Clin. Pharmacokinet. 2006, 45, 931–956. [Google Scholar] [CrossRef]
- Edginton, A.N.; Schmitt, W.; Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharmacokinet. 2006, 45, 1013–1034. [Google Scholar] [CrossRef]
- Ginsberg, G.; Hattis, D.; Russ, A.; Sonawane, B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: Implications for assessing children’s risks from environmental agents. J. Toxicol. Environ. Health A 2004, 67, 297–329. [Google Scholar] [CrossRef]
- Parrott, N.; Davies, B.; Hoffmann, G.; Koerner, A.; Lave, T.; Prinssen, E.; Theogaraj, E.; Singer, T. Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin. Pharmacokinet. 2011, 50, 613–623. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Summary Minutes of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology 2012. Available online: https://wayback.archive-it.org/7993/20170403224110/https://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/ucm286697.htm (accessed on 15 September 2022).
- Olafuyi, O.; Badhan, R.K.S. Dose Optimization of Chloroquine by Pharmacokinetic Modeling During Pregnancy for the Treatment of Zika Virus Infection. J. Pharm. Sci. 2019, 108, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Batchelor, H.; Project oboS-U. Evidence of acceptability of oral paediatric medicines: A review*. J. Pharm. Pharmacol. 2016, 69, 361–376. [Google Scholar] [CrossRef] [PubMed]
- van Riet-Nales, D.A.; de Neef, B.J.; Schobben, A.F.; Ferreira, J.A.; Egberts, T.C.; Rademaker, C.M. Acceptability of different oral formulations in infants and preschool children. Arch. Dis. Child. 2013, 98, 725–731. [Google Scholar] [CrossRef] [PubMed]
- van Riet–Nales, D.A.; Römkens, E.G.; Saint-Raymond, A.; Kozarewicz, P.; Schobben, A.F.; Egberts, T.C.; Rademaker, C.M. Oral medicines for children in the European paediatric investigation plans. PLoS ONE 2014, 9, e98348. [Google Scholar] [CrossRef]
- Bakowski, M.T.; Prescott, P. Patterns of use of pancreatic enzyme supplements in fibrosing colonopathy: Implications for pathogenesis. Pharmacoepidemiol. Drug Saf. 1997, 6, 347–358. [Google Scholar] [CrossRef]
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Di Domenico, A.; Dusemund, B.; Filipič, M.; Jose Frutos, M.; Galtier, P.; et al. Re-evaluation of celluloses E 460(i), E 460(ii), E 461, E 462, E 463, E 464, E 465, E 466, E 468 and E 469 as food additives. EFSA J. 2018, 16, e05047. [Google Scholar]
- Dürig, T.; Karan, K. Chapter 9—Binders in Wet Granulation. In Handbook of Pharmaceutical Wet Granulation; Narang, A.S., Badawy, S.I.F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 317–349. [Google Scholar]
- Handa, T.; Singh, S.; Singh, I.P. Characterization of a new degradation product of nifedipine formed on catalysis by atenolol: A typical case of alteration of degradation pathway of one drug by another. J. Pharm. Biomed. Anal. 2014, 89, 6–17. [Google Scholar] [CrossRef]
- Li, C.L.; Martini, L.G.; Ford, J.L.; Roberts, M. The use of hypromellose in oral drug delivery. J. Pharm. Pharmacol. 2010, 57, 533–546. [Google Scholar] [CrossRef]
- Salsa, T.; Veiga, F.; Teixeira-Dias, J.J.; Pina, M.E. Effect of polymer hydration on the kinetic release of drugs: A study of ibuprofen and ketoprofen in HPMC matrices. Drug Dev. Ind. Pharm. 2003, 29, 289–297. [Google Scholar] [CrossRef]
- EMA. EMAAnnex to the European Commission guideline on ‘Excipients in the Labelling and Package Leaflet of Medicinal Products for Human Use’ 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/annex-european-commission-guideline-excipients-labelling-package-leaflet-medicinal-products-human_en.pdf (accessed on 15 September 2022).
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Hiremath, P.S.; Saha, R.N. Controlled release hydrophilic matrix tablet formulations of isoniazid: Design and in vitro studies. AAPS PharmSciTech 2008, 9, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Mamani, P.L.; Ruiz-Caro, R.; Veiga, M.D. Matrix tablets: The effect of hydroxypropyl methylcellulose/anhydrous dibasic calcium phosphate ratio on the release rate of a water-soluble drug through the gastrointestinal tract I. In vitro tests. AAPS PharmSciTech 2012, 13, 1073–1083. [Google Scholar] [CrossRef]
- BNF. Nifedipine: The National Institute for Health and Care Excellence. Available online: https://bnfc.nice.org.uk/drug/nifedipine.html (accessed on 15 September 2022).
- Johnson, C.E.; Beekman, R.H.; Kostyshak, D.A.; Nguyen, T.; Oh, D.M.; Amidon, G.L. Pharmacokinetics and pharmacodynamics of nifedipine in children with bronchopulmonary dysplasia and pulmonary hypertension. Pediatr. Res. 1991, 29, 500–503. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Equation |
|---|---|
| Zero order | Q1 = Q0 + K0t |
| First order | Log Ct = Log C0 − k t/2.303 |
| Higuchi | Q = KH t1/2 |
| Korsmeyer–Peppas | Qt/Q∞ = Ktn |
| Composition | F1 (% w/w) | F2 (% w/w) | F3 (% w/w) | F4 (% w/w) |
|---|---|---|---|---|
| Nifedipine | 10 | 10 | 10 | 10 |
| Lactose | 89.25 | 59.25 | 39.25 | 19.25 |
| HPMC (K4M) | - | 30 | 50 | 70 |
| Mg stearate | 0.5 | 0.5 | 0.5 | 0.5 |
| AEROSIL® | 0.25 | 0.25 | 0.25 | 0.25 |
| Formulation | Zero Order | First Order | Higuchi | Korsmeyer–Peppas | (n) |
|---|---|---|---|---|---|
| F1 (immediate) | 0.645 | 0.987 | 0.348 | 0.331 | 0.05 |
| F2 (30% HPMC) | 0.387 | 0.717 | 0.638 | 0.895 | 0.15 |
| F3 (50% HPMC) | 0.936 | 0.985 | 0.993 | 0.984 | 0.58 |
| F4 (70% HPMC) | 0.932 | 0.971 | 0.994 | 0.990 | 0.53 |
| Study | Dose | Cmax (ng/mL) | tmax (h) | AUC (ng/mL·h) | Mean Cmax Ratio | Mean tmax Ratio | Mean AUC Ratio | |
|---|---|---|---|---|---|---|---|---|
| Ohashi et al. (1993) [37] | Single (20 mg) | Predicted | 245 ± 152 | 0.6 ± 0.2 | 501 ± 124 | - | - | - |
| Observed G1 | nr | nr | 680 ± 135 | - | - | 0.74 | ||
| Observed G1 | nr | nr | 809 ± 318 | - | - | 0.62 | ||
| Observed G1 | nr | nr | 579 ± 191 | - | - | 0.87 | ||
| Tateishi et al. (1989) [38] | Single (20 mg) | Predicted | 245 ± 152 | 0.6 ± 0.2 | 501 ± 124 | - | - | - |
| Observed | 236 ± 70 | 1 ± 0.9 | 623 ± 139 | 1.04 | 0.6 | 0.8 | ||
| Ohashi et al. (1990) [39] | Single (20 mg) | Predicted | 238 ± 142 | 1.1 ± 0.8 | 357.1 ± 124.9 | - | - | - |
| Observed | 421 ± 177 | nr | 453.6 ± 176.2 | 0.57 | - | 0.79 | ||
| Smith et al. (1987) [40] | Single (20 mg) | Predicted | 101 (62–225) | 1 ± 0.5 | 279 (307–435) | - | - | - |
| Observed | 131 (97–179) | 0.5 | 266 (322–415) | 0.77 | 2 | 1.04 | ||
| Multiple (10 mg) | Predicted | 82 (62–225) | 1 ± 0.5 | 201 (142–235) | - | - | - | |
| Observed | 56 (42–74) | 0.5 | 134 (113–158) | 1.46 | 2 | 1.5 | ||
| Wonnerman et al. (2008) [42] | Single OROS (30 mg) | Predicted | 21.8 (15.9–42) | 5 (3–17) | 319 (214–517) | - | - | - |
| Observed | 17.6 (11.6–30.8) | 6 (5–24) | 358 (160–906) | 1.23 | 0.83 | 0.89 |
| Formulation | Dose | Cmax (ng/mL) | tmax (h) | AUC (ng/mL·h) |
|---|---|---|---|---|
| F1 | 30 mg BD | 229 ± 94.2 | 1.2 ± 0.15 | 703 ± 425.6 |
| 60 mg OD | 466 ± 187 | 1.2 ± 0.24 | 1459 ± 876.8 | |
| F2 | 30 mg BD | 167.1 ± 70.16 | 1.23 ± 0.18 | 617.9 ± 366.8 |
| 60 mg OD | 331 ± 133 | 1.25 ± 0.26 | 1270.7 ± 751 | |
| F3 | 30 mg BD | 76.68 ± 39.5 | 2.25 ± 1.99 | 711.6 ± 407 |
| 60 mg OD | 126.8 ± 62 | 5.5 ± 1.34 | 1432.8 ± 817 |
| Formulation | Dosing | Age Range (Years) | Cmax (ng/mL) | tmax (h) | AUC (ng/mL·h) |
|---|---|---|---|---|---|
| F1 | OD | 5–7 | 113 | 1.2 | 398.2 |
| 7–11 | 101.6 | 1.2 | 300 | ||
| BD | 5–7 | 113 | 1.6 | 397.7 | |
| 7–11 | 105.8 | 1.36 | 300 | ||
| TDS | 5–7 | 113 | 1.43 | 398.1 | |
| 7–11 | 107.1 | 1.38 | 301 | ||
| F2 | OD | 5–7 | 113 | 2.2 | 364 |
| 7–11 | 101 | 2.16 | 274 | ||
| BD | 5–7 | 111 | 2.2 | 363 | |
| 7–11 | 102 | 2.16 | 275 | ||
| TDS | 5–7 | 123 | 2.15 | 362 | |
| 7–11 | 106 | 2.13 | 274 | ||
| F3 | OD | 5–7 | 39 | 5.3 | 418 |
| 7–11 | 31 | 4.7 | 314 | ||
| BD | 5–7 | 46 | 3 | 417 | |
| 7–11 | 37 | 2.45 | 314 | ||
| TDS | 5–7 | 63 | 2.4 | 418 | |
| 7–11 | 49 | 2.16 | 313 |
| Dose | Age Range (Years) | Cmax (ng/mL) | Cmin (ng/mL) | tmax (h) | AUC (ng/mL·h) | <0 ng/mL a (%) | Dose (mg) |
|---|---|---|---|---|---|---|---|
| 350 µg/kg | 3–5 | 62.48 ± 35 | 31.76 ± 25.9 | 3.35 ± 2 | 563.3 ± 376 | 36 | 5.75 ± 0.87 |
| 5–7 | 53.97 ± 25.3 | 25.6 ± 15.6 | 2.59 ± 2 | 458.9 ± 248.3 | 46 | 7.03 ± 1.37 | |
| 450 µg/kg | 3–5 | 80.45 ± 45.8 | 40.85 ± 33 | 3.35 ± 2 | 726 ± 485.2 | 23 | 7.4 ± 1.1 |
| 5–7 | 69.5 ± 32.6 | 32.8 ± 20.1 | 2.59 ± 2 | 590.8 ± 319 | 20 | 9.04 ± 1.7 |
| Dosing Strategy | Average Weight (kg) | Dose (mg) | 5 mg Tablets Required |
|---|---|---|---|
| 250 μg/kg | 3 years: 14 kg 5 years: 18 kg 7 years: 23 kg | 3.5 ≈ 5 4.5 ≈ 5 5.8 ≈ 5 | 1 1 1 |
| 350 μg/kg | 3 years: 14 kg 5 years: 18 kg 7 years: 23 kg | 4.9 ≈ 5 6.3 ≈ 5 8.1 ≈ 10 | 1 1 2 |
| 450 μg/kg | 3 years: 14 kg 5 years: 18 kg 7 years: 23 kg | 6.3 ≈ 5 8.1 ≈ 10 10.4 ≈ 10 | 1 2 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, D.; Badhan, R.; Kirby, D.J.; Bryson, S.; Shah, M.; Mohammed, A.R. Virtual Clinical Trials Guided Design of an Age-Appropriate Formulation and Dosing Strategy of Nifedipine for Paediatric Use. Pharmaceutics 2023, 15, 556. https://doi.org/10.3390/pharmaceutics15020556
Khan D, Badhan R, Kirby DJ, Bryson S, Shah M, Mohammed AR. Virtual Clinical Trials Guided Design of an Age-Appropriate Formulation and Dosing Strategy of Nifedipine for Paediatric Use. Pharmaceutics. 2023; 15(2):556. https://doi.org/10.3390/pharmaceutics15020556
Chicago/Turabian StyleKhan, Dilawar, Raj Badhan, Daniel J. Kirby, Simon Bryson, Maryam Shah, and Afzal Rahman Mohammed. 2023. "Virtual Clinical Trials Guided Design of an Age-Appropriate Formulation and Dosing Strategy of Nifedipine for Paediatric Use" Pharmaceutics 15, no. 2: 556. https://doi.org/10.3390/pharmaceutics15020556
APA StyleKhan, D., Badhan, R., Kirby, D. J., Bryson, S., Shah, M., & Mohammed, A. R. (2023). Virtual Clinical Trials Guided Design of an Age-Appropriate Formulation and Dosing Strategy of Nifedipine for Paediatric Use. Pharmaceutics, 15(2), 556. https://doi.org/10.3390/pharmaceutics15020556