
The Neuroprotective Effects of Flavonoid Fisetin against Corticosterone-Induced Cell Death through Modulation of ERK, p38, and PI3K/Akt/FOXO3a-Dependent Pathways in PC12 Cells

 , , and
, , and 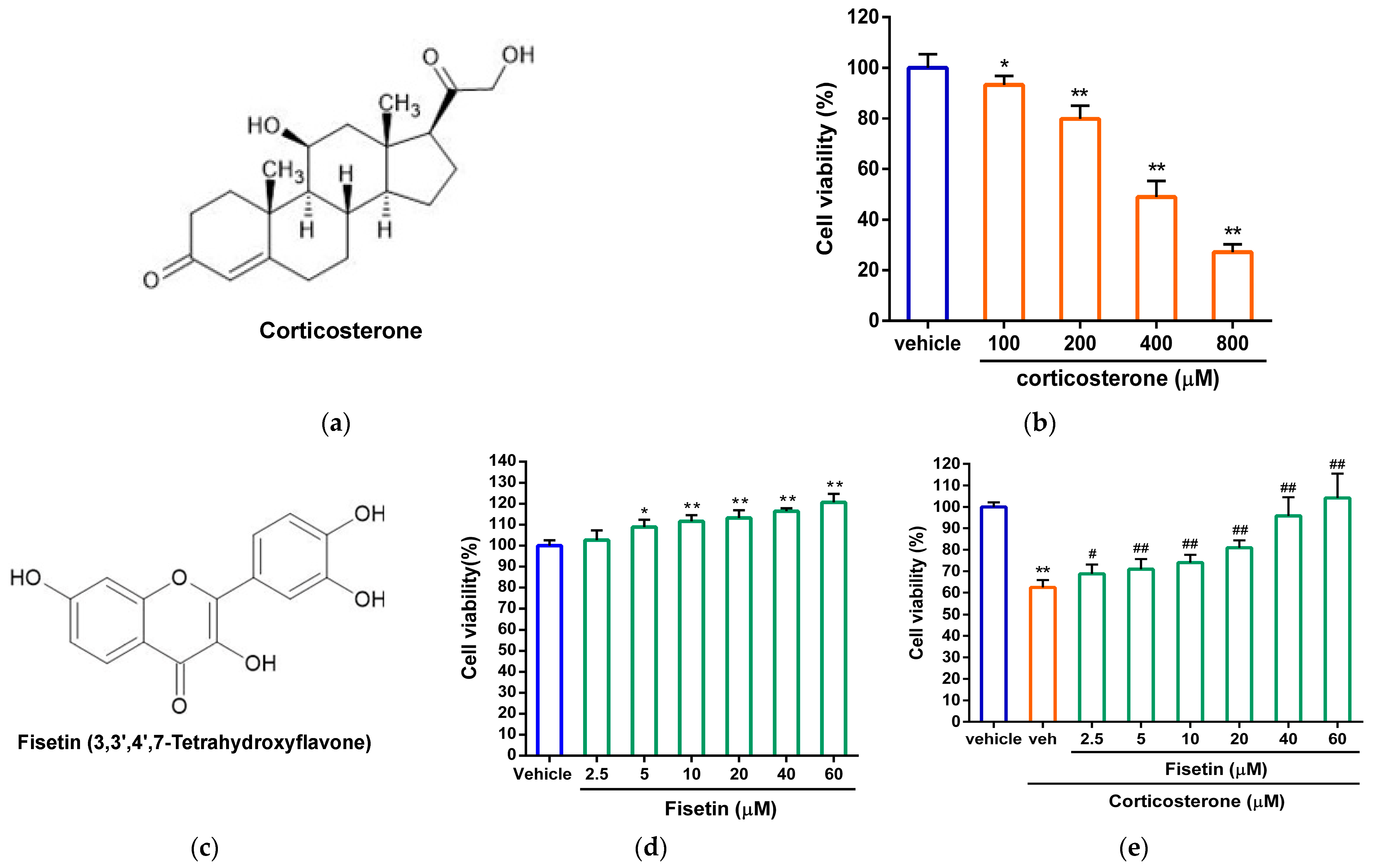{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Compounds Treatment
2.4. Analysis of Cell Viability
2.5. Analysis of ROS Generation
2.6. Analysis of Cell Apoptosis by Fluorescent Microscopy
2.7. Analysis of Cell Apoptosis by Flow Cytometry
2.8. Proteins Preparation and Western Blot Analysis
2.9. Transfection of FOXO3a-GFP Expression Plasmid
2.10. Statistical Analysis
3. Results
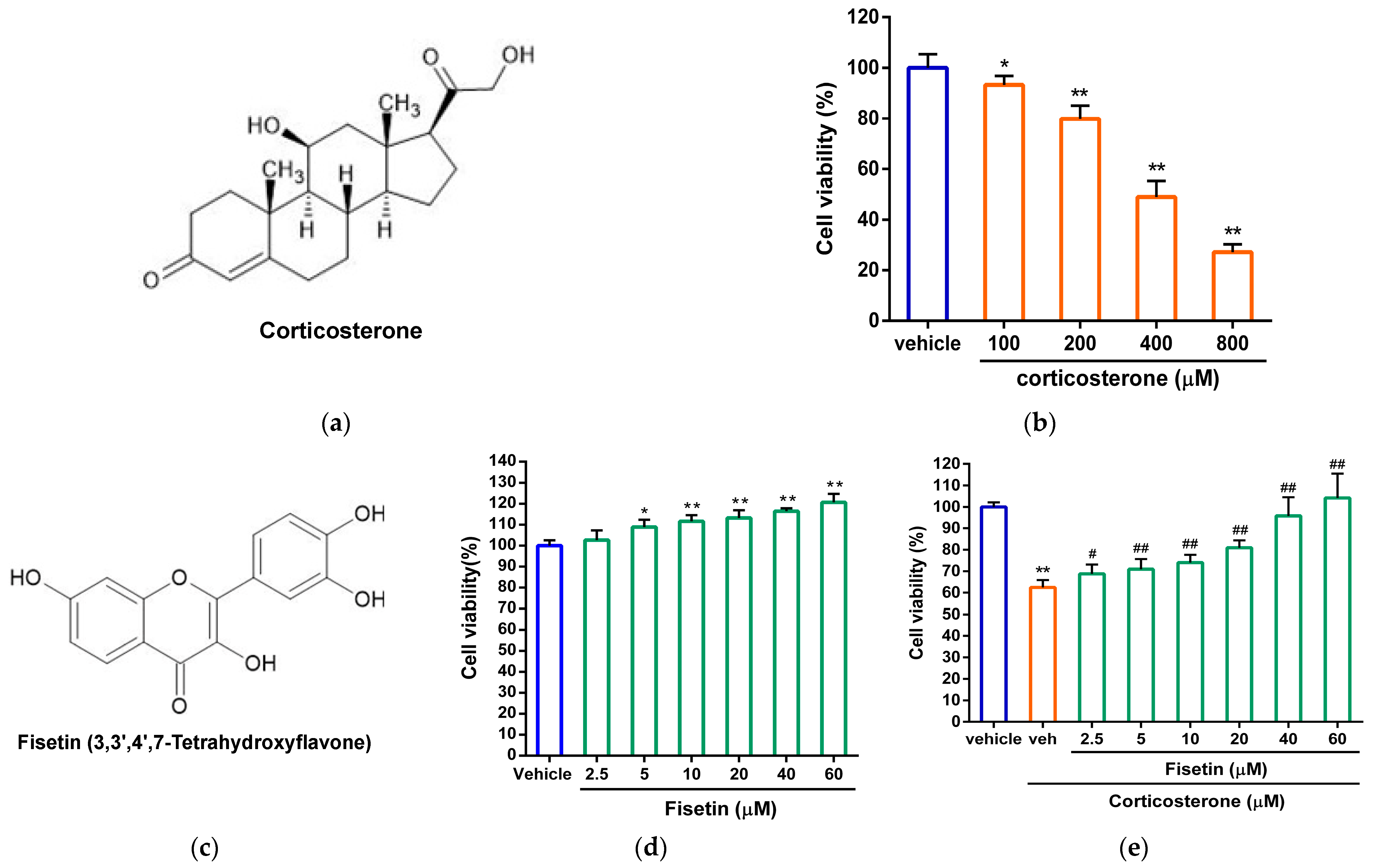
3.1. Effects of Fisetin on Cell Viability in Corticosterone-Treated PC12 Cells
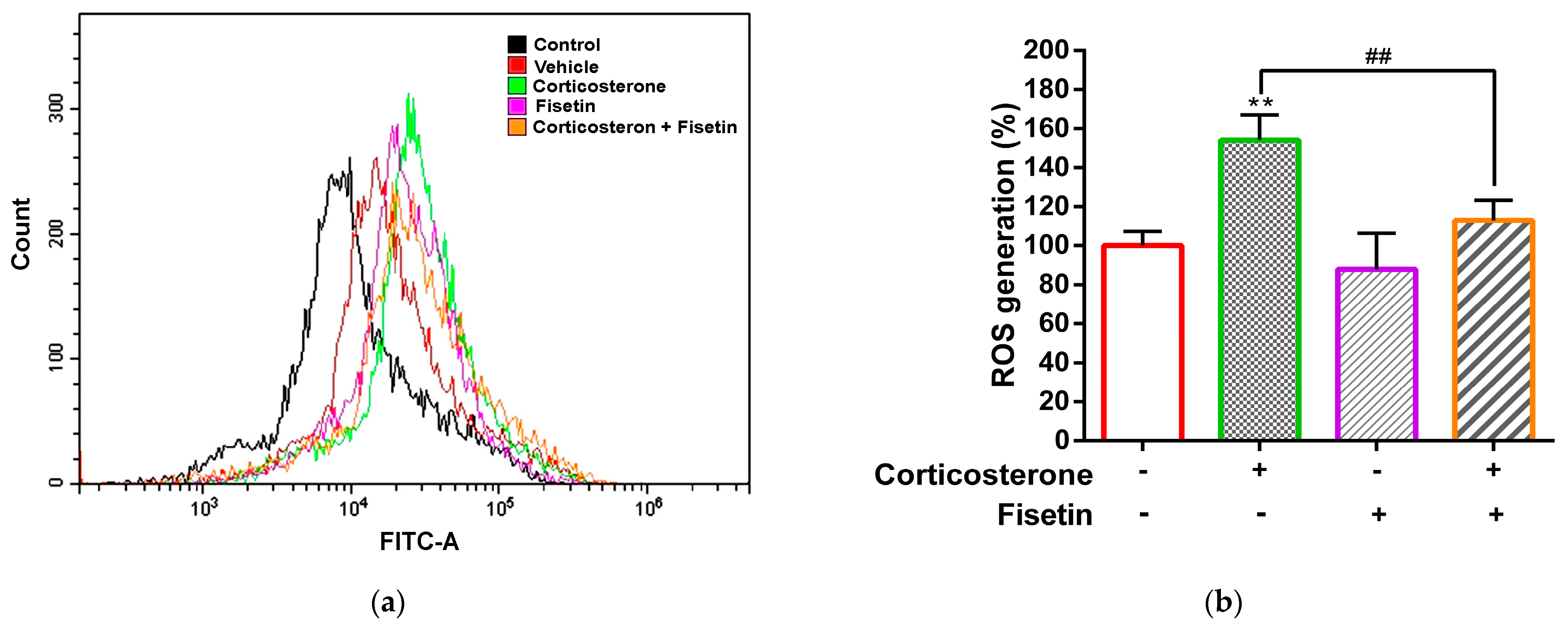
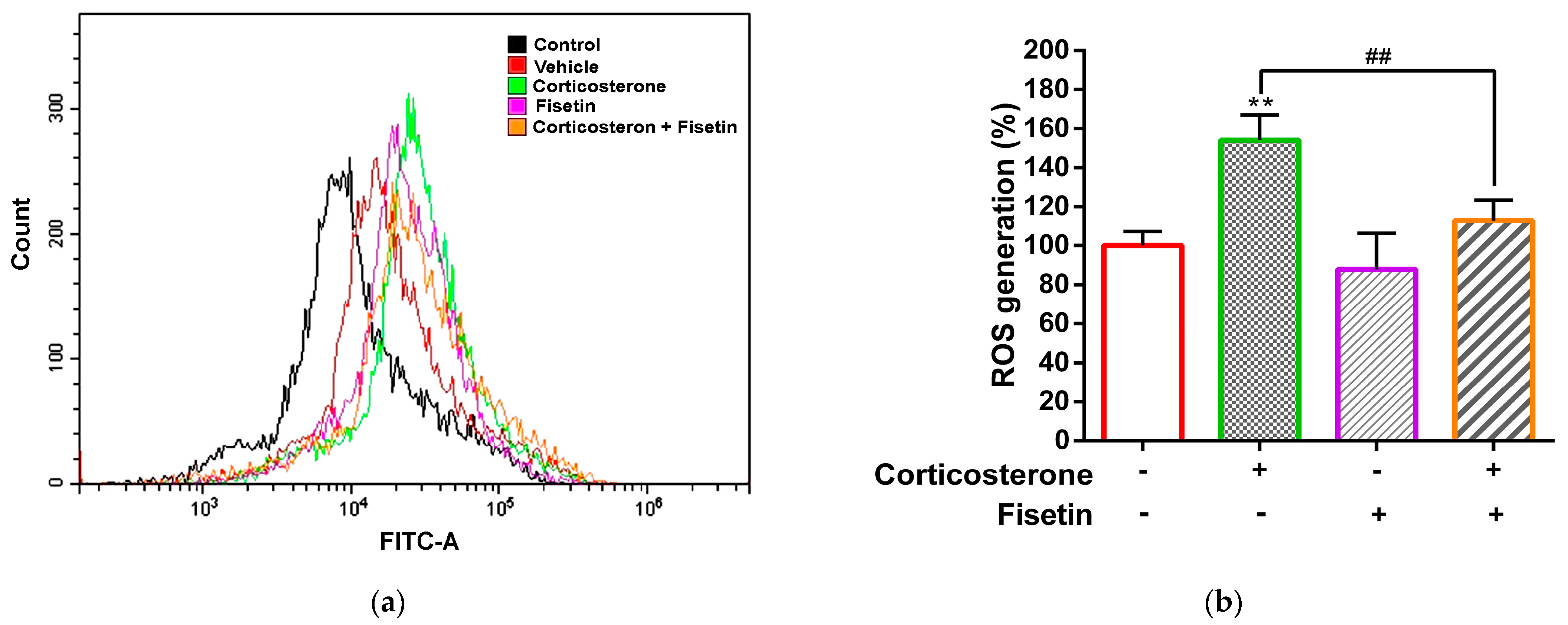
3.2. Effects of Fisetin on ROS Production in Corticosterone-Treated PC12 Cells
3.3. Effects of Fisetin on Cell Apoptosis in Corticosterone-Treated PC12 Cells
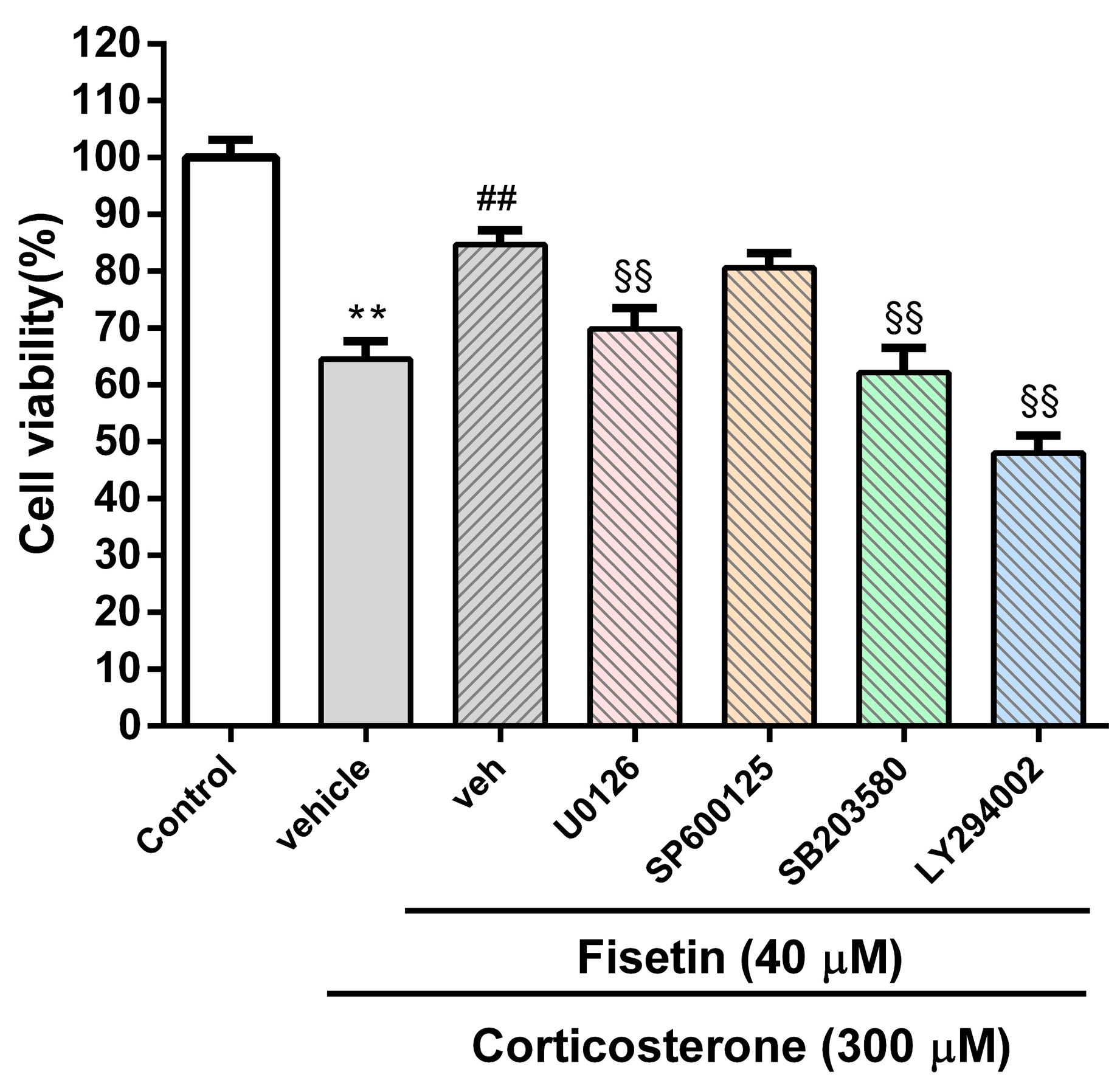
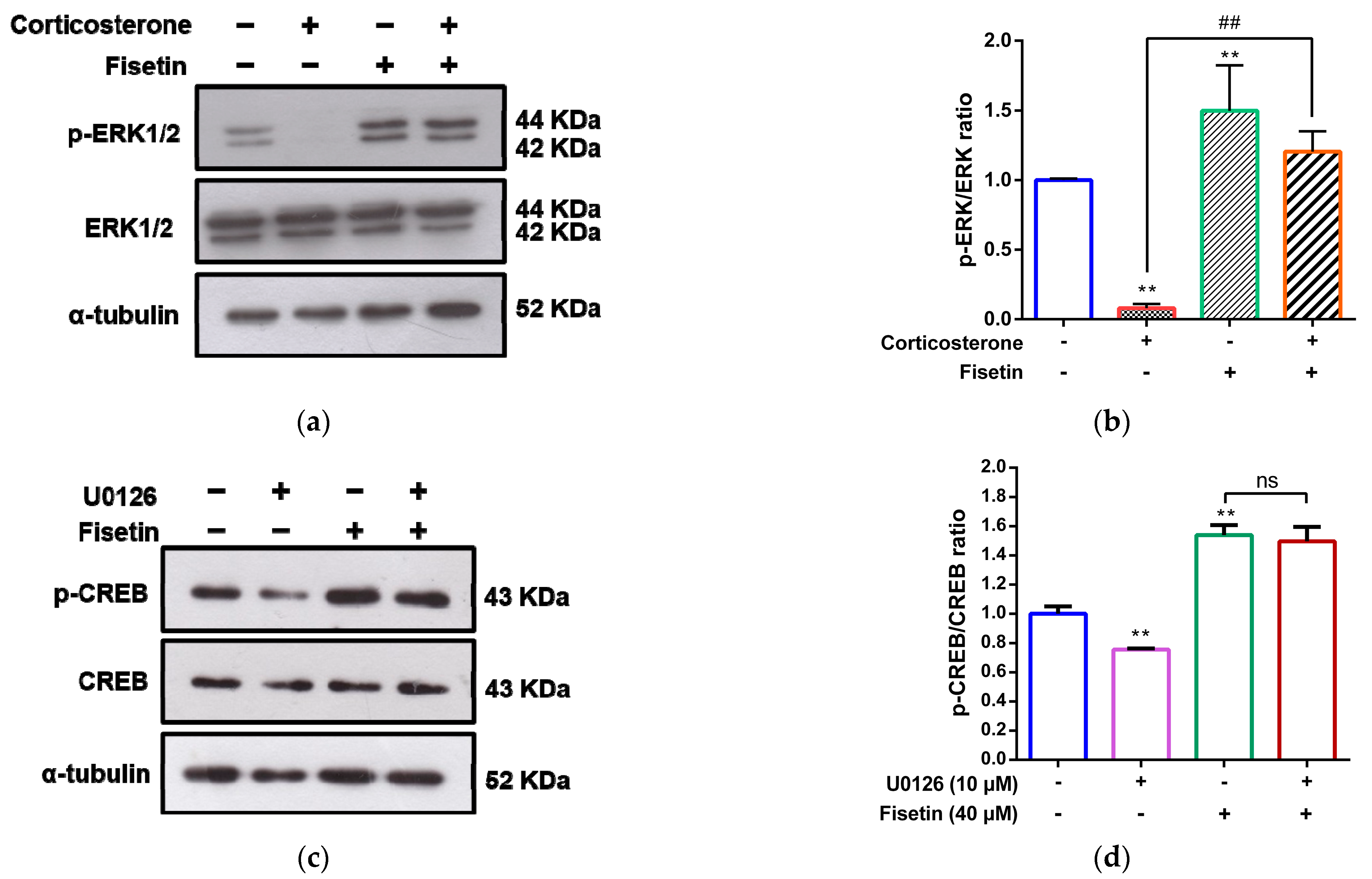
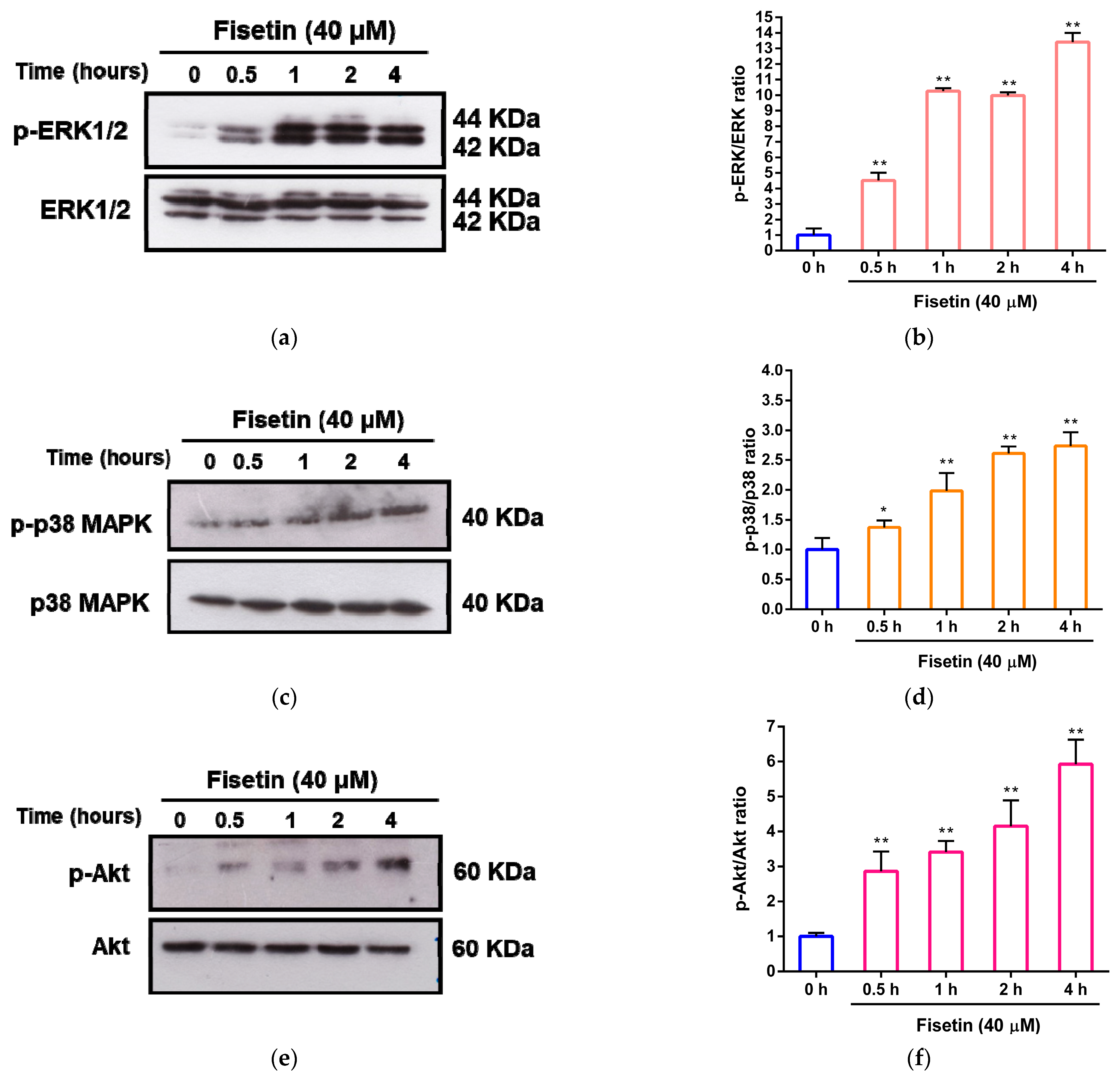
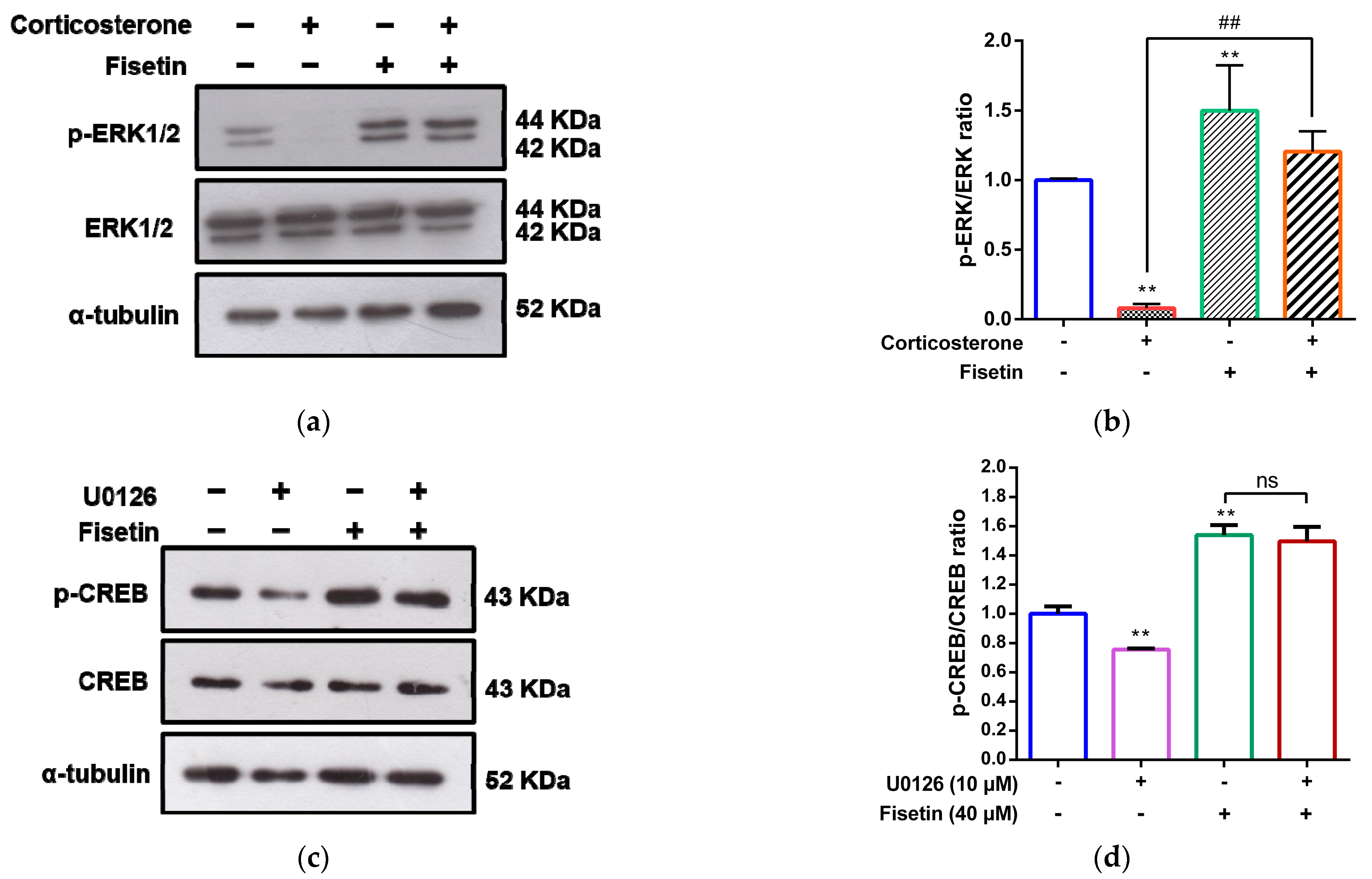
3.4. Involvement of MAPK/ERK, p38 MAPK and PI3/Akt Pathways in the Cytoprotective Effect of Fisetin against Corticosterone-Induced Cell Death
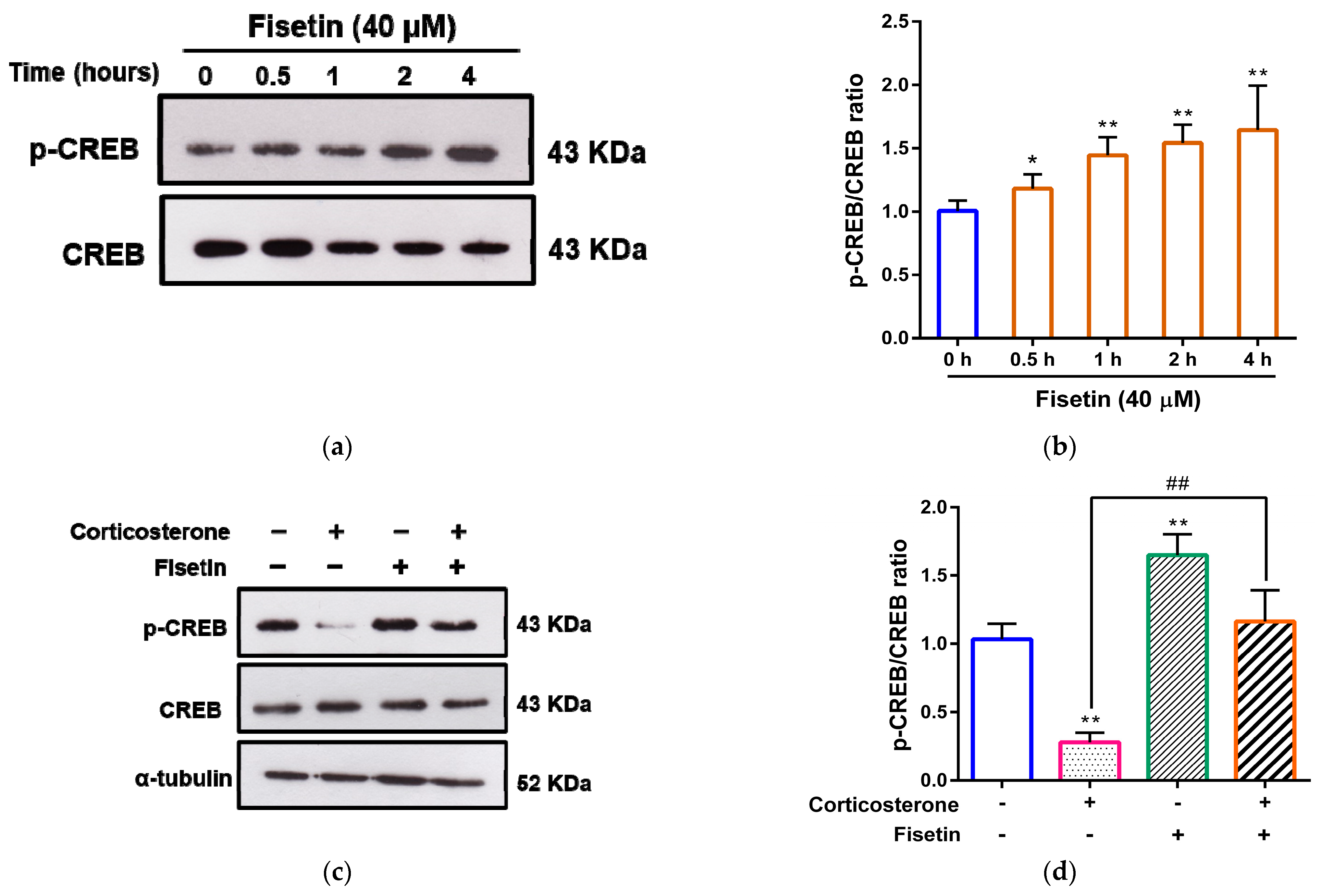
3.5. Effects of Fisetin on CREB Activation in Corticosterone-Treated PC12 Cells
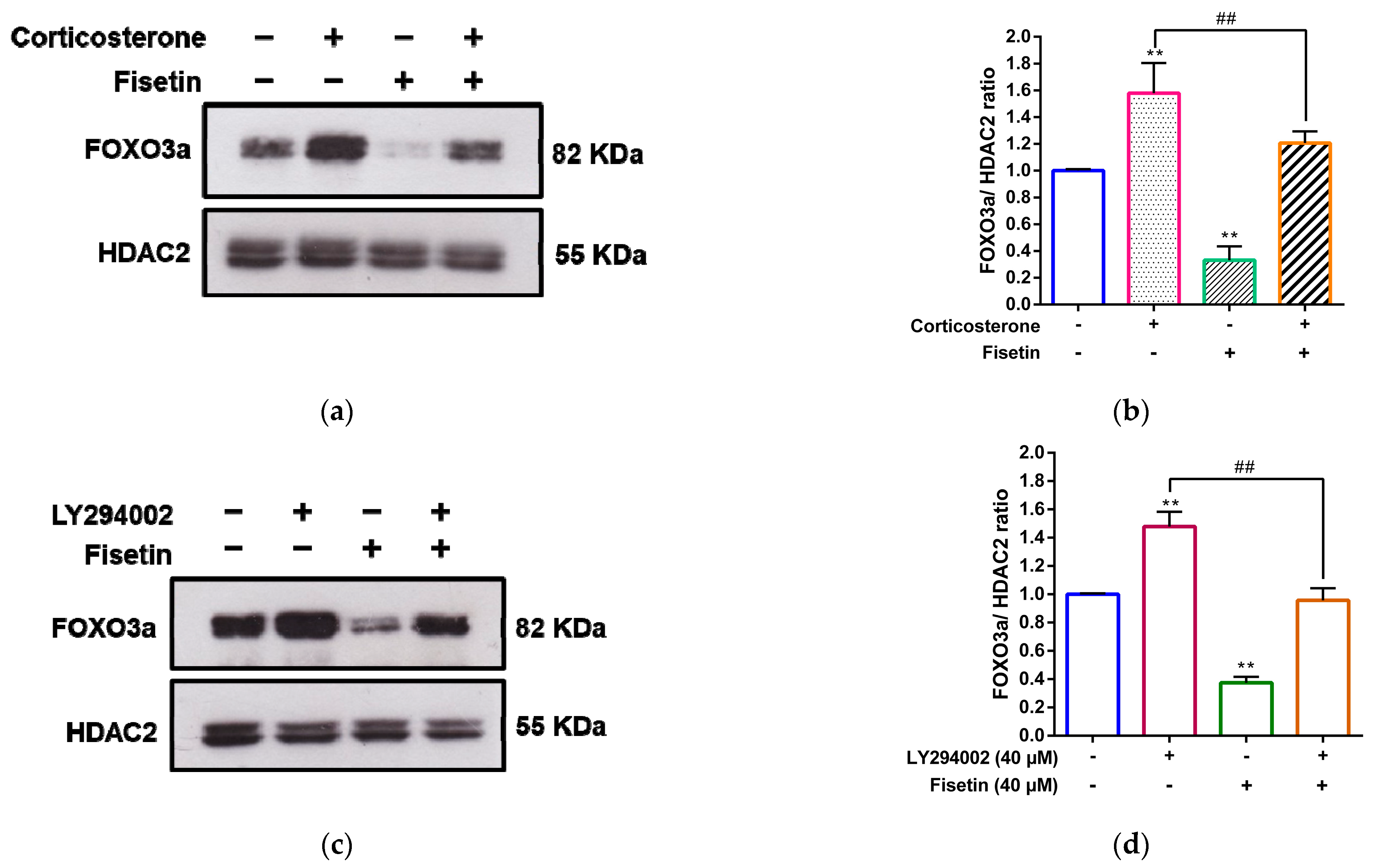
3.6. Fisetin Increases FOXO3a Phosphorylation and Promotes Cytosolic Localization of FOXO3a in PC12 Cells
3.7. Fisetin Counteracts the Corticosterone-Induced Nuclear Accumulation of FOXO3a through the PI3K/Akt Pathway
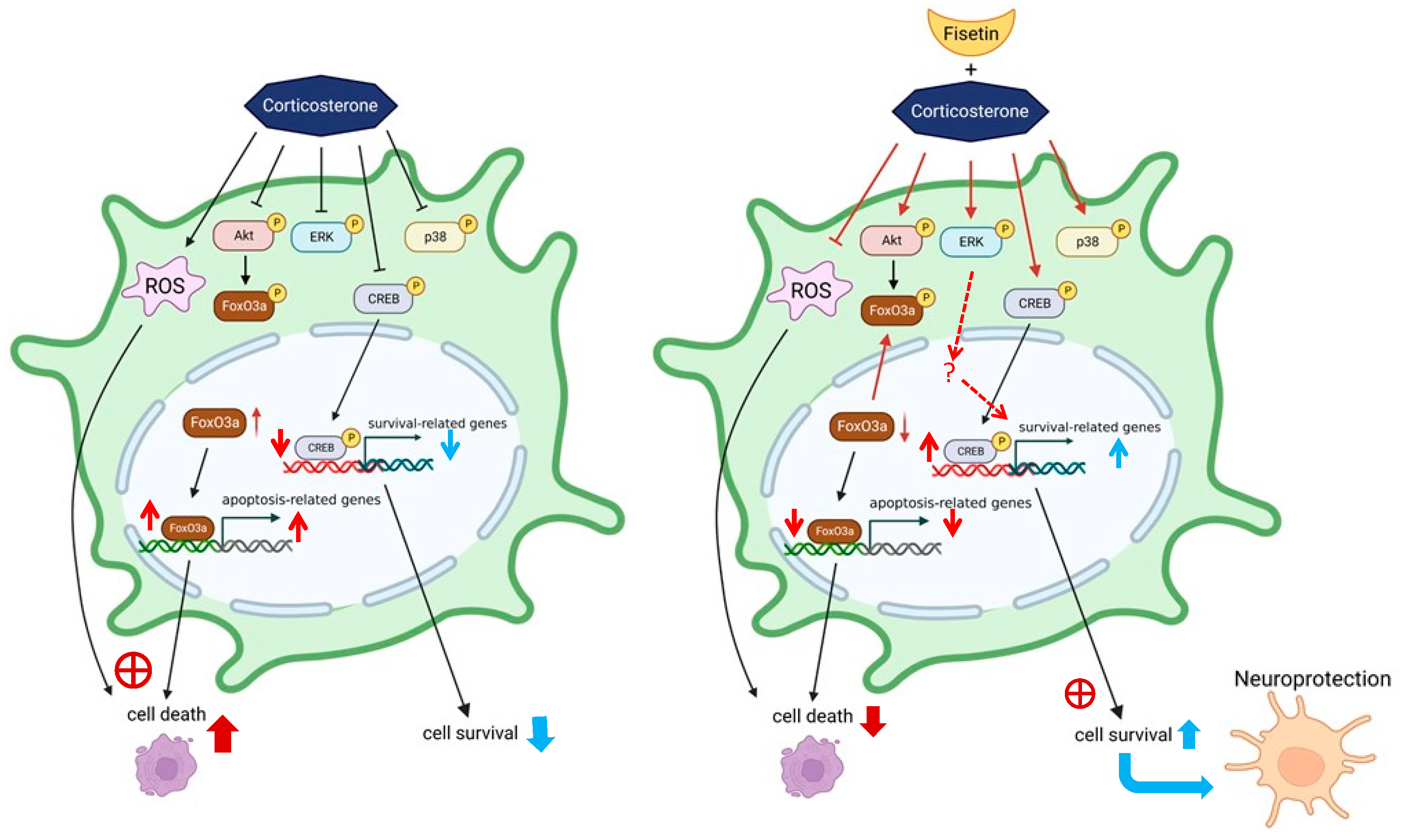
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vyas, S.; Rodrigues, A.J.; Silva, J.M.; Tronche, F.; Almeida, O.F.; Sousa, N.; Sotiropoulos, I. Chronic Stress and Glucocorticoids: From Neuronal Plasticity to Neurodegeneration. Neural Plast. 2016, 2016, 6391686. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar]
- Peng, G.J.; Tian, J.S.; Gao, X.X.; Zhou, Y.Z.; Qin, X.M. Research on the Pathological Mechanism and Drug Treatment Mechanism of Depression. Curr. Neuropharmacol. 2015, 13, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Ising, M.; Horstmann, S.; Kloiber, S.; Lucae, S.; Binder, E.B.; Kern, N.; Kunzel, H.E.; Pfennig, A.; Uhr, M.; Holsboer, F. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression—A potential biomarker? Biol. Psychiatry 2007, 62, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.B.; Pinna, G.; Barros, H.M.T. The Role of HPA Axis and Allopregnanolone on the Neurobiology of Major Depressive Disorders and PTSD. Int. J. Mol. Sci. 2021, 22, 5495. [Google Scholar] [CrossRef] [PubMed]
- Pearson Murphy, B.E. Antiglucocorticoid therapies in major depression: A review. Psychoneuroendocrinology 1997, 22, S125–S132. [Google Scholar] [CrossRef]
- Joseph, J.J.; Golden, S.H. Cortisol dysregulation: The bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2017, 1391, 20–34. [Google Scholar] [CrossRef]
- Pretorius, E. Corticosteroids, depression and the role of serotonin. Rev. Neurosci. 2004, 15, 109–116. [Google Scholar] [CrossRef]
- Spiers, J.G.; Chen, H.J.; Sernia, C.; Lavidis, N.A. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front. Neurosci. 2014, 8, 456. [Google Scholar] [CrossRef]
- Picard, K.; St-Pierre, M.K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M.E. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987. [Google Scholar] [CrossRef]
- Hill, A.S.; Sahay, A.; Hen, R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology 2015, 40, 2368–2378. [Google Scholar] [CrossRef]
- Podgorny, O.V.; Gulyaeva, N.V. Glucocorticoid-mediated mechanisms of hippocampal damage: Contribution of subgranular neurogenesis. J. Neurochem. 2021, 157, 370–392. [Google Scholar] [CrossRef]
- Kwon, M.S.; Kim, M.H.; Kim, S.H.; Park, K.D.; Yoo, S.H.; Oh, I.U.; Pak, S.; Seo, Y.J. Erythropoietin exerts cell protective effect by activating PI3K/Akt and MAPK pathways in C6 Cells. Neurol. Res. 2014, 36, 215–223. [Google Scholar] [CrossRef]
- Owuor, E.D.; Kong, A.N. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.; Devasahayam, G. Neurodegeneration by oxidative stress: A review on prospective use of small molecules for neuroprotection. Mol. Biol. Rep. 2020, 47, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Albert-Gascó, H.; Ros-Bernal, F.; Castillo-Gómez, E.; Olucha-Bordonau, F.E. MAP/ERK Signaling in Developing Cognitive and Emotional Function and Its Effect on Pathological and Neurodegenerative Processes. Int. J. Mol. Sci. 2020, 21, 4471. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Teng, Y.; Chen, X.; Liu, Z.; Geng, F.; Liu, Y.; Jiang, H.; Wang, Z.; Yang, L. Platelet-derived growth factor (PDGF)-BB protects dopaminergic neurons via activation of Akt/ERK/CREB pathways to upregulate tyrosine hydroxylase. CNS Neurosci. Ther. 2021, 27, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef]
- Monje, P.; Hernández-Losa, J.; Lyons, R.J.; Castellone, M.D.; Gutkind, J.S. Regulation of the transcriptional activity of c-Fos by ERK. A novel role for the prolyl isomerase PIN1. J. Biol. Chem. 2005, 280, 35081–35084. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Man, H.Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef]
- Sharma, A.; Mehan, S. Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 2021, 147, 105067. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Hergert, P. FoxO3a and disease progression. World J. Biol. Chem. 2014, 5, 346–354. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Martins, J.; Brijesh, S. Phytochemistry and pharmacology of anti-depressant medicinal plants: A review. Biomed. Pharmacother. 2018, 104, 343–365. [Google Scholar] [CrossRef]
- Khan, A.; Jahan, S.; Imtiyaz, Z.; Alshahrani, S.; Antar Makeen, H.; Mohammed Alshehri, B.; Kumar, A.; Arafah, A.; Rehman, M.U. Neuroprotection: Targeting Multiple Pathways by Naturally Occurring Phytochemicals. Biomedicines 2020, 8, 284. [Google Scholar] [CrossRef]
- Pal, H.C.; Pearlman, R.L.; Afaq, F. Fisetin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 213–244. [Google Scholar]
- Kubina, R.; Krzykawski, K.; Kabala-Dzik, A.; Wojtyczka, R.D.; Chodurek, E.; Dziedzic, A. Fisetin, a Potent Anticancer Flavonol Exhibiting Cytotoxic Activity against Neoplastic Malignant Cells and Cancerous Conditions: A Scoping, Comprehensive Review. Nutrients 2022, 14, 2604. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.H.; Almatroudi, A.; Allemailem, K.S.; Khan, A.A.; Almatroodi, S.A. The Potential Role of Fisetin, a Flavonoid in Cancer Prevention and Treatment. Molecules 2022, 27, 9009. [Google Scholar] [CrossRef]
- Prem, P.N.; Sivakumar, B.; Boovarahan, S.R.; Kurian, G.A. Recent advances in potential of Fisetin in the management of myocardial ischemia-reperfusion injury-A systematic review. Phytomedicine 2022, 101, 154123. [Google Scholar] [CrossRef]
- Ravula, A.R.; Teegala, S.B.; Kalakotla, S.; Pasangulapati, J.P.; Perumal, V.; Boyina, H.K. Fisetin, potential flavonoid with multifarious targets for treating neurological disorders: An updated review. Eur. J. Pharmacol. 2021, 910, 174492. [Google Scholar] [CrossRef]
- Hassan, S.S.U.; Samanta, S.; Dash, R.; Karpinski, T.M.; Habibi, E.; Sadiq, A.; Ahmadi, A.; Bunagu, S. The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: Focus on the role of oxidative stress. Front. Pharmacol. 2022, 13, 1015835. [Google Scholar] [CrossRef]
- Jiang, Y.; Tang, X.; Deng, P.; Jiang, C.; He, Y.; Hao, D.; Yang, H. The Neuroprotective Role of Fisetin in Different Neurological Diseases: A Systematic Review. Mol. Neurobiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Ho, Y.R.; Wu, M.J.; Huang, S.P.; Chen, P.K.; Tai, M.H.; Ho, C.T.; Yen, J.H. Cytoprotective effects of fisetin against hypoxia-induced cell death in PC12 cells. Food Funct. 2015, 6, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jiang, X.; Zhang, X.; Chen, Z.; Xu, L.; Chen, L.; Wang, G.; Pan, J. The effects of fisetin on lipopolysaccharide-induced depressive-like behavior in mice. Metab. Brain Dis. 2016, 31, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, L.; Kandhare, A.D.; Mukherjee-Kandhare, A.A.; Bodhankar, S.L. Attenuation of reserpine-induced fibromyalgia via ROS and serotonergic pathway modulation by fisetin, a plant flavonoid polyphenol. Exp. Ther. Med. 2020, 19, 1343–1355. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Lu, J.; Shi, H.; Gong, S.; Wang, Y.; Hamdy, R.C.; Chua, B.H.L.; Yang, L.; Xu, X. Fisetin provides antidepressant effects by activating the tropomyosin receptor kinase B signal pathway in mice. J. Neurochem. 2017, 143, 561–568. [Google Scholar] [CrossRef]
- Chen, P.Y.; Wang, C.Y.; Tsao, E.C.; Chen, Y.T.; Wu, M.J.; Ho, C.T.; Yen, J.H. 5-Demethylnobiletin Inhibits Cell Proliferation, Downregulates ID1 Expression, Modulates the NF-kappaB/TNF-alpha Pathway and Exerts Antileukemic Effects in AML Cells. Int. J. Mol. Sci. 2022, 23, 7392. [Google Scholar] [CrossRef]
- Lin, R.; Liu, L.; Silva, M.; Fang, J.; Zhou, Z.; Wang, H.; Xu, J.; Li, T.; Zheng, W. Hederagenin Protects PC12 Cells Against Corticosterone-Induced Injury by the Activation of the PI3K/AKT Pathway. Front. Pharmacol. 2021, 12, 712876. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, N.; Cheng, J.; Shi, X.; Huang, H.; Zhou, M.; Zhu, H. Chlorogenic acid protects PC12 cells against corticosterone-induced neurotoxicity related to inhibition of autophagy and apoptosis. BMC Pharmacol. Toxicol. 2019, 20, 56. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Qi, X.; Lin, W.; Li, J.; Li, H.; Wang, W.; Wang, D.; Sun, M. Fluoxetine increases the activity of the ERK-CREB signal system and alleviates the depressive-like behavior in rats exposed to chronic forced swim stress. Neurobiol. Dis. 2008, 31, 278–285. [Google Scholar] [CrossRef]
- Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells 2019, 8, 1110. [Google Scholar] [CrossRef]
- Tian, J.S.; Liu, S.B.; He, X.Y.; Xiang, H.; Chen, J.L.; Gao, Y.; Zhou, Y.Z.; Qin, X.M. Metabolomics studies on corticosterone-induced PC12 cells: A strategy for evaluating an in vitro depression model and revealing the metabolic regulation mechanism. Neurotoxicol. Teratol. 2018, 69, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Oprea, D.; Sanz, C.G.; Barsan, M.M.; Enache, T.A. PC-12 Cell Line as a Neuronal Cell Model for Biosensing Applications. Biosensors 2022, 12, 500. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, R.; Shen, J.; Su, H.; Xing, D.; Du, L. A mouse model of depression induced by repeated corticosterone injections. Eur. J. Pharmacol. 2008, 581, 113–120. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhao, Y.N.; Wang, Z.L.; Huang, Y.F. Chronic corticosterone exposure reduces hippocampal glycogen level and induces depression-like behavior in mice. J. Zhejiang Univ. Sci. B 2015, 16, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.; Benvenuto, M.; Masuelli, L.; Frajese, G.V.; Tresoldi, I.; Modesti, A.; Bei, R. In vitro and in vivo antitumoral effects of combinations of polyphenols, or polyphenols and anticancer drugs: Perspectives on cancer treatment. Int. J. Mol. Sci. 2015, 16, 9236–9282. [Google Scholar] [CrossRef]
- Calis, Z.; Mogulkoc, R.; Baltaci, A.K. The Roles of Flavonols/Flavonoids in Neurodegeneration and Neuroinflammation. Mini-Rev. Med. Chem. 2020, 20, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, R.; Azam, S.; Cho, D.Y.; Su-Kim, I.; Choi, D.K. Natural Phytochemicals as Novel Therapeutic Strategies to Prevent and Treat Parkinson’s Disease: Current Knowledge and Future Perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 6680935. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Khan, H. Anti-Parkinson Potential of Silymarin: Mechanistic Insight and Therapeutic Standing. Front. Pharmacol. 2018, 9, 422. [Google Scholar] [CrossRef]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.P.; Wu, M.J.; Chen, P.Y.; Ho, Y.R.; Tai, M.H.; Ho, C.T.; Yen, J.H. Neurotrophic action of 5-hydroxylated polymethoxyflavones: 5-demethylnobiletin and gardenin A stimulate neuritogenesis in PC12 cells. J. Agric. Food Chem. 2013, 61, 9453–9463. [Google Scholar] [CrossRef]
- Lin, L.F.; Chiu, S.P.; Wu, M.J.; Chen, P.Y.; Yen, J.H. Luteolin induces microRNA-132 expression and modulates neurite outgrowth in PC12 cells. PLoS ONE 2012, 7, e43304. [Google Scholar] [CrossRef]
- Inkielewicz-Stepniak, I.; Radomski, M.W.; Wozniak, M. Fisetin prevents fluoride- and dexamethasone-induced oxidative damage in osteoblast and hippocampal cells. Food Chem. Toxicol. 2012, 50, 583–589. [Google Scholar] [CrossRef]
- Madalena, K.M.; Lerch, J.K. The Effect of Glucocorticoid and Glucocorticoid Receptor Interactions on Brain, Spinal Cord, and Glial Cell Plasticity. Neural Plast. 2017, 2017, 8640970. [Google Scholar] [CrossRef]
- McEwen, B.S. Corticosteroids and hippocampal plasticity. Ann. N. Y. Acad. Sci. 1994, 746, 134–142; discussion 142-4, 178–179. [Google Scholar] [CrossRef]
- Gite, S.; Ross, R.P.; Kirke, D.; Guiheneuf, F.; Aussant, J.; Stengel, D.B.; Dinan, T.G.; Cryan, J.F.; Stanton, C. Nutraceuticals to promote neuronal plasticity in response to corticosterone-induced stress in human neuroblastoma cells. Nutr. Neurosci. 2019, 22, 551–568. [Google Scholar] [CrossRef]
- Kowianski, P.; Lietzau, G.; Czuba, E.; Waskow, M.; Steliga, A.; Morys, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Frebel, K.; Wiese, S. Signalling molecules essential for neuronal survival and differentiation. Biochem. Soc. Trans. 2006, 34 Pt 6, 1287–1290. [Google Scholar] [CrossRef]
- Hossain, M.S.; Ifuku, M.; Take, S.; Kawamura, J.; Miake, K.; Katafuchi, T. Plasmalogens rescue neuronal cell death through an activation of AKT and ERK survival signaling. PLoS ONE 2013, 8, e83508. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.H.; Wu, P.S.; Chen, S.F.; Wu, M.J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 852. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Esvald, E.E.; Tuvikene, J.; Sirp, A.; Patil, S.; Bramham, C.R.; Timmusk, T. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J. Neurosci. 2020, 40, 1405–1426. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, T.; Huang, P. Post-translational modifications of FOXO family proteins (Review). Mol. Med. Rep. 2016, 14, 4931–4941. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, P.-R.; Liou, J.-W.; Chen, P.-Y.; Gao, W.-Y.; Wu, C.-L.; Wu, M.-J.; Yen, J.-H. The Neuroprotective Effects of Flavonoid Fisetin against Corticosterone-Induced Cell Death through Modulation of ERK, p38, and PI3K/Akt/FOXO3a-Dependent Pathways in PC12 Cells. Pharmaceutics 2023, 15, 2376. https://doi.org/10.3390/pharmaceutics15102376
Chang P-R, Liou J-W, Chen P-Y, Gao W-Y, Wu C-L, Wu M-J, Yen J-H. The Neuroprotective Effects of Flavonoid Fisetin against Corticosterone-Induced Cell Death through Modulation of ERK, p38, and PI3K/Akt/FOXO3a-Dependent Pathways in PC12 Cells. Pharmaceutics. 2023; 15(10):2376. https://doi.org/10.3390/pharmaceutics15102376
Chicago/Turabian StyleChang, Pei-Rong, Je-Wen Liou, Pei-Yi Chen, Wan-Yun Gao, Chia-Ling Wu, Ming-Jiuan Wu, and Jui-Hung Yen. 2023. "The Neuroprotective Effects of Flavonoid Fisetin against Corticosterone-Induced Cell Death through Modulation of ERK, p38, and PI3K/Akt/FOXO3a-Dependent Pathways in PC12 Cells" Pharmaceutics 15, no. 10: 2376. https://doi.org/10.3390/pharmaceutics15102376
APA StyleChang, P.-R., Liou, J.-W., Chen, P.-Y., Gao, W.-Y., Wu, C.-L., Wu, M.-J., & Yen, J.-H. (2023). The Neuroprotective Effects of Flavonoid Fisetin against Corticosterone-Induced Cell Death through Modulation of ERK, p38, and PI3K/Akt/FOXO3a-Dependent Pathways in PC12 Cells. Pharmaceutics, 15(10), 2376. https://doi.org/10.3390/pharmaceutics15102376