
Chimeric Peptide Engineered Nanomedicine for Synergistic Suppression of Tumor Growth and Therapy-Induced Hyperlipidemia by mTOR and PCSK9 Inhibition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
2.2. Synthesis of Tumor-Targeted Peptide
2.3. Preparation and Characterization of PRS
2.4. Cell Culture and Cellular Internalization
2.5. MTT Assay
2.6. Live/Dead Cell Staining and Cell Apoptosis Analysis
2.7. Western Blot
2.8. Fluorescence Imaging In Vivo
2.9. Tumor Growth Inhibition
3. Results
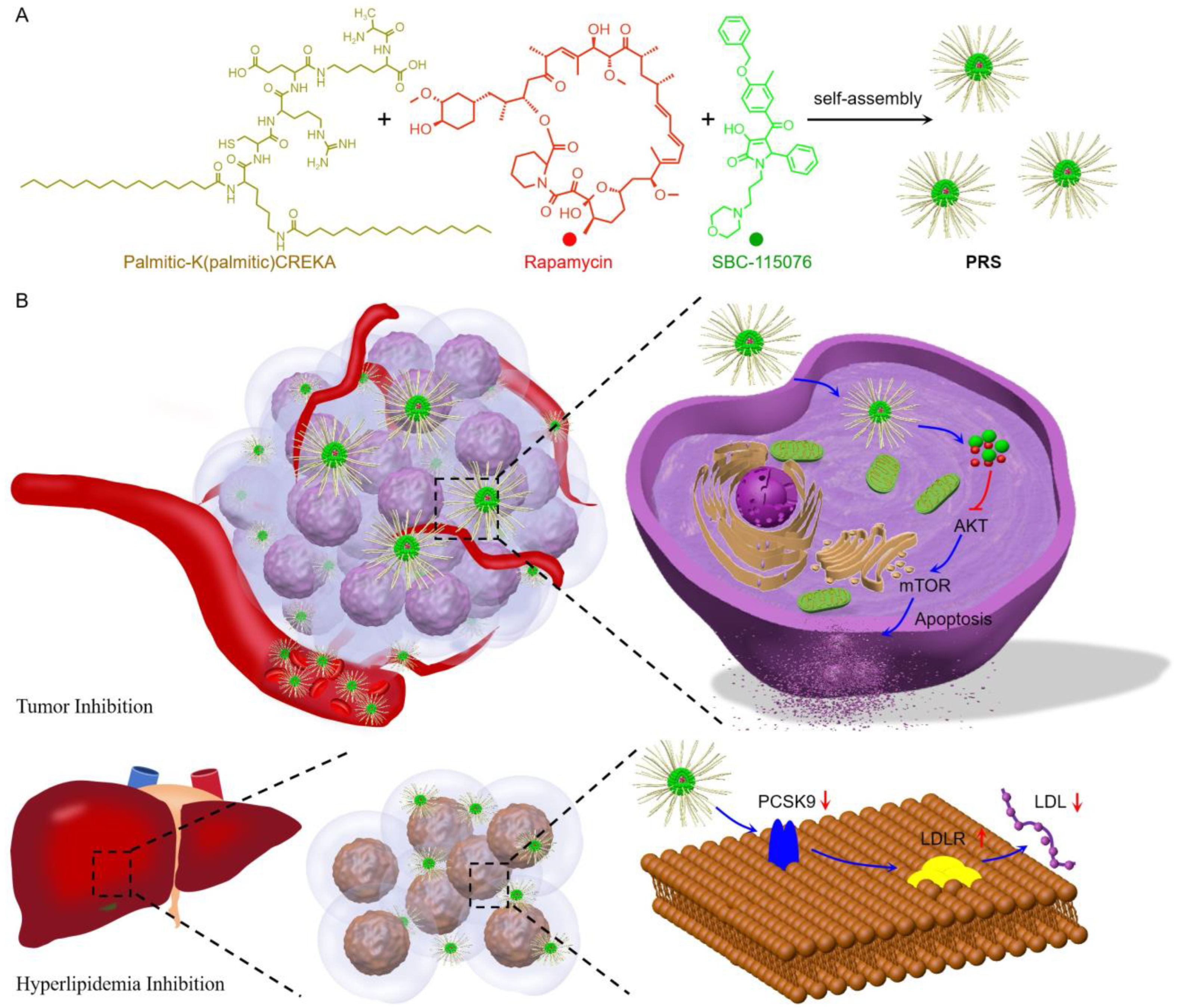
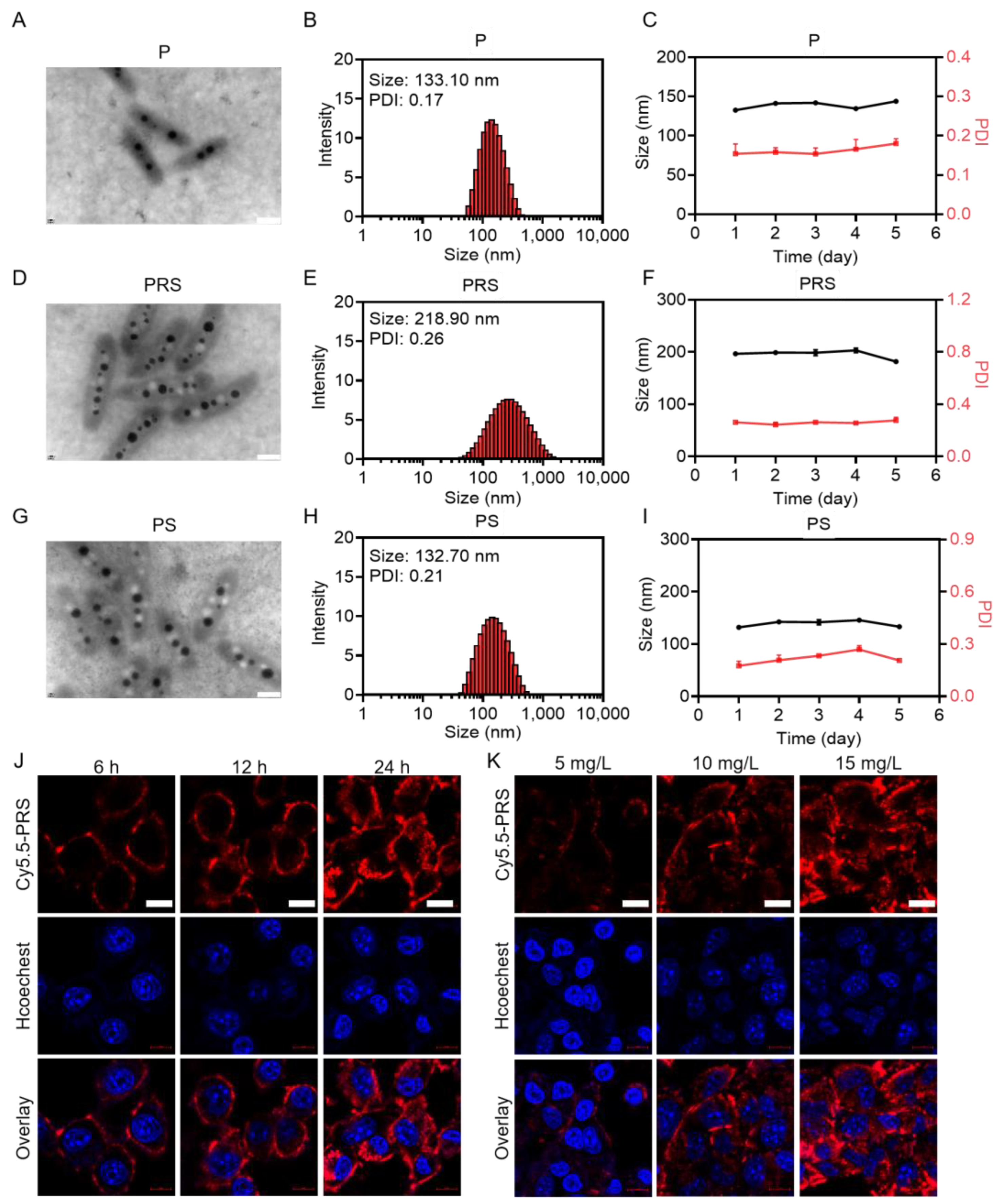
3.1. Synthesis and Characterization of PRS Nanomedicine
3.2. Cytotoxicity of PRS In Vitro
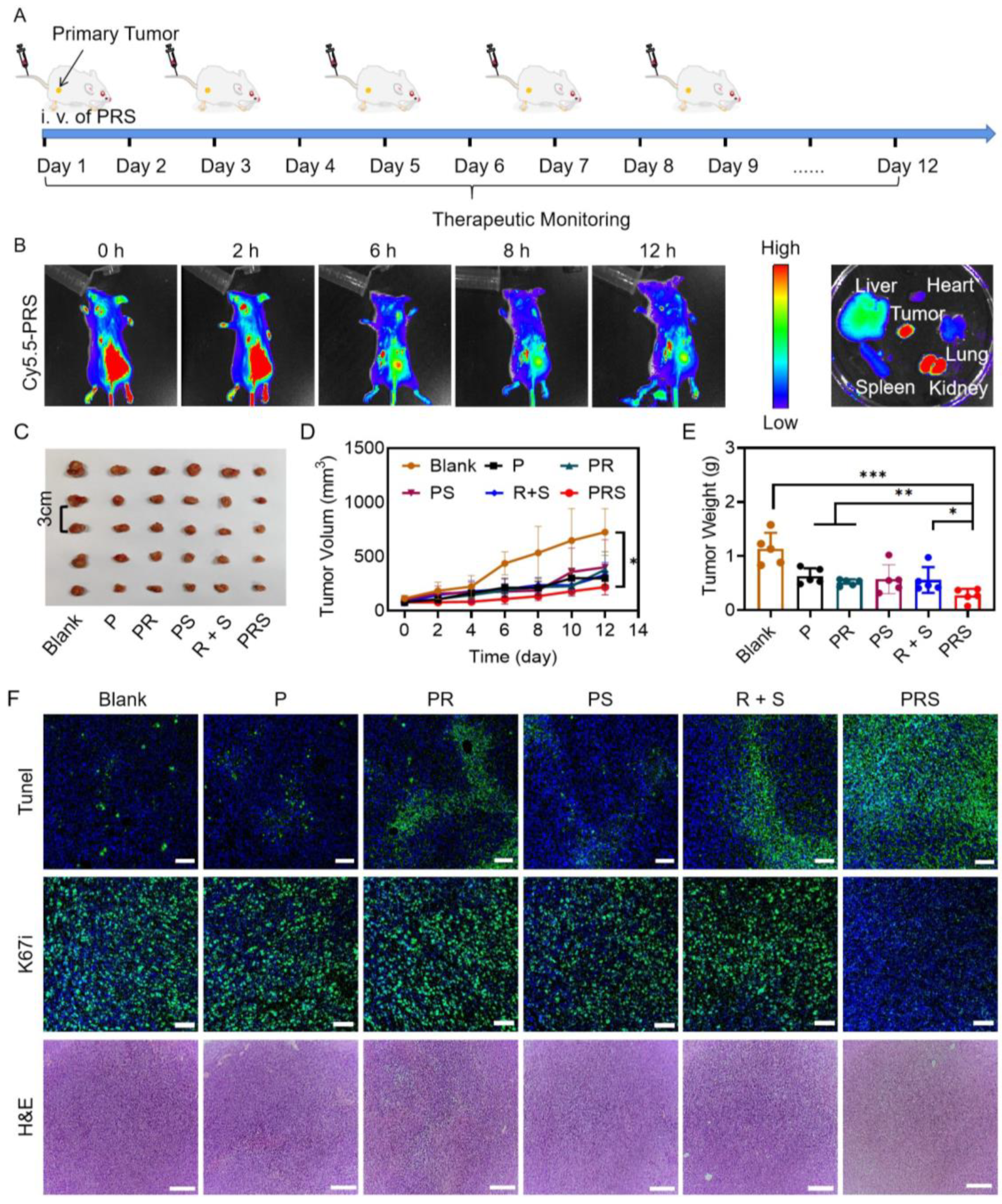
3.3. Anti-Tumor Study In Vivo
3.4. Effect of PRS on LDLR Expression In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, D.S.; Podolsky, S.H.; Greene, J.A. The burden of disease and the changing task of medicine. N. Engl. J. Med. 2012, 366, 2333–2338. [Google Scholar] [CrossRef]
- Curigliano, G.; Lenihan, D.; Fradley, M.; Ganatra, S.; Barac, A.; Blaes, A.; Herrmann, J.; Porter, C.; Lyon, A.R.; Lancellotti, P.; et al. ESMO Guidelines Committee. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. 2020, 31, 171–190. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for practice guidelines: The task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Karlstaedt, A.; Moslehi, J.; de Boer, R.A. Cardio-onco-metabolism: Metabolic remodelling in cardiovascular disease and cancer. Nat. Rev. Cardiol. 2022, 19, 414–425. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.; Chen, L.; Geng, Y.; Yu, S.; Chen, Z. Long-term cardiovascular disease mortality among 160,834 5-year survivors of adolescent and young adult cancer: An American population-based cohort study. Eur. Heart J. 2021, 42, 101–109. [Google Scholar] [CrossRef]
- Chao, C.; Xu, L.; Bhatia, S.; Cooper, R.; Brar, S.; Wong, F.L.; Armenian, S.H. Cardiovascular disease risk profiles in survivors of adolescent and young adult (AYA) cancer: The kaiser permanente AYA cancer survivors study. J. Clin. Oncol. 2016, 34, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Reidy Lagunes, D.; Yao, J. Potential synergies for combined targeted therapy in the treatment of neuroendocrine cancer. Drugs 2011, 71, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.; Halpern, E.F.; Thiele, E.A. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, A.; González, G.; Carbonero, I.G.; Frade, J.G.; Munárriz, J.; Aguiar, D.; Cuevas, B.; Cuevas, M.V.; Aporta, R.; Bárez, A.; et al. Efficacy and safety of temsirolimus in patients with relapsed or refractory mantle cell lymphoma: Results from the spanish experience. Blood 2013, 122, 5117. [Google Scholar] [CrossRef]
- Svoboda, J.; Barta, S.K.; Landsburg, D.; Nasta, S.D.; Hwang, W.T.; Delp, G.; Amundsen, B.; Ballard, H.J.; Gerson, J.N.; Chong, E.A.; et al. Everolimus plus Itacitinib in relapsed/refractory classical hodgkin lymphoma: Results of a phase I/II investigator initiated trial (EVITA Study). Blood 2020, 136, 20–21. [Google Scholar] [CrossRef]
- Cohen, E.E.; Wu, K.; Hartford, C.; Kocherginsky, M.; Eaton, K.N.; Zha, Y.; Nallari, A.; Maitland, M.L.; Fox-Kay, K.; Moshier, K.; et al. Phase I studies of sirolimus alone or in combination with pharmacokinetic modulators in advanced cancer patients. Clin. Cancer Res. 2012, 18, 4785–4793. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hudes, G.R.; Curti, B.D.; McDermott, D.F.; Escudier, B.J.; Negrier, S.; Duclos, B.; Moore, L.; O’Toole, T.; Boni, J.P.; et al. Phase I/II trial of temsirolimus combined with interferon alfa for advanced renal cell carcinoma. J. Clin. Oncol. 2007, 25, 3958–3964. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Bouillet, B.; Buffier, P.; Smati, S.; Archambeaud, F.; Cariou, B.; Vergès, B. Expert opinion on the metabolic complications of mTOR inhibitors. Ann. Endocrinol. 2018, 79, 583–590. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Windecker, S.; Pedrazzini, G.; Mueller, C.; Cook, S.; Matter, C.M.; Muller, O.; Häner, J.; Gencer, B.; Crljenica, C.; et al. Evolocumab for early reduction of LDL cholesterol levels in patients with acute coronary syndromes (EVOPACS). J. Am. Coll. Cardiol. 2019, 74, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Nallari, A.S.; Karrison, T.; Rosner, G.L.; Levine, M.R.; Sit, L.; Wu, K.W.; Stadler, M.M.; Ratain, J.E.; Cohen, E.M.; Maitland, L. Fasting glucose and triglycerides as biomarkers of mTOR inhibition, evidence of a categorical response. J. Clin. Oncol. 2010, 28, 3091-3091. [Google Scholar] [CrossRef]
- Chakrabarti, P.; English, T.; Shi, J.; Smas, C.M.; Kandror, K.V. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 2010, 59, 775–781. [Google Scholar] [CrossRef]
- Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration; Sarwar, N.; Sandhu, M.S.; Ricketts, S.L.; Butterworth, A.S.; Di Angelantonio, E.; Boekholdt, S.M.; Ouwehand, W.; Watkins, H.; Samani, N.J.; et al. Triglyceride-mediated pathways and coronary disease: Collaborative analysis of 101 studies. Lancet 2010, 375, 1634–1639. [Google Scholar] [PubMed]
- Do, R.; Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Gao, C.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 2013, 45, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, L.; Lian, J.; Schauer, S.; Vesely, P.W.; Kratky, D.; Hoefler, G.; Lehner, R. Tumor-induced hyperlipidemia contributes to tumor growth. Cell Rep. 2016, 15, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Havel, R.J. Role of the liver in hyperlipidemia. Semin. Liver Dis. 1992, 12, 356–363. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The Multifaceted Biology of PCSK9. Endocr. Rev. 2022, 43, 558–582. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Ai, D.; Chen, C.; Han, S.; Ganda, A.; Murphy, A.J.; Haeusler, R.; Thorp, E.; Accili, D.; Horton, J.D.; Tall, A.R. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J. Clin. Investig. 2012, 122, 1262–1270. [Google Scholar] [CrossRef]
- Fan, G.L.; Deng, F.A.; Zhou, X.; Liu, L.S.; Huang, J.Q.; Zhang, D.W.; Sun, Y.X.; Chen, A.L.; Cheng, H.; Li, S.Y. Plasma membrane targeted photodynamic O2 economizer for hypoxic tumor therapy. Biomaterials 2021, 273, 120854. [Google Scholar] [CrossRef]
- Nakamura, O.; Hitora, T.; Yamagami, Y.; Mori, M.; Nishimura, H.; Horie, R.; Yamaguchi, K.; Yamamoto, T. The combination of rapamycin and MAPK inhibitors enhances the growth inhibitory effect on Nara-H cells. Int. J. Mol. Med. 2014, 33, 1491–1497. [Google Scholar] [CrossRef]
- Guan, S.; Deng, Z.; Huang, T.; Wen, W.; Zhao, Y.; Chen, A. Light-triggered reversible slimming of azobenzene-containing wormlike nanoparticles synthesized by polymerization-induced self-assembly for nanofiltration switches. ACS Macro Lett. 2019, 8, 460–465. [Google Scholar] [CrossRef]
- Chu, R.; Wang, N.; Bi, Y.; Nan, G. Rapamycin prevents lung injury related to acute spinal cord injury in rats. Sci. Rep. 2023, 13, 10674. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Jiang, X.M.; Qian, W.C.; Zhang, J. Inhibition of PCSK9 improves the development of pulmonary arterial hypertension via down-regulating notch 3 expression. Cardiovasc. Drugs Ther. 2023; Online ahead of print. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yan, Y.; Lv, H.; Li, J.; Wang, Z.; Wang, K.; Wang, L.; Li, Y.; Jiang, H.; Zhang, Y. Rapamycin targets STAT3 and impacts c-Myc to suppress tumor growth. Cell Chem. Biol. 2022, 29, 373–385. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Zhu, X.D.; Feng, L.H.; Li, X.L.; Liu, X.F.; Sun, H.C.; Tang, Z.Y. PCSK9 promotes tumor growth by inhibiting tumor cell apoptosis in hepatocellular carcinoma. Exp. Hematol. Oncol. 2021, 10, 25. [Google Scholar] [CrossRef]
- Xu, X.; Cui, Y.; Cao, L.; Zhang, Y.; Yin, Y.; Hu, X. PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling. Exp. Ther. Med. 2017, 13, 1993–1999. [Google Scholar] [CrossRef]
- Sun, X.; Essalmani, R.; Day, R.; Khatib, A.M.; Seidah, N.G.; Prat, A. Proprotein convertase subtilisin/kexin type 9 deficiency reduces melanoma metastasis in liver. Neoplasia 2012, 14, 1122–1131. [Google Scholar] [CrossRef]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Gao, X.; Yi, L.; Jiang, C.; Li, S.; Wang, X.; Yang, B.; Li, W.; Che, N.; Wang, J.; Zhang, H.; et al. PCSK9 regulates the efficacy of immune checkpoint therapy in lung cancer. Front. Immunol. 2023, 14, 1142428. [Google Scholar] [CrossRef]
- Gu, Y.; Lin, X.; Dong, Y.; Wood, G.; Seidah, N.G.; Werstuck, G.; Major, P.; Bonert, M.; Kapoor, A.; Tang, D. PCSK9 facilitates melanoma pathogenesis via a network regulating tumor immunity. J. Exp. Clin. Cancer Res. 2023, 42, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, S.; Luo, H.; Lu, Q.; Yu, S. PCSK9 promotes the progression and metastasis of colon cancer cells through regulation of EMT and PI3K/AKT signaling in tumor cells and phenotypic polarization of macrophages. J. Exp. Clin. Cancer Res. 2022, 41, 303. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cai, T.; Zheng, X.; Ren, Y.; Qi, J.; Lu, X.; Chen, H.; Lin, H.; Chen, Z.; Liu, M.; et al. Potentiating CD8+ T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell 2021, 12, 240–260. [Google Scholar] [CrossRef]
- Wang, W.; Shen, T.; Dong, B.; Creighton, C.J.; Meng, Y.; Zhou, W.; Shi, Q.; Zhou, H.; Zhang, Y.; Moore, D.D.; et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J. Clin. Investig. 2019, 129, 1015–1029. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Lasithiotakis, K.G.; Sinnberg, T.W.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Garbe, C.; Meier, F.E. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J. Investig. Dermatol. 2008, 128, 2013–2023. [Google Scholar] [CrossRef]
- Tortora, G.; Bianco, R.; Daniele, G.; Ciardiello, F.; McCubrey, J.A.; Ricciardi, M.R.; Ciuffreda, L.; Cognetti, F.; Tafuri, A.; Milella, M. Overcoming resistance to molecularly targeted anticancer therapies: Rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug Resist. Updat. 2007, 10, 81–100. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef]
- Xu, B.; Li, S.; Fang, Y.; Zou, Y.; Song, D.; Zhang, S.; Cai, Y. Proprotein convertase subtilisin/kexin type 9 promotes gastric cancer metastasis and suppresses apoptosis by facilitating MAPK signaling pathway through HSP70 up-regulation. Front. Oncol. 2021, 10, 609–663. [Google Scholar] [CrossRef] [PubMed]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E., Jr.; Stark, G.R.; O’Shea, J.J. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhu, J.; Luo, H.H.; Yu, S.W.; Wang, L. Pro-protein convertase subtilisin/kexin type 9 promotes intestinal tumor development by activating janus kinase 2/signal transducer and activator of transcription 3/SOCS3 signaling in ApcMin/+ mice. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211038345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.W.; Sun, K.X.; Zheng, R.S.; Zeng, H.M.; Wang, S.M.; Chen, R.; Wei, W.Q.; He, J.; Zhang, S.W.; Sun, K.X.; et al. Cancer incidence and mortality in China, 2015. J. Natl. Cancer Cent. 2021, 1, 2–11. [Google Scholar] [CrossRef]
- Andrade, M.L.; Gilio, G.R.; Perandini, L.A.; Peixoto, A.S.; Moreno, M.F.; Castro, É.; Oliveira, T.E.; Vieira, T.S.; Ortiz-Silva, M.; Thomazelli, C.A.; et al. PPARγ-induced upregulation of subcutaneous fat adiponectin secretion, glyceroneogenesis and BCAA oxidation requires mTORC1 activity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158967. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar]
- Koskinas, K.C.; Siontis, G.C.M.; Piccolo, R.; Mavridis, D.; Räber, L.; Mach, F.; Windecker, S. Effect of statins and non-statin LDL-lowering medications on cardiovascular outcomes in secondary prevention: A meta-analysis of randomized trials. Eur. Heart J. 2018, 39, 1172–1180. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, H.; Zheng, R.; Wu, N.; Hu, J.; Wang, R.; Chi, J.; Zhang, W.; Zhao, L.; Cheng, H.; Chen, A.; et al. Chimeric Peptide Engineered Nanomedicine for Synergistic Suppression of Tumor Growth and Therapy-Induced Hyperlipidemia by mTOR and PCSK9 Inhibition. Pharmaceutics 2023, 15, 2377. https://doi.org/10.3390/pharmaceutics15102377
Cai H, Zheng R, Wu N, Hu J, Wang R, Chi J, Zhang W, Zhao L, Cheng H, Chen A, et al. Chimeric Peptide Engineered Nanomedicine for Synergistic Suppression of Tumor Growth and Therapy-Induced Hyperlipidemia by mTOR and PCSK9 Inhibition. Pharmaceutics. 2023; 15(10):2377. https://doi.org/10.3390/pharmaceutics15102377
Chicago/Turabian StyleCai, Hua, Rongrong Zheng, Ningxia Wu, Jiaman Hu, Ruixin Wang, Jianing Chi, Wei Zhang, Linping Zhao, Hong Cheng, Ali Chen, and et al. 2023. "Chimeric Peptide Engineered Nanomedicine for Synergistic Suppression of Tumor Growth and Therapy-Induced Hyperlipidemia by mTOR and PCSK9 Inhibition" Pharmaceutics 15, no. 10: 2377. https://doi.org/10.3390/pharmaceutics15102377
APA StyleCai, H., Zheng, R., Wu, N., Hu, J., Wang, R., Chi, J., Zhang, W., Zhao, L., Cheng, H., Chen, A., Li, S., & Xu, L. (2023). Chimeric Peptide Engineered Nanomedicine for Synergistic Suppression of Tumor Growth and Therapy-Induced Hyperlipidemia by mTOR and PCSK9 Inhibition. Pharmaceutics, 15(10), 2377. https://doi.org/10.3390/pharmaceutics15102377