
Insights into the Currently Available Drugs and Investigational Compounds Against RSV with a Focus on Their Drug-Resistance Profiles

 , ,
, ,  ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Epidemiology and RSV Genotype Distribution
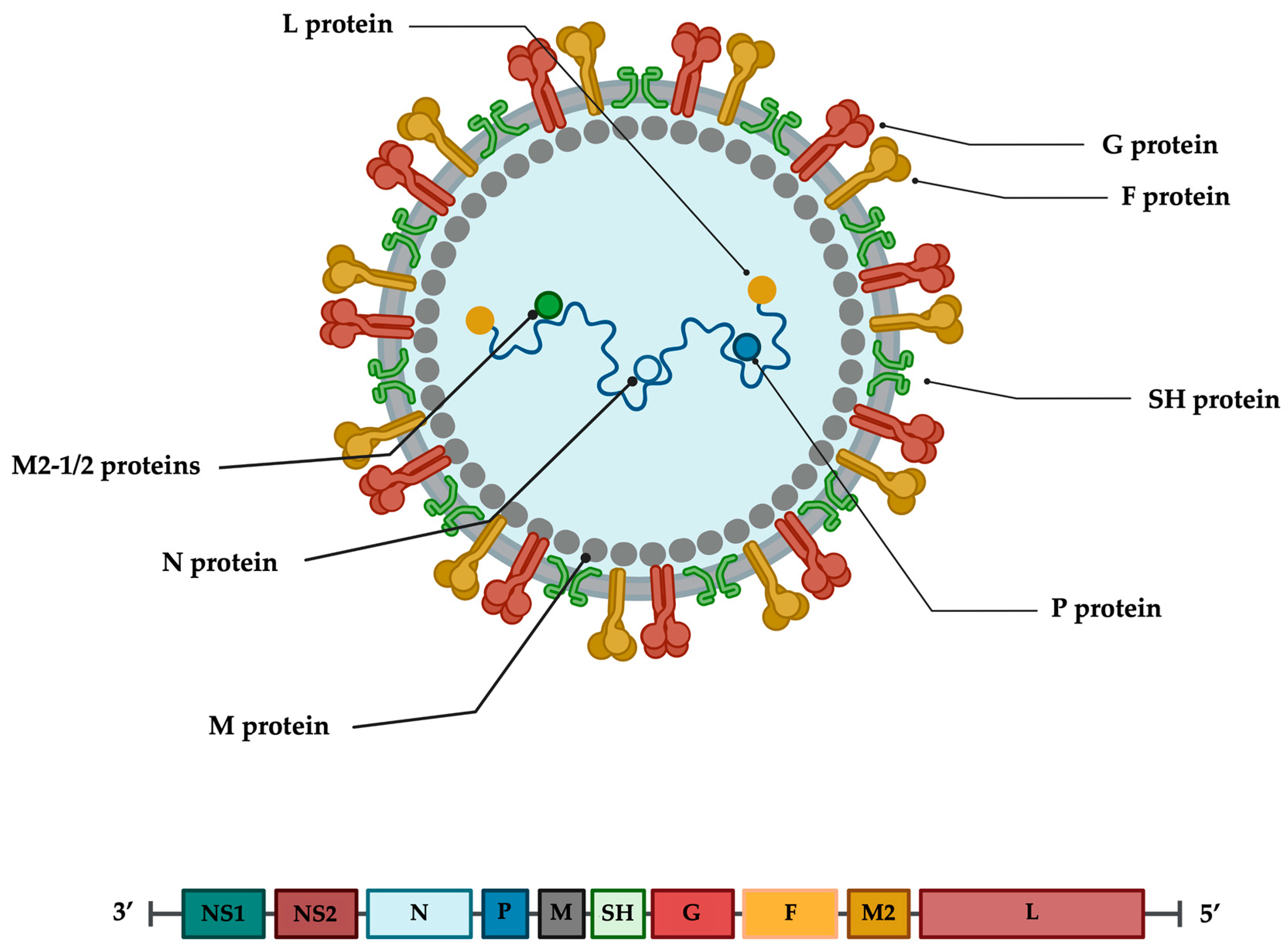
3. RSV Virion and Genome
4. Replication Cycle
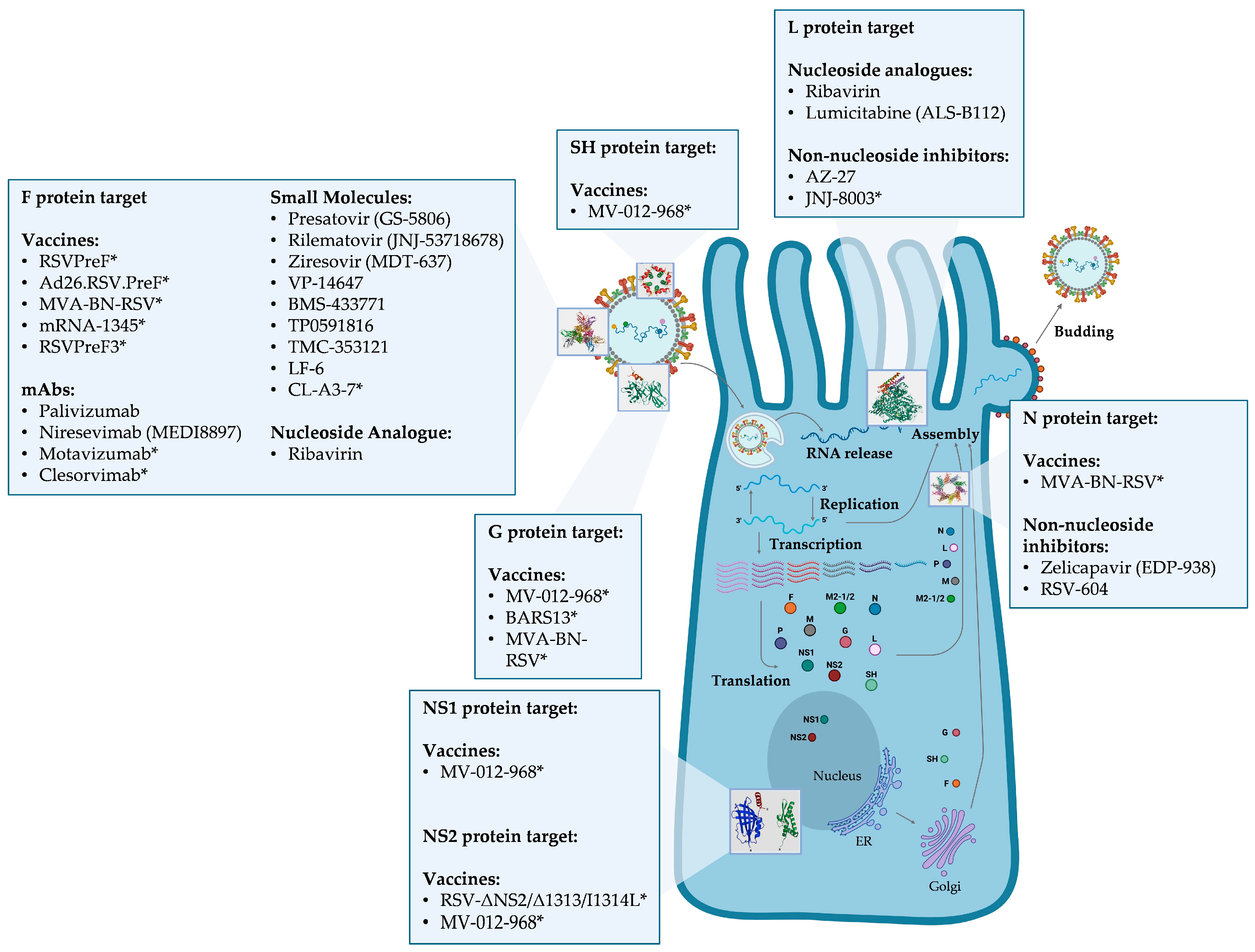
5. RSV Proteins as Targets of RSV Inhibitors and Related Drug-Resistance Mutations
6. Materials and Methods
6.1. Search Strategy and Eligibility Criteria
6.2. Structural Analysis
7. Fusion Glycoprotein (F)
7.1. Monoclonal Antibodies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Drug Class | Drug Name | Amino Acid Substitution | Domain | Strain a | Fold Change Resistance Value b | Reference |
|---|---|---|---|---|---|---|---|
| F | Monoclonal | Palivizumab | S275L c | Antigenic site A | A2 | >25,000 | [99] |
| Antibodies | S275F c | Antigenic site A | A2 | >25,000 | [99] | ||
| K272E c | Antigenic site A | B | >25,000 | [99] | |||
| K272M c | Antigenic site A | B | >25,000 | [99] | |||
| K272T c | Antigenic site A | A2 | >25,000 | [99] | |||
| K272N c | Antigenic site A | B | 5164 | [99] | |||
| K272Q d | Antigenic site A | A2 | >3000 | [100] | |||
| N262Y d | Antigenic site A | A2 | 512 | [100] | |||
| T400A c | Cysteine-Rich | A2 | 3.3 | [101] | |||
| L138F d | Fusion Peptide | A2 | 2.4 | [100] | |||
| K399I d | Cysteine-Rich | A2 | 2.3 | [100] | |||
| F488L d | Heptad Repeat 2 | B | 1.4 | [100] | |||
| F488S d | Heptad Repeat 2 | B | 1.1 | [100] | |||
| F488Y d | Heptad Repeat 2 | A2 | 1.1 | [100] | |||
| E487D d | Heptad Repeat 2 | A2 | 1 | [100] | |||
| D486N d | Heptad Repeat 2 | A2 | 0.8 | [100] | |||
| F140L/N517I d | F1 | A2 | 0.6 | [100] | |||
| N262D d | Antigenic site A | A2 | NA | [102] | |||
| S276N d | Antigenic site A | B | NA | [103] | |||
| Nirsevimab | N208D c | F1 | B9320 | >90,000 | [82] | ||
| (MEDI8897) | K68N/N208S c | F1/F2 | B9320 | >90,000 | [82] | ||
| N208S c | F1 | B9320 | 24,618 | [82] | |||
| K68N/N201S c | F1/F2 | B9320 | 13,438 | [82] | |||
| N67I/N208Y c | F1/F2 | A2 | 102.5 | [82] | |||
| N201S c | Heptad Repeat 1 | B9320 | 64.5 | [82] | |||
| K68N c | F2 | B9320 | 3.8 | [82] | |||
| N67I c | F2 | A2 | 1.5 | [82] | |||
| N208Y c | F1 | A2 | 1.1 | [82] | |||
| Small Molecules | Presatovir | F488L d | Heptad Repeat 2 | B | >5000 | [100] | |
| (GS-5806) | F488S d | Heptad Repeat 2 | B | >5000 | [100] | ||
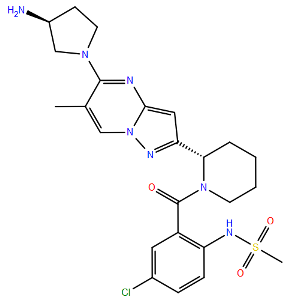 | L138F d | Fusion Peptide | A2 | >2000 | [100] | ||
| F140L/N517I d | F1 | A2 | >2000 | [100] | |||
| D486N d | Heptad Repeat 2 | A2 | 1193 | [100] | |||
| T400I d | Cysteine-Rich | A2 | 410 | [101] | |||
| T400A d | Cysteine-Rich | A2 | 214 | [100] | |||
| K399I d | Cysteine-Rich | A2 | 87 | [100] | |||
| F488Y d | Heptad Repeat 2 | A2 | 75 | [100] | |||
| E487D d | Heptad Repeat 2 | A2 | 35 | [100] | |||
| K394R c | Cysteine-Rich | A2 | 4 | [104] | |||
| K272Q d | Antigenic site A | A2 | 2 | [100] | |||
| N262Y d | Antigenic site A | A2 | 0.2 | [100] | |||
| Rilematovir | K394R c | Cysteine-Rich | A2 | 6024 | [100] | ||
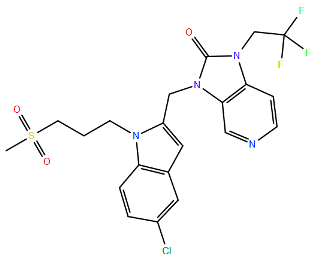 | |||||||
| VP-14637 | T400A d | Cysteine-Rich | A2 | >3200 | [100] | ||
| (MDT-637) | D486N d | Heptad Repeat 2 | A2 | >3200 | [100] | ||
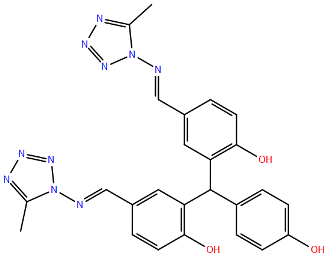 | F488L d | Heptad Repeat 2 | B | >2500 | [100] | ||
| F488S d | Heptad Repeat 2 | B | >2500 | [100] | |||
| L138F d | Fusion Peptide | A2 | >2000 | [100] | |||
| F140L/N517I d | F1 | A2 | >2000 | [100] | |||
| E487D d | Heptad Repeat 2 | A2 | 75 | [100] | |||
| F488Y d | Heptad Repeat 2 | A2 | 52 | [100] | |||
| K399I d | Cysteine-Rich | A2 | 45 | [100] | |||
| N262Y d | Antigenic site A | A2 | 0.6 | [100] | |||
| K272Q d | Antigenic site A | A2 | 0.6 | [100] | |||
| BMS-433771 | K394R c | Cysteine-Rich | A2 | 1902 | [104] | ||
 | L141F c | Fusion Peptide | A2 | >18.3 | [105] | ||
| TP0591816 | L141F c | Fusion Peptide | A2 | >4720 | [105] | ||
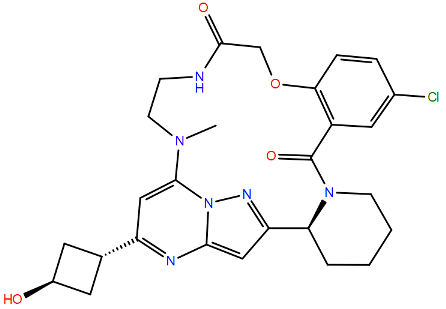 | |||||||
| TMC-353121 | K394R c | Cysteine-Rich | A2 | 1033 | [104] | ||
 | |||||||
| Ziresovir | K394R c | Cysteine-Rich | A2 | 355 | [104] | ||
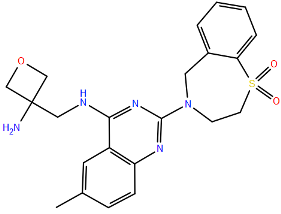 | |||||||
| LF-6 | K394R c | Cysteine-Rich | A2 | >71 | [104] | ||
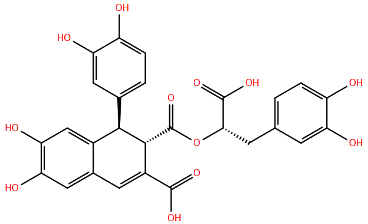 | |||||||
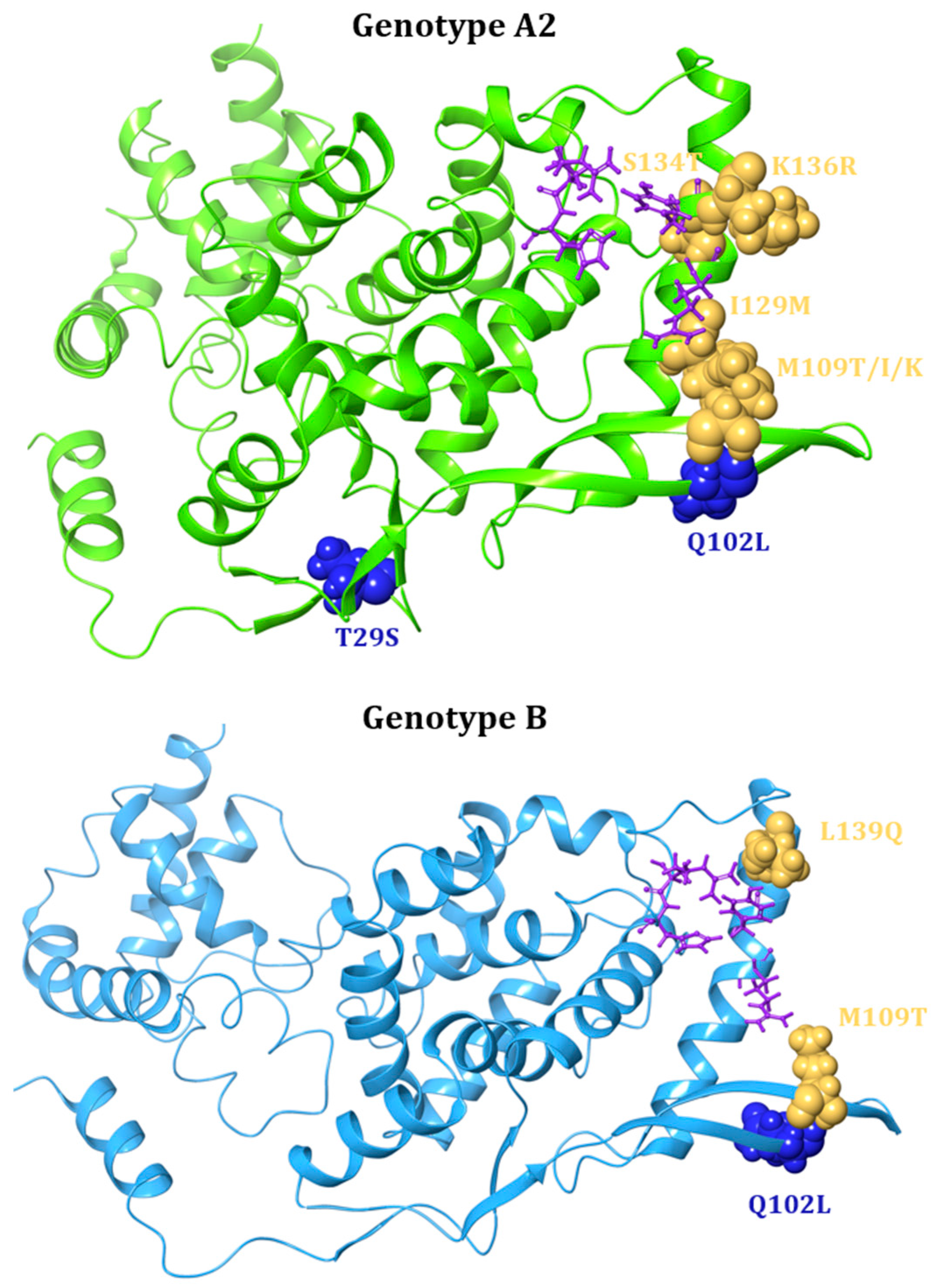
| N | Small Molecules | Zelicapavir | Q102L/M109T/I129M c | A2 | 42.4 | [106] | |
| (EDP-938) | L139Q c | B | 42 | [106] | |||
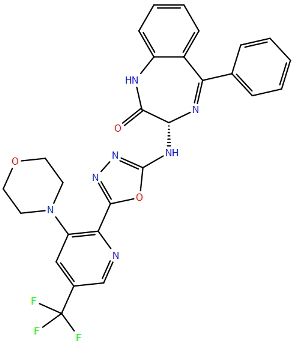 | M109K c | A2 | 26.9 | [106] | |||
| M109T c | A2 | 5.4 | [106] | ||||
| I129M c | A2 | 3.7 | [106] | ||||
| T29S/S134T c | A2 | 3.3 | [106] | ||||
| K136R c | A2 | 2.7 | [106] | ||||
| S134T c | A2 | 2.2 | [106] | ||||
| Q102L c | A2 | 2 | [106] | ||||
| M109I c | A2 | 1.6 | [106] | ||||
| G | Small Molecules | Zelicapavir | K205G/K213G/T219A c | A2 | 60 | [106] | |
| (EDP-938) | R8H c | A2 | 3.1 | [106] | |||
| L | Nucleoside | ALS-8112 | A789V f | Motif B | A2 | Km = 61.0 ± 26.9 μM | [107] |
| Analogue | 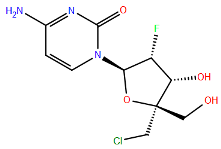 | M628L f | Motif F | A2 | Km = 23.1 ± 0.07 μM | [107] | |
| I796V f | Motif B | A2 | Km = 22.0 ± 9.9 μM | [107] | |||
| L795I f | Motif B | A2 | Km = 16.5 ± 6.4 μM | [107] | |||
| NN inhibitor | AZ-27 | Y1631H e | Capping domain | A2 | 940 | [107] | |
 |
7.1.1. Mutations Associated with Resistance to Palivizumab
7.1.2. Mutations Associated with Resistance to Nirsevimab
7.2. F Protein Inhibitors
7.2.1. TMC-353121 and BMS-433771 and Their Resistance Profiles
7.2.2. Presatovir and Its Resistance Profiles
7.2.3. Rilematovir (JNJ-53718678) and Ziresovir (RO-0529, AK0529) and Their Resistance Profiles
7.2.4. VP-14637 and Its Resistance Profiles
7.2.5. TP0591816 and Its Resistance Profiles
7.2.6. Salvianolic Acid R (LF-6) and Its Resistance Profiles
7.3. Multi-Drug Resistance Mutations
7.4. Structural Characterisation of Drug-Resistance Mutations
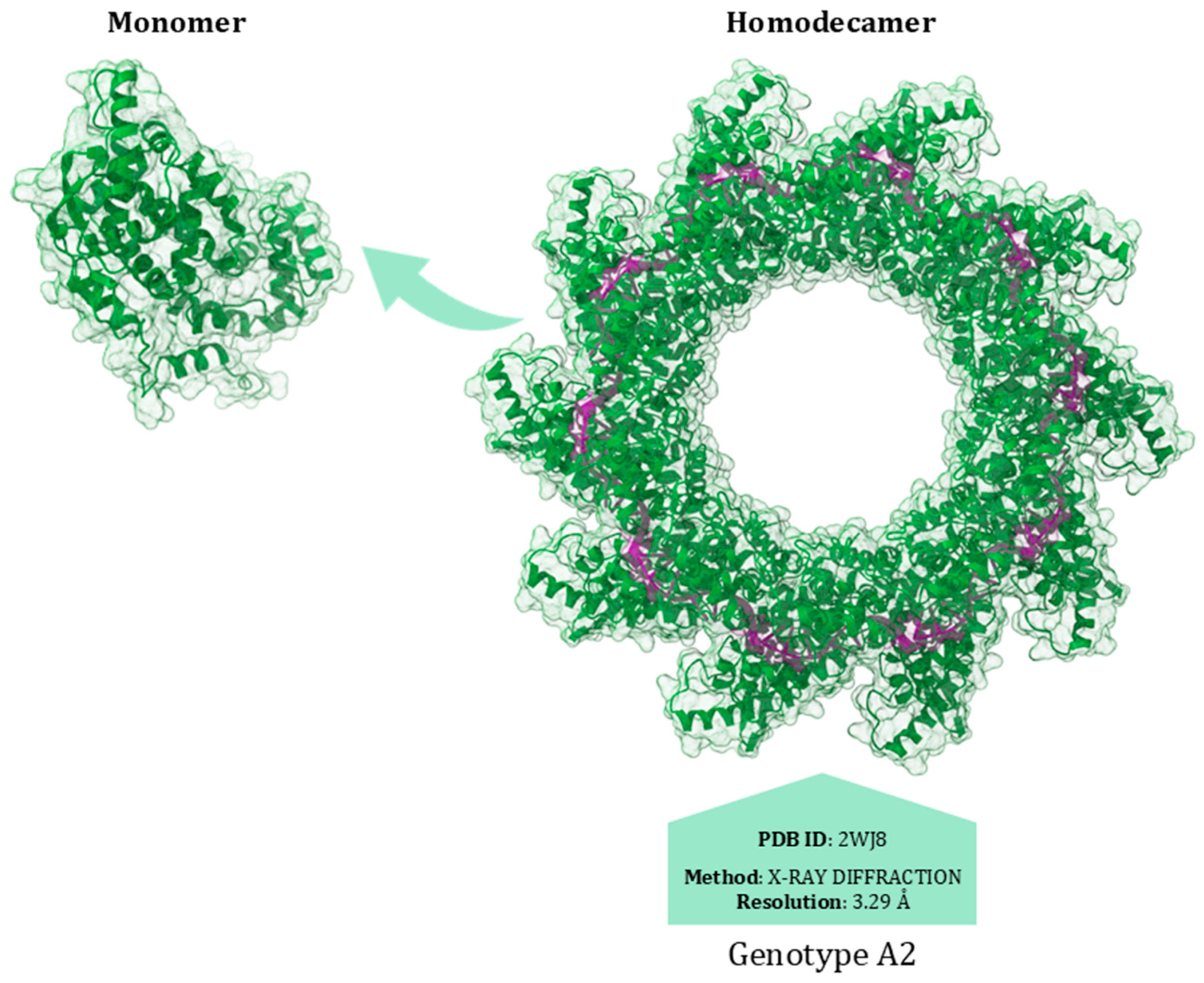
8. Nucleoprotein (N)
N Protein Inhibitors
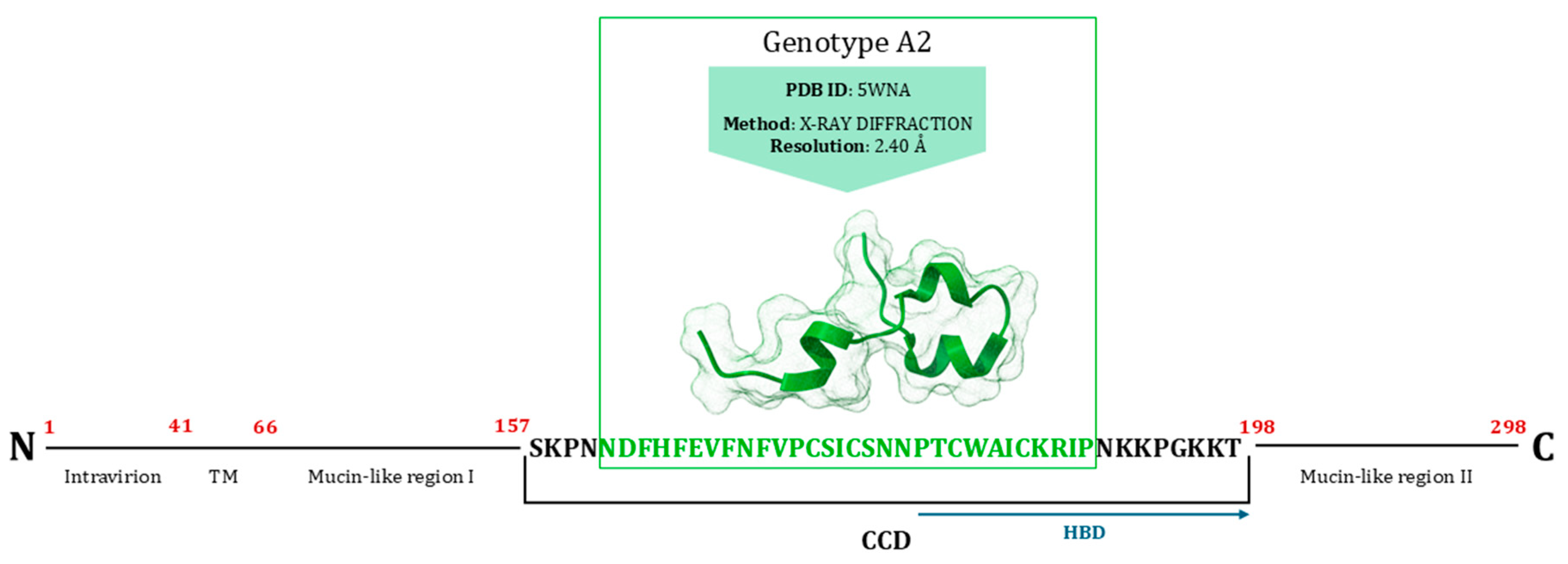
9. Major Surface Glycoprotein (G)
G Protein Inhibitors
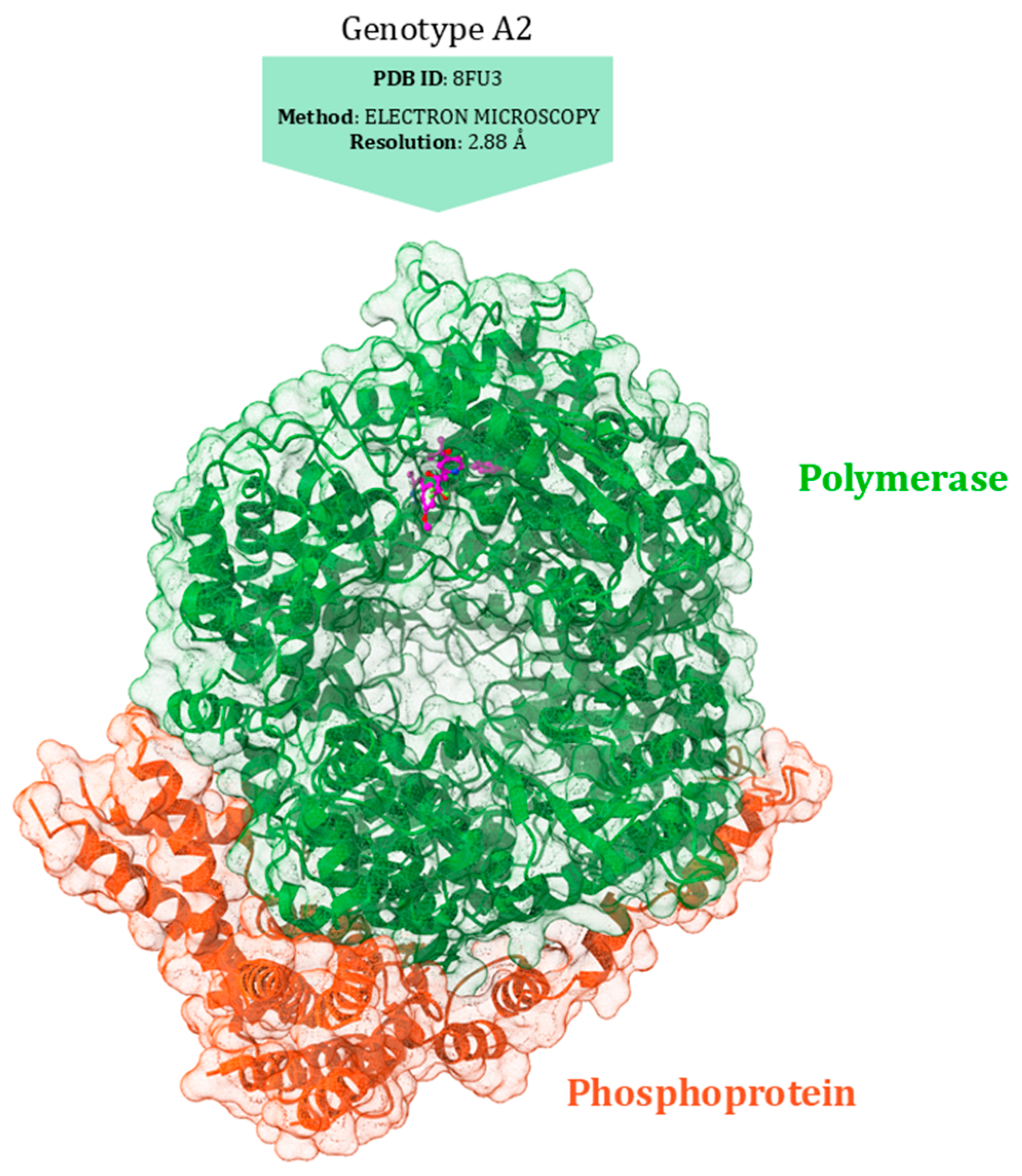
10. Polymerase (L)
L Protein Inhibitors
11. Discussion
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chanock, R.; Roizman, B.; Myers, R. Recovery from infants with respiratory illness of a virus related to Chimpanzee Coryza Agent (CCA). I. Isolation, properties and characterization. Am. J. Epidemiol. 1957, 66, 281–290. [Google Scholar] [CrossRef]
- Blount, R.E., Jr.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc. Soc. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar] [CrossRef]
- Cantú-Flores, K.; Rivera-Alfaro, G.; Carlos Muñoz-Escalante, J.; Noyola, D.E. Global distribution of Respiratory Syncytial Virus A and B infections: A Systematic Review. Pathog. Glob. Health 2022, 116, 398–409. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.P.S.; Martinón-Torres, F.; Drysdale, S.B.; Faust, S.N. The disease burden of respiratory syncytial virus in Infants. Curr. Opin. Infect. Dis. 2023, 36, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, Q.; Liu, D.; Zu, W.; Zhang, D.; Chen, L. The global burden of lower respiratory infections attributable to respiratory syncytial virus in 204 countries and territories, 1990–2019: Findings from the Global Burden of Disease Study 2019. Intern. Emerg. Med. 2024, 19, 59–70. [Google Scholar] [CrossRef]
- Gatt, D.; Martin, I.; Alfouzan, R.; Moraes, T.J. Prevention and treatment strategies for Respiratory Syncytial Virus (RSV). Pathogens 2023, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Rodríguez-Fernández, R.; Oliva, S.; Peeples, M.E.; Ramilo, O. The journey to an RSV vaccine. Ann. Allergy Asthma Immunol. 2020, 125, 36–46. [Google Scholar] [CrossRef]
- Killikelly, A.M.; Kanekiyo, M.; Graham, B.S. Pre-fusion F is absent on the surface of formalin-inactivated Respiratory Syncytial Virus. Sci. Rep. 2016, 6, 34108. [Google Scholar] [CrossRef]
- Qiu, X.; Xu, S.; Lu, Y.; Luo, Z.; Yan, Y.; Wang, C.; Ji, J. Development of mRNA vaccines against Respiratory Syncytial Virus (RSV). Cytokine Growth Factor Rev. 2022, 68, 37–53. [Google Scholar] [CrossRef]
- Vidal Valero, M. “A Good Day”: FDA approves world’s first RSV vaccine. Nature 2023, 617, 234–235. [Google Scholar] [CrossRef]
- Venkatesan, P. First RSV vaccine approvals. Lancet Microbe 2023, 4, e577. [Google Scholar] [CrossRef]
- Fleming-Dutra, K.E.; Jones, J.M.; Roper, L.E.; Prill, M.M.; Ortega-Sanchez, I.R.; Moulia, D.L.; Wallace, M.; Godfrey, M.; Broder, K.R.; Tepper, N.K.; et al. Use of the Pfizer Respiratory Syncytial Virus vaccine during pregnancy for the prevention of respiratory syncytial virus-associated lower respiratory tract disease in infants: Recommendations of the advisory committee on immunization practices—United States, 2023. MMWR. Morb. Mortal. Wkly. Rep. 2023, 72, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves mRNA-based RSV vaccine. Nat. Rev. Drug. Discov. 2024, 23, 487. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Lamb, H.M. Palivizumab. Drugs 1999, 58, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, T.; Bergamasco, A.; Cristarella, T.; Goyer, C.; Wojdyla, M.; Oladapo, A.; Sawicky, J.; Yee, J.; Moride, Y. Effectiveness and safety of palivizumab for the prevention of serious lower respiratory tract infection caused by Respiratory Syncytial Virus: A systematic review. Am. J. Perinatol. 2024, 41, e1107–e1115. [Google Scholar] [CrossRef]
- Moline, H.L.; Tannis, A.; Toepfer, A.P.; Williams, J.V.; Boom, J.A.; Englund, J.A.; Halasa, N.B.; Staat, M.A.; Weinberg, G.A.; Selvarangan, R.; et al. Early estimate of nirsevimab effectiveness for prevention of Respiratory Syncytial Virus–associated hospitalization among infants entering their first Respiratory Syncytial Virus season—New vaccine surveillance network, October 2023–February 2024. MMWR. Morb. Mortal. Wkly. Rep. 2024, 73, 209–214. [Google Scholar] [CrossRef]
- Jones, J.M.; Fleming-Dutra, K.E.; Prill, M.M.; Roper, L.E.; Brooks, O.; Sánchez, P.J.; Kotton, C.N.; Mahon, B.E.; Meyer, S.; Long, S.S.; et al. Use of Nirsevimab for the prevention of Respiratory Syncytial Virus disease among infants and young children: Recommendations of the advisory committee on immunization practices—United States, 2023. MMWR. Morb. Mortal. Wkly. Rep. 2023, 72, 920–925. [Google Scholar] [CrossRef]
- Tejada, S.; Martinez-Reviejo, R.; Karakoc, H.N.; Peña-López, Y.; Manuel, O.; Rello, J. Ribavirin for Treatment of Subjects with Respiratory Syncytial Virus-Related Infection: A Systematic Review and Meta-Analysis. Adv. Ther. 2022, 39, 4037–4051. [Google Scholar] [CrossRef]
- Glanville, A.R.; Scott, A.I.R.; Morton, J.M.; Aboyoun, C.L.; Plit, M.L.; Carter, I.W.; Malouf, M.A. Intravenous Ribavirin is a safe and cost-effective treatment for Respiratory Syncytial Virus infection after lung transplantation. J. Heart Lung Transpl. 2005, 24, 2114–2119. [Google Scholar] [CrossRef]
- Cockerill, G.S.; Good, J.A.D.; Mathews, N. State of the art in Respiratory Syncytial Virus drug discovery and development. J. Med. Chem. 2019, 62, 3206–3227. [Google Scholar] [CrossRef] [PubMed]
- Domachowske, J.B.; Anderson, E.J.; Goldstein, M. The future of Respiratory Syncytial Virus disease prevention and treatment. Infect. Dis. Ther. 2021, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, T.; Sarnari, C.; Gaito, R.; Tabarrini, O.; Manfroni, G. Recent progress toward the discovery of small molecules as novel anti-Respiratory Syncytial Virus agents. J. Med. Chem. 2024, 67, 11543–11579. [Google Scholar] [CrossRef]
- Shi, T.; Denouel, A.; Tietjen, A.K.; Campbell, I.; Moran, E.; Li, X.; Campbell, H.; Demont, C.; Nyawanda, B.O.; Chu, H.Y.; et al. RESCEU Investigators. Global disease burden estimates of Respiratory Syncytial Virus-associated acute respiratory infection in older adults in 2015: A systematic review and meta-analysis. J. Infect. Dis. 2020, 222, 577–583. [Google Scholar] [CrossRef]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, detection, and new options for prevention and treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef] [PubMed]
- Pebody, R.; Moyes, J.; Hirve, S.; Campbell, H.; Jackson, S.; Moen, A.; Nair, H.; Simões, E.A.F.; Smith, P.G.; Wairagkar, N.; et al. Approaches to use the WHO respiratory syncytial virus surveillance platform to estimate disease burden. Influenza Other Respir. Viruses 2020, 14, 615–621. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to Respiratory Syncytial Virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Zheng, Z.; Warren, J.L.; Shapiro, E.D.; Pitzer, V.E.; Weinberger, D.M. Estimated incidence of respiratory hospitalizations attributable to RSV infections across age and socioeconomic groups. Pneumonia 2022, 14, 6. [Google Scholar] [CrossRef]
- Alfano, F.; Bigoni, T.; Caggiano, F.P.; Papi, A. Respiratory Syncytial Virus Infection in Older Adults: An Update. Drugs Aging 2024, 41, 487–505. [Google Scholar] [CrossRef]
- Savic, M.; Penders, Y.; Shi, T.; Branche, A.; Pirçon, J.Y. Respiratory Syncytial Virus disease burden in adults aged 60 years and older in high-income countries: A systematic literature review and meta-analysis. Influenza Other Respir. Viruses 2023, 17, e13031. [Google Scholar] [CrossRef]
- Obando-Pacheco, P.; Justicia-Grande, A.J.; Rivero-Calle, I.; Rodríguez-Tenreiro, C.; Sly, P.; Ramilo, O.; Mejías, A.; Baraldi, E.; Papadopoulos, N.G.; Nair, H.; et al. Respiratory Syncytial Virus seasonality: A global overview. J. Inf. Dis. 2018, 217, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Winn, A.; Parikh, R.; Jones, J.M.; McMorrow, M.; Prill, M.M.; Silk, B.J.; Scobie, H.M.; Hall, A.J. Seasonality of Respiratory Syncytial Virus—United States, 2017–2023. MMWR. Morb. Mortal. Wkly. Rep. 2023, 72, 355–361. [Google Scholar] [CrossRef]
- Bardsley, M.; Morbey, R.A.; Hughes, H.E.; Beck, C.R.; Watson, C.H.; Zhao, H.; Ellis, J.; Smith, G.E.; Elliot, A.J. Epidemiology of Respiratory Syncytial Virus in children younger than 5 years in England during the COVID-19 pandemic, measured by laboratory, clinical, and syndromic surveillance: A retrospective observational study. Lancet Infect. Dis. 2023, 23, 56–66. [Google Scholar] [CrossRef]
- Lastrucci, V.; Pacifici, M.; Puglia, M.; Alderotti, G.; Berti, E.; Riccio, M.D.; Bonaccorsi, G.; Moriondo, M.; Resti, M.; Peroni, D.; et al. Seasonality and severity of Respiratory Syncytial Virus during the COVID-19 Pandemic: A dynamic cohort study. J. Inf. Dis. 2024, 148, 107231. [Google Scholar] [CrossRef]
- Abu-Raya, B.; Viñeta Paramo, M.; Reicherz, F.; Lavoie, P.M. Why Has the Epidemiology of RSV Changed during the COVID-19 pandemic? eClinicalMedicine 2023, 61, 102089. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.M.; Fu, Y.H.; Peng, X.L.; Zheng, Y.P. Genetic diversity and molecular evolution of human Respiratory Syncytial Virus A and B. Sci. Rep. 2021, 11, 12941. [Google Scholar] [CrossRef]
- Muñoz-Escalante, J.C.; Comas-García, A.; Bernal-Silva, S.; Noyola, D.E. Respiratory Syncytial Virus B sequence analysis reveals a novel early genotype. Sci. Rep. 2021, 11, 3452. [Google Scholar] [CrossRef]
- Jafri, H.S.; Wu, X.; Makari, D.; Henrickson, K.J. Distribution of Respiratory Syncytial Virus subtypes A and B among infants presenting to the emergency department with lower respiratory tract infection or apnea. Pediatr. Infect. Dis. J. 2013, 32, 335–340. [Google Scholar] [CrossRef]
- Battles, M.B.; McLellan, J.S. Respiratory Syncytial Virus Entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Yegambaram, K.; Todd, E.J.; Richard, C.A.; Dods, R.L.; Pangratiou, G.M.; Trinh, C.H.; Moul, S.L.; Murphy, J.C.; Mankouri, J.; et al. The Structure of the Human Respiratory Syncytial Virus M2-1 Protein Bound to the Interaction Domain of the Phosphoprotein P Defines the Orientation of the Complex. mBio 2018, 9, 1110–1128. [Google Scholar] [CrossRef]
- Scudero, O.B.; Santiago, V.F.; Palmisano, G.; Simabuco, F.M.; Ventura, A.M. The respiratory syncytial virus M2-2 protein is targeted for proteasome degradation and inhibits translation and stress granules assembly. PLoS ONE 2023, 18, e0289100. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, A.C.; Bont, L.J. Respiratory Syncytial Virus infection and novel interventions. Nat. Rev. Microbiol. 2023, 21, 734–749. [Google Scholar] [CrossRef]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory Syncytial Virus: Virology, reverse genetics, and pathogenesis of disease. Curr. Top. Microbiol. Immunol. 2013, 372, 3–38. [Google Scholar] [CrossRef]
- Kiss, G.; Holl, J.M.; Williams, G.M.; Alonas, E.; Vanover, D.; Lifland, A.W.; Gudheti, M.; Guerrero-Ferreira, R.C.; Nair, V.; Yi, H.; et al. Structural Analysis of Respiratory Syncytial Virus Reveals the position of M2-1 between the matrix protein and the ribonucleoprotein complex. J. Virol. 2014, 88, 7602–7617. [Google Scholar] [CrossRef]
- Okura, T.; Takahashi, T.; Kameya, T.; Mizukoshi, F.; Nakai, Y.; Kakizaki, M.; Nishi, M.; Otsuki, N.; Kimura, H.; Miyakawa, K.; et al. MARCH8 Restricts RSV Replication by Promoting Cellular Apoptosis Through Ubiquitin-Mediated Proteolysis of Viral SH Protein. Viruses 2024, 16, 1935. [Google Scholar] [CrossRef]
- Anderson, C.S.; Chirkova, T.; Slaunwhite, C.G.; Qiu, X.; Walsh, E.E.; Anderson, L.J.; Mariani, T.J. CX3CR1 engagement by Respiratory Syncytial Virus leads to induction of nucleolin and dysregulation of cilium-related genes. J. Virol. 2021, 95, e00095. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.A.; Audet, S.; Beeler, J.A. The fusion glycoprotein of human Respiratory Syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 2000, 74, 6442–6447. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.D.; Bilawchuk, L.M.; McDonough, J.E.; Jamieson, K.C.; Elawar, F.; Cen, Y.; Duan, W.; Lin, C.; Song, H.; Casanova, J.L.; et al. IGF1R is an entry receptor for Respiratory Syncytial Virus. Nature 2020, 583, 615–619. [Google Scholar] [CrossRef]
- Hallak, L.K.; Spillmann, D.; Collins, P.L.; Peeples, M.E. Glycosaminoglycan sulfation requirements for Respiratory Syncytial Virus infection. J. Virol. 2000, 74, 10508–10513. [Google Scholar] [CrossRef]
- Lingemann, M.; Mccarty, T.; Liu, X.; Buchholz Id, U.J.; Surman, S.; Martin, S.E.; Collins, P.L.; Munir Id, S. The alpha-1 subunit of the Na+, K+-ATPase (ATP1A1) is required for macropinocytic entry of Respiratory Syncytial Virus (RSV) in human respiratory epithelial cells. PLoS Pathog. 2019, 15, e1007963. [Google Scholar] [CrossRef]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human Respiratory Syncytial Virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef]
- Feng, Z.; Xu, L.; Xie, Z. Receptors for Respiratory Syncytial Virus infection and host factors regulating the life cycle of Respiratory Syncytial Virus. Front. Cell. Infect. Microbiol. 2022, 12, 858629. [Google Scholar] [CrossRef] [PubMed]
- Techaarpornkul, S.; Barretto, N.; Peeples, M.E. Functional analysis of recombinant Respiratory Syncytial Virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 2001, 75, 6825–6834. [Google Scholar] [CrossRef]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of Respiratory Syncytial Virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef]
- Tripp, R.A.; Jones, L.P.; Haynes, L.M.; Zheng, H.; Murphy, P.M.; Anderson, L.J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2001, 8, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M.E. Respiratory Syncytial Virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 31, 1113–1117. [Google Scholar] [CrossRef]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.Y.; et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 14, 7. [Google Scholar] [CrossRef]
- Tang, W.; Li, M.; Liu, Y.; Liang, N.; Yang, Z.; Zhao, Y.; Wu, S.; Lu, S.; Li, Y.; Liu, F. Small molecule inhibits respiratory syncytial virus entry and infection by blocking the interaction of the viral fusion protein with the cell membrane. FASEB J. 2019, 33, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, S.; La Frazia, S.; Riccio, A.; Pedersen, J.Z.; Topai, A.; Nicolotti, O.; Rossignol, J.F.; Santoro, M.G. Nitazoxanide inhibits paramyxovirus replication by targeting the Fusion protein folding: Role of glycoprotein-specific thiol oxidoreductase ERp57. Sci. Rep. 2018, 8, 10425. [Google Scholar] [CrossRef]
- Gamiño-Arroyo, A.E.; Guerrero, M.L.; McCarthy, S.; Ramírez-Venegas, A.; Llamosas-Gallardo, B.; Galindo-Fraga, A.; Moreno-Espinosa, S.; Roldán-Aragón, Y.; Araujo-Meléndez, J.; Hunsberger, S.; et al. Efficacy and Safety of Nitazoxanide in Addition to Standard of Care for the Treatment of Severe Acute Respiratory Illness. Clin. Infect. Dis. 2019, 69, 1903–1911. [Google Scholar] [CrossRef]
- Piacentini, S.; Riccio, A.; Santopolo, S.; Pauciullo, S.; La Frazia, S.; Rossi, A.; Rossignol, J.F.; Santoro, M.G. The FDA-approved drug nitazoxanide is a potent inhibitor of human seasonal coronaviruses acting at postentry level: Effect on the viral spike glycoprotein. Front. Microbiol. 2023, 14, 1206951. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.S.; Easton, A.J. Coupled translation of the second open reading frame of M2 mRNA is sequence dependent and differs significantly within the subfamily Pneumovirinae. J. Virol. 2007, 81, 8488–8496. [Google Scholar] [CrossRef]
- Bakker, S.E.; Duquerroy, S.; Galloux, M.; Loney, C.; Conner, E.; Eléouët, J.F.; Rey, F.A.; Bhella, D. The Respiratory Syncytial Virus nucleoprotein-rna complex forms a left-handed helical nucleocapsid. J. Gen. Virol. 2013, 94, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Blondot, M.-L.; Dubosclard, V.; Fix, J.; Lassoued, S.; Aumont-Nicaise, M.; Bontems, F.; Eléouët, J.F.; Sizun, C. Structure and functional analysis of the RNA-and viral phosphoprotein-binding domain of Respiratory Syncytial Virus M2-1 protein. PLoS Pathog. 2012, 8, e1002734. [Google Scholar] [CrossRef] [PubMed]
- Hastie, M.L.; Headlam, M.J.; Patel, N.B.; Bukreyev, A.A.; Buchholz, U.J.; Dave, K.A.; Norris, E.L.; Wright, C.L.; Spann, K.M.; Collins, P.L.; et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol. Cell. Proteom. 2012, 11, 108–127.59. [Google Scholar] [CrossRef]
- Pei, J.; Wagner, N.D.; Zou, A.J.; Chatterjee, S.; Borek, D. Structural basis for IFN antagonism by human respiratory syncytial virus nonstructural protein 2. Proc. Natl. Acad. Sci. USA 2021, 118, e2020587118. [Google Scholar] [CrossRef]
- Thornhill, E.M.; Verhoeven, D. Respiratory syncytial virus’s non-structural proteins: Masters of interference. Front. Cell. Infect. Microbiol. 2020, 10, 225. [Google Scholar] [CrossRef]
- Low, K.W.; Tan, T.; Ng, K.; Tan, B.H.; Sugrue, R.J. The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem. Biophys. Res. Commun. 2008, 366, 308–313. [Google Scholar] [CrossRef]
- Huong, T.N.; Lee, Z.Q.; Lai, S.K.; Lee, H.Y.; Tan, B.H.; Sugrue, R.J. Evidence that an interaction between the respiratory syncytial virus F and G proteins at the distal ends of virus filaments mediates efficient multiple cycle infection. Virology 2024, 591, 109985. [Google Scholar] [CrossRef]
- Gan, S.W.; Tan, E.; Lin, X.; Yu, D.; Wang, J.; Tan, G.M.; Vararattanavech, A.; Yeo, C.Y.; Soon, C.H.; Soong, T.W.; et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J. Biol. Chem. 2012, 287, 24671–24689. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, T.; Rossey, I.; Sedeyn, K.; Schepens, B.; Saelens, X. How RSV Proteins Join Forces to Overcome the Host Innate Immune Response. Viruses 2022, 14, 419. [Google Scholar] [CrossRef] [PubMed]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Clements-Mann, M.L.; Harris, D.O.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory Syncytial Virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 13961–13966. [Google Scholar] [CrossRef]
- Krzyzaniak, M.A.; Zumstein, M.T.; Gerez, J.A.; Picotti, P.; Helenius, A. Host cell entry of Respiratory Syncytial Virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog. 2013, 9, e1003309. [Google Scholar] [CrossRef] [PubMed]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.M.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 15, 563. [Google Scholar] [CrossRef]
- Noton, S.L.; Fearns, R. Initiation and regulation of paramyxovirus transcription and replication. Virology 2015, 479–480, 545–554. [Google Scholar] [CrossRef]
- Shang, Z.; Tan, S.; Ma, D. Respiratory Syncytial Virus: From pathogenesis to potential therapeutic strategies. Int. J. Biol. Sci. 2021, 17, 4073–4091. [Google Scholar] [CrossRef]
- Vanover, D.; Smith, D.V.; Blanchard, E.L.; Alonas, E.; Kirschman, J.L.; Lifland, A.W.; Zurla, C.; Santangelo, P.J. RSV glycoprotein and genomic RNA dynamics reveal filament assembly prior to the plasma membrane. Nat. Commun. 2017, 8, 667. [Google Scholar] [CrossRef]
- Lewis, F.A.; Rae, M.L.; Lehmann, N.I.; Ferris, A.A. A syncytial virus associated with epidemic disease of the lower respiratory tract in infants and young children. Med. J. Aust. 1961, 2, 932–933. [Google Scholar] [CrossRef]
- Ahani, B.; Tuffy, K.M.; Aksyuk, A.A.; Wilkins, D.; Abram, M.E.; Dagan, R.; Domachowske, J.B.; Guest, J.D.; Ji, H.; Kushnir, A.; et al. Molecular and phenotypic characteristics of RSV infections in infants during two nirsevimab randomized clinical trials. Nat. Commun. 2024, 15, 3026. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lu, B.; Mctamney, P.; Palaszynski, S.; Diallo, S.; Ren, K.; Ulbrandt, N.D.; Kallewaard, N.; Wang, W.; Fernandes, F.; et al. Prevalence and significance of substitutions in the fusion protein of Respiratory Syncytial Virus resulting in neutralization escape from antibody MEDI8897. J. Infect. Dis. 2018, 218, 572–580. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (NCBI) [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 1988. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 19 May 2025).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2025 update. Nucleic Acids Res. 2025, 53, D1516–D1525. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-4: Maestro; Schrödinger LLC: New York, NY, USA, 2020.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Orlando, G.; Raimondi, D.; Codicè, F.; Tabaro, F.; Vranken, W. Prediction of disordered regions in proteins with recurrent neural networks and protein dynamics. J. Mol. Biol. 2022, 434, 167579. [Google Scholar] [CrossRef]
- Swanson, K.A.; Settembre, E.C.; Shaw, C.A.; Dey, A.K.; Rappuoli, R.; Mandl, C.W.; Dormitzer, P.R.; Carfi, A. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. USA 2011, 108, 9619–9624. [Google Scholar] [CrossRef]
- Roymans, D.; De Bondt, H.L.; Arnoult, E.; Geluykens, P.; Gevers, T.; Van Ginderen, M.; Verheyen, N.; Kim, H.; Willebrords, R.; Bonfanti, J.F.; et al. Binding of a potent small-molecule inhibitor of six-helix bundle formation requires interactions with both heptad-repeats of the RSV fusion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 308–313. [Google Scholar] [CrossRef]
- Leemans, A.; Boeren, M.; Van der Gucht, W.; Martinet, W.; Caljon, G.; Maes, L.; Cos, P.; Delputte, P. Characterization of the role of N-glycosylation sites in the respiratory syncytial virus fusion protein in virus replication, syncytium formation and antigenicity. Virus Res. 2019, 266, 58–68. [Google Scholar] [CrossRef]
- Hotard, A.L.; Lee, S.; Currier, M.G.; Crowe, J.E.; Sakamoto, K., Jr.; Newcomb, D.C.; Peebles, R.S.; Plemper, R.K., Jr.; Moore, M.L. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J. Virol. 2015, 89, 512–522. [Google Scholar] [CrossRef]
- Cabán, M.; Rodarte, J.V.; Bibby, M.; Gray, M.D.; Taylor, J.J.; Pancera, M.; Boonyaratanakornkit, J. Cross-protective antibodies against common endemic respiratory viruses. Nat. Commun. 2023, 14, 798. [Google Scholar] [CrossRef]
- Walsh, E.E.; Hruska, J. Monoclonal antibodies to Respiratory Syncytial Virus proteins: Identification of the fusion protein. J. Virol. 1983, 47, 171–177. [Google Scholar] [CrossRef]
- McLellan, J.S. Neutralizing epitopes on the respiratory syncytial virus fusion glycoprotein. Cur. Opin. Virol. 2015, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Alansari, K.; Toaimah, F.H.; Almatar, D.H.; El Tatawy, D.H.; Davidson, B.L.; Qusad, M.I.M. Monoclonal antibody treatment of RSV bronchiolitis in young infants: A randomized trial. Pediatrics 2019, 143, e20182308. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat. Struct. Mol. Biol. 2010, 17, 248–250. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, S.; Galway, N.; Shields, M.D.; Mallett, P.; Groves, H.E. Review of the Safety, Efficacy and Tolerability of Palivizumab in the Prevention of Severe Respiratory Syncytial Virus (RSV) Disease. Drug Healthc. Patient Saf. 2023, 15, 103–112. [Google Scholar] [CrossRef]
- Zhu, Q.; Mcauliffe, J.M.; Patel, N.K.; Palmer-Hill, F.J.; Yang, C.-F.; Liang, B.; Su, L.; Zhu, W.; Wachter, L.; Wilson, S.; et al. Analysis of Respiratory Syncytial Virus preclinical and clinical variants resistant to neutralization by monoclonal antibodies palivizumab and/or motavizumab. J. Infect. Dis. 2011, 203, 674–682. [Google Scholar] [CrossRef]
- Perron, M.; Stray, K.; Kinkade, A.; Theodore, D.; Lee, G.; Eisenberg, E.; Sangi, M.; Gilbert, B.E.; Jordan, R.; Piedra, P.A.; et al. GS-5806 inhibits a broad range of Respiratory Syncytial Virus clinical isolates by blocking the virus-cell fusion process. Antimicrob. Agents Chemother. 2016, 60, 1264–1273. [Google Scholar] [CrossRef]
- Stray, K.; Perron, M.; Porter, D.P.; Anderson, F.; Lewis, S.A.; Perry, J.; Miller, M.; Cihlar, T.; Devincenzo, J.; Chien, J.W.; et al. Drug resistance assessment following administration of Respiratory Syncytial Virus (RSV) fusion inhibitor presatovir to participants experimentally infected with RSV. J. Infect. Dis. 2020, 222, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Patel, N.K.; McAuliffe, J.M.; Zhu, W.; Wachter, L.; McCarthy, M.P.; Suzich, J.A. Natural Polymorphisms and resistance-associated mutations in the fusion protein of Respiratory Syncytial Virus (RSV): Effects on RSV susceptibility to palivizumab. J. Infect. Dis. 2012, 205, 635–638. [Google Scholar] [CrossRef]
- Hashimoto, K.; Hosoya, M. Neutralizing epitopes of RSV and palivizumab resistance in Japan. J. Med. Sci. 2017, 63, 127–134. [Google Scholar] [CrossRef]
- Tang, W.; Li, Y.; Song, Q.; Wang, Z.; Li, M.; Zhang, Q.; Wang, Y.; Ye, W.; Li, Y. Mechanism of cross-resistance to fusion inhibitors conferred by the K394R mutation in Respiratory Syncytial Virus Fusion protein. J. Virol. 2021, 95, e0120521. [Google Scholar] [CrossRef]
- Yoshida, I.; Arikawa, K.; Honma, Y.; Inatani, S.; Yoshinaga, M.; Sugiyama, H. Pharmacological characterization of TP0591816, a novel macrocyclic Respiratory Syncytial Virus fusion inhibitor with antiviral activity against F protein mutants. Antimicrob. Agents Chemother. 2021, 65, e1407–e01420. [Google Scholar] [CrossRef] [PubMed]
- Rhodin, M.H.J.; McAllister, N.V.; Castillo, J.; Noton, S.L.; Fearns, R.; Kim, I.J.; Yu, J.; Blaisdell, T.P.; Panarese, J.; Shook, B.C.; et al. EDP-938, a novel nucleoprotein inhibitor of Respiratory Syncytial Virus, demonstrates potent antiviral activities in vitro and in a non-human primate model. PLoS Pathog. 2021, 17, e1009428. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Hong, J.; Wang, G.; Taylor, J.; Smith, L.K.; Fung, A.; Stevens, S.K.; Liu, H.; Jin, Z.; Dyatkina, N.; et al. Molecular Basis for the selective inhibition of Respiratory Syncytial Virus RNA polymerase by 2′-fluoro-4′-chloromethyl-cytidine triphosphate. PLoS Pathog. 2015, 11, e1004995. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Day, N.D.; Branigan, P.J.; Gutshall, L.L.; Sarisky, R.T.; Del Vecchio, A.M. Relationship between the loss of neutralizing antibody binding and fusion activity of the F protein of human Respiratory Syncytial Virus. Virol. J. 2007, 4, 71. [Google Scholar] [CrossRef]
- Fedechkin, S.O.; George, N.L.; Wolff, J.T.; Kauvar, L.M.; DuBois, R.M. Structures of respiratory syncytial virus G antigen bound to broadly neutralizing antibodies. Sci. Immunol. 2018, 3, eaar3534. [Google Scholar] [CrossRef]
- Zhu, Q.; McLellan, J.S.; Kallewaard, N.L.; Ulbrandt, N.D.; Palaszynski, S.; Zhang, J.; Moldt, B.; Khan, A.; Svabek, C.; McAuliffe, J.M.; et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci. Transl. Med. 2017, 9, eaaj1928. [Google Scholar] [CrossRef]
- Bates, J.T.; Keefer, C.J.; Slaughter, J.C.; Kulp, D.W.; Schief, W.R.; Crowe, J.E. Escape from neutralization by the Respiratory Syncytial Virus-specific neutralizing monoclonal antibody palivizumab is driven by changes in on-rate of binding to the fusion protein. Virology 2014, 454–455, 139–144. [Google Scholar] [CrossRef]
- Battles, M.B.; Langedijk, J.P.; Furmanova-Hollenstein, P.; Chaiwatpongsakorn, S.; Costello, H.M.; Kwanten, L.; Vranckx, L.; Vink, P.; Jaensch, S.; Jonckers, T.H.M.; et al. Molecular mechanism of Respiratory Syncytial Virus fusion inhibitors. Nat. Chem. Biol. 2016, 12, 87–93. [Google Scholar] [CrossRef]
- Chemaly, R.F.; Dadwal, S.S.; Bergeron, A.; Ljungman, P.; Kim, Y.J.; Cheng, G.S.; Pipavath, S.N.; Limaye, A.P.; Blanchard, E.; Winston, D.J.; et al. A phase 2, randomized, double-blind, placebo-controlled trial of presatovir for the treatment of Respiratory Syncytial Virus upper respiratory tract infection in hematopoietic-cell transplant recipients. Clin. Infect. Dis. 2020, 71, 2777–2786. [Google Scholar] [CrossRef]
- Vendeville, S.; Tahri, A.; Hu, L.; Demin, S.; Cooymans, L.; Vos, A.; Kwanten, L.; Van Den Berg, J.; Battles, M.B.; Mclellan, J.S.; et al. Discovery of 3-({5-Chloro-1-[3-(methylsulfonylpropyl]-1 H-indol-2-Yl}methyl)-1-(2,2,2-trifluoroethyl)-1,3-dihydro-2 H-imidazo [4,5- c]pyridin-2-one (JNJ-53718678), a potent and orally bioavailable fusion inhibitor of Respiratory Syncytial Virus. J. Med. Chem. 2020, 63, 8046–8058. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Gao, L.; Wang, L.; Liang, C.; Wang, B.; Liu, Y.; Feng, S.; Zhang, B.; Zhou, M.; Yu, X.; et al. Discovery of ziresovir as a potent, selective, and orally bioavailable Respiratory Syncytial Virus fusion protein inhibitor. J. Med. Chem. 2019, 62, 6003–6014. [Google Scholar] [CrossRef]
- Song, Q.; Zhu, H.; Qiu, M.; Cai, J.; Hu, Y.; Yang, H.; Rao, S.; Li, Y.; Li, M.; Hu, L.; et al. A new mechanism of respiratory syncytial virus entry inhibition by small-molecule to overcome K394R-associated resistance. mBio 2024, 15, e0138524. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Panis, M.L.; Ho, E.; Lin, K.-Y.; Krawczyk, S.H.; Grant, D.M.; Cai, R.; Swaminathan, S.; Cihlar, T. Inhibition of Respiratory Syncytial Virus fusion by the small molecule VP-14637 via specific interactions with F protein. J. Virol. 2003, 77, 5054–5064. [Google Scholar] [CrossRef]
- Amaral, M.; Kokh, D.B.; Bomke, J.; Wegener, A.; Buchstaller, H.P.; Eggenweiler, H.M.; Matias, P.; Sirrenberg, C.; Wade, R.C.; Frech, M. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017, 8, 2276. [Google Scholar] [CrossRef]
- Purohit, R.; Rajasekaran, R.; Sudandiradoss, C.; George Priya Doss, C.; Ramanathan, K.; Rao, S. Studies on flexibility and binding affinity of Asp25 of HIV-1 protease mutants. Int. J. Biol. Macromol. 2008, 42, 386–891. [Google Scholar] [CrossRef]
- Fiorani, P.; Bruselles, A.; Falconi, M.; Chillemi, G.; Desideri, A.; Benedetti, P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003, 278, 43268–43275. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Tata, R.B.; Tastan Bishop, Ö. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef]
- Braz, A.S.; Tufanetto, P.; Perahia, D.; Scott, L.P. Relation between flexibility and positively selected HIV-1 protease mutants against inhibitors. Proteins 2012, 80, 2680–2691. [Google Scholar] [CrossRef]
- Kagami, L.P.; Orlando, G.; Raimondi, D.; Ancien, F.; Dixit, B.; Gavaldá-Garciá, J.; Ramasamy, P.; Roca-Martínez, J.; Tzavella, K.; Vranken, W. B2bTools: Online predictions for protein biophysical features and their conservation. Nucleic Acids Res. 2021, 49, W52–W59. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Tarus, B.; Blazevic, I.; Fix, J.; Duquerroy, S.; Eléouët, J.-F. Characterization of a viral phosphoprotein binding site on the surface of the Respiratory Syncytial nucleoprotein. J. Virol. 2012, 86, 8375–8387. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Gao, Y.; Liang, B. Structural Insights into the Respiratory Syncytial Virus RNA Synthesis Complexes. Viruses 2021, 13, 834. [Google Scholar] [CrossRef]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagne, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of Respiratory Syncytial Virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef]
- Ouizougun-Oubari, M.; Pereira, N.; Tarus, B.; Galloux, M.; Lassoued, S.; Fix, J.; Tortorici, M.A.; Hoos, S.; Baron, B.; England, P.; et al. A druggable pocket at the nucleocapsid/phosphoprotein interaction site of human Respiratory Syncytial Virus. J. Virol. 2015, 89, 11129–11143. [Google Scholar] [CrossRef]
- Bonneux, B.; Jacoby, E.; Ceconi, M.; Stobbelaar, K.; Delputte, P.; Herschke, F. Direct-acting antivirals for RSV treatment, a review. Antivir. Res. 2024, 229, 105948. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Jadhao, S.J.; Paden, C.R.; Tong, S. Functional features of the Respiratory Syncytial Virus G protein. Viruses 2021, 13, 1214. [Google Scholar] [CrossRef]
- Kauvar, L.M.; Harcourt, J.L.; Haynes, L.M.; Tripp, R.A. Therapeutic targeting of Respiratory Syncytial Virus G-protein. Immunotherapy 2010, 2, 655–661. [Google Scholar] [CrossRef]
- Borochova, K.; Niespodziana, K.; Hammar, K.S.; van Hage, M.; Hedlin, G.; Söderhäll, C.; Focke-Tejkl, M.; Valenta, R. Features of the human antibody response against the Respiratory Syncytial Virus surface glycoprotein G. Vaccines 2020, 8, 337. [Google Scholar] [CrossRef]
- Chirkova, T.; Lin, S.; Oomens, A.G.P.; Gaston, K.A.; Boyoglu-Barnum, S.; Meng, J.; Stobart, C.C.; Cotton, C.U.; Hartert, T.V.; Moore, M.L.; et al. CX3CR1 is an important surface molecule for Respiratory Syncytial Virus infection in human airway epithelial cells. J. Gen. Virol. 2015, 96, 2543–2556. [Google Scholar] [CrossRef]
- Feldman, S.A.; Hendry, R.M.; Beeler, J.A. Identification of a linear heparin binding domain for human Respiratory Syncytial Virus attachment glycoprotein G. J. Virol. 1999, 73, 6610–6617. [Google Scholar] [CrossRef]
- Bukreyev, A.; Yang, L.; Fricke, J.; Cheng, L.; Ward, J.M.; Murphy, B.R.; Collins, P.L. The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on Fc receptor-bearing leukocytes. J. Virol. 2008, 82, 12191–12204. [Google Scholar] [CrossRef]
- Boyoglu-Barnum, S.; Todd, S.O.; Chirkova, T.; Barnum, T.R.; Gaston, K.A.; Haynes, L.M.; Tripp, R.A.; Moore, M.L.; Anderson, L.J. An anti-G protein monoclonal antibody treats RSV disease more effectively than an anti-F monoclonal antibody in BALB/c mice. Virology 2015, 483, 117–125. [Google Scholar] [CrossRef]
- Radu, G.U.; Caidi, H.; Miao, C.; Tripp, R.A.; Anderson, L.J.; Haynes, L.M. Prophylactic treatment with a G glycoprotein monoclonal antibody reduces pulmonary inflammation in Respiratory Syncytial Virus (RSV)-challenged naïve and formalin-inactivated RSV-immunized BALB/c mice. J. Virol. 2010, 84, 9632–9636. [Google Scholar] [CrossRef]
- Caidi, H.; Miao, C.; Thornburg, N.J.; Tripp, R.A.; Anderson, L.J.; Haynes, L.M. Anti-Respiratory Syncytial Virus (RSV) G monoclonal antibodies reduce lung inflammation and viral lung titers when delivered therapeutically in a BALB/c mouse model. Antivir. Res. 2018, 154, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M.; Caidi, H.; Radu, G.U.; Miao, C.; Harcourt, J.L.; Tripp, R.A.; Anderson, L.J. Therapeutic monoclonal antibody treatment targeting Respiratory Syncytial Virus (RSV) G protein mediates viral clearance and reduces the pathogenesis of RSV infection in BALB/c mice. J. Infect. Dis. 2009, 200, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, Y.; Burdick, R.C.; Patro, S.C.; Penrose, K.J.; Clark, E.; Ablan, S.D.; Mellors, J.W.; Parikh, U.M.; Wu, X.; Pathak, V.K.; et al. Elucidating the Mechanism by Which Nucleocapsid Mutations Confer Resistance to InSTIs. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, USA, 9–12 March 2025. Abstract Number 155. [Google Scholar]
- Gilman, M.S.A.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eléouët, J.F.; et al. Structure of the Respiratory Syncytial Virus polymerase complex. Cell 2019, 179, 193–204.e14. [Google Scholar] [CrossRef] [PubMed]
- Valle, C.; Martin, B.; Debart, F.; Vasseur, J.-J.; Imbert, I.; Canard, B.; Coutard, B.; Decroly, E. The C-terminal domain of the Sudan Ebolavirus L protein is essential for RNA binding and methylation. J. Virol. 2020, 94, e00520-20. [Google Scholar] [CrossRef]
- Sutto-Ortiz, P.; Eléouët, J.F.; Ferron, F.; Decroly, E. Biochemistry of the Respiratory Syncytial Virus L protein embedding RNA polymerase and capping activities. Viruses 2023, 15, 341. [Google Scholar] [CrossRef]
- Cao, D.; Gao, Y.; Roesler, C.; Rice, S.; D’Cunha, P.; Zhuang, L.; Slack, J.; Domke, M.; Antonova, A.; Romanelli, S.; et al. Cryo-EM structure of the Respiratory Syncytial Virus RNA polymerase. Nat. Commun. 2020, 11, 368. [Google Scholar] [CrossRef]
- Yu, X.; Abeywickrema, P.; Bonneux, B.; Behera, I.; Anson, B.; Jacoby, E.; Fung, A.; Adhikary, S.; Bhaumik, A.; Carbajo, R.J.; et al. Structural and mechanistic insights into the inhibition of Respiratory Syncytial Virus polymerase by a non-nucleoside inhibitor. Commun. Biol. 2023, 6, 1074. [Google Scholar] [CrossRef]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef]
- Noton, S.L.; Nagendra, K.; Dunn, E.F.; Mawhorter, M.E.; Yu, Q.; Fearns, R. Respiratory Syncytial Virus inhibitor AZ-27 differentially inhibits different polymerase activities at the promoter. J. Virol. 2015, 89, 7786–7798. [Google Scholar] [CrossRef] [PubMed]
- Tiong-Yip, C.L.; Aschenbrenner, L.; Johnson, K.D.; McLaughlin, R.E.; Fan, J.; Challa, S.R.; Xiong, H.; Yu, Q. Characterization of a Respiratory Syncytial Virus L protein inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3867–3873. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Kirkpatrick, C.M.; Nieforth, K.A.; Chanda, S.; Zhang, Q.; McClure, M.; Fry, J.; Symons, J.A.; Blatt, L.M.; Beigelman, L.; et al. Respiratory Syncytial Virus-A dynamics and the effects of lumicitabine, a nucleoside viral replication inhibitor, in experimentally infected humans. J. Antimicrob. Chemother. 2019, 74, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Garriga, D.; Ferrer-Orta, C.; Querol-Audí, J.; Oliva, B.; Verdaguer, N. Role of motif B loop in allosteric regulation of RNA-dependent RNA polymerization activity. J. Mol. Biol. 2013, 425, 2279–2287. [Google Scholar] [CrossRef]
- Noton, S.L.; Aljabr, W.; Hiscox, J.A.; Matthews, D.A.; Fearns, R. Factors affecting de novo RNA synthesis and back-priming by the Respiratory Syncytial Virus polymerase. Virology 2014, 462–463, 318–327. [Google Scholar] [CrossRef]
- Deval, J.; Fung, A.; Stevens, S.K.; Jordan, P.C.; Gromova, T.; Taylor, J.S.; Hong, J.; Meng, J.; Wang, G.; Dyatkina, N.; et al. Biochemical effect of resistance mutations against synergistic inhibitors of RSV RNA polymerase. PLoS ONE 2016, 11, e0154097. [Google Scholar] [CrossRef]
- Liang, Y.; Shao, S.; Li, X.Y.; Zhao, Z.X.; Liu, N.; Liu, Z.M.; Shen, F.J.; Zhang, H.; Hou, J.W.; Zhang, X.F.; et al. Mutating a flexible region of the RSV F protein can stabilize the prefusion conformation. Science 2024, 385, 1484–1491. [Google Scholar] [CrossRef]
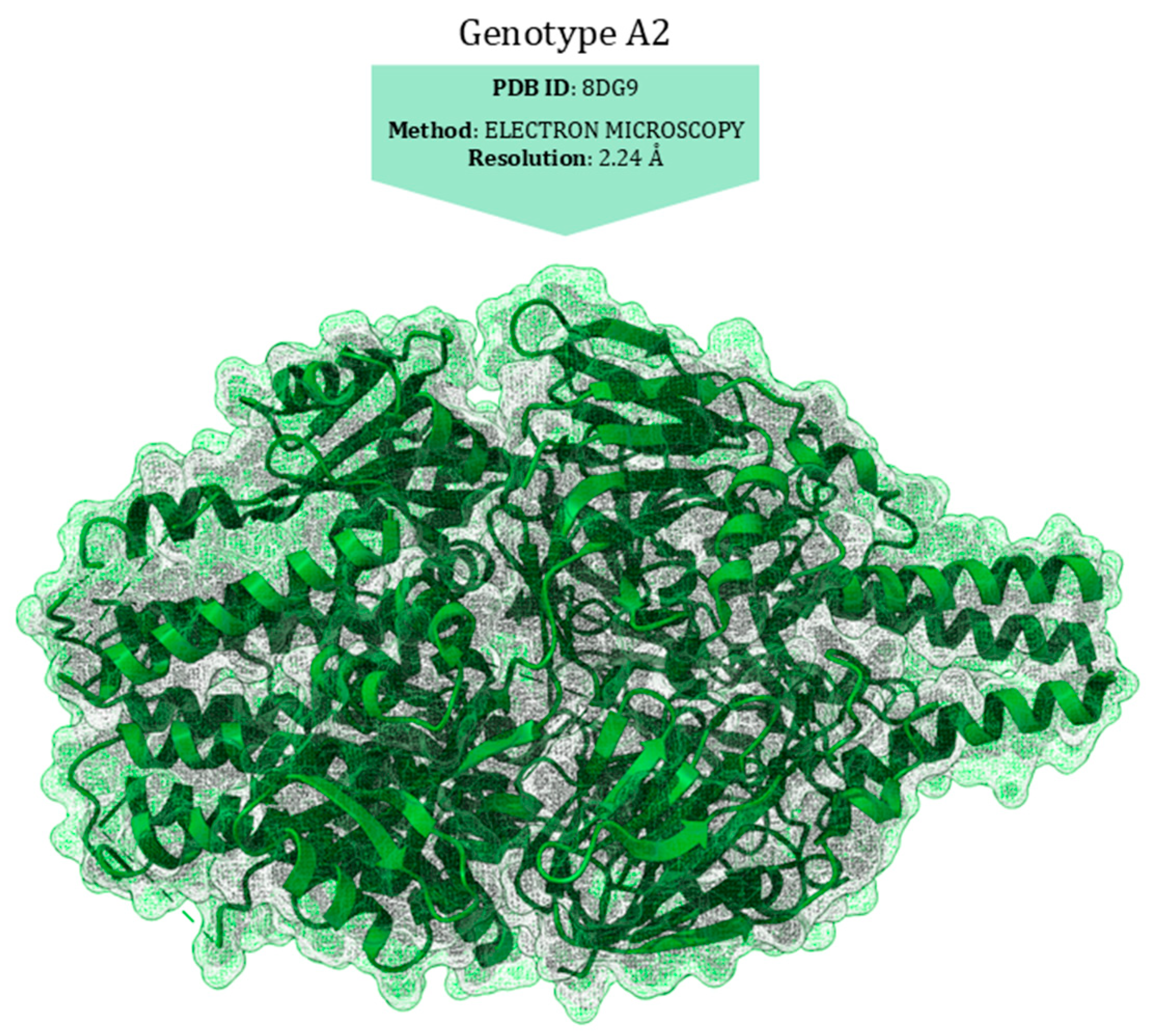


| Genotype A2 | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| N208 | 0.863 | |
| Y208 | 0.938 | +75 |
| N67 | 0.812 | |
| I67 | 0.892 | +80 |
| N262 | 0.852 | |
| Y262 | 0.937 | +85 |
| D262 | 0.888 | +36 |
| K272 | 0.814 | |
| T272 | 0.880 | +66 |
| Q272 | 0.825 | +11 |
| S275 | 0.807 | |
| L275 | 0.896 | +89 |
| F275 | 0.897 | +90 |
| L138 | 0.896 | |
| F138 | 0.909 | +13 |
| F140 | 0.881 | |
| L140 | 0.888 | +7 |
| L141 | 0.864 | |
| F141 | 0.873 | +9 |
| K394 | 0.920 | |
| R394 | 0.957 | +37 |
| K399 | 0.826 | |
| I399 | 0.881 | +55 |
| T400 | 0.832 | |
| A400 | 0.878 | +46 |
| I400 | 0.878 | +46 |
| D486 | 0.804 | |
| N486 | 0.781 | −23 |
| E487 | 0.812 | |
| D487 | 0.803 | −9 |
| F488 | 0.827 | |
| Y488 | 0.820 | −7 |
| N517 | 0.794 | |
| I517 | 0.874 | +80 |
| Genotype B | ||
| AA | Backbone Dynamics | Δ WT/Mutant |
| K272 | 0.802 | |
| E272 | 0.813 | +11 |
| Q272 | 0.799 | −3 |
| N272 | 0.778 | −24 |
| M272 | 0.818 | +16 |
| S276 | 0.797 | |
| N276 | 0.807 | +10 |
| F488 | 0.827 | |
| L488 | 0.809 | −18 |
| S488 | 0.738 | −89 |
| Genotype B9320 | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| K68 | 0.821 | |
| N68 | 0.797 | −24 |
| N201 | 0.876 | |
| S201 | 0.861 | −25 |
| N208 | 0.872 | |
| D208 | 0.896 | +24 |
| S208 | 0.863 | −9 |
| Genotype A2 | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| T29 | 0.666 | |
| S29 | 0.625 | −41 |
| S134 | 0.816 | |
| T134 | 0.857 | +41 |
| M109 | 0.844 | |
| T109 | 0.837 | −7 |
| I109 | 0.883 | +39 |
| K109 | 0.827 | −17 |
| Q102 | 0.830 | |
| L102 | 0.873 | +43 |
| I129 | 0.868 | |
| M129 | 0.828 | −40 |
| K136 | 0.818 | |
| R136 | 0.822 | +4 |
| Genotype B | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| M109 | 0.849 | |
| T109 | 0.843 | −6 |
| L139 | 0.792 | |
| Q139 | 0.794 | +2 |
| Genotype A2 | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| K205 | 0.671 | |
| G205 | 0.614 | −57 |
| R8 | 0.756 | |
| H8 | 0.748 | −8 |
| K213 | 0.635 | |
| G213 | 0.586 | −49 |
| T219 | 0.627 | |
| A219 | 0.626 | −1 |
| Genotype A2 | ||
|---|---|---|
| AA | Backbone Dynamics | Δ WT/Mutant |
| A789 | 1017 | |
| V789 | 1053 | +36 |
| M628 | 0.819 | |
| L628 | 0.842 | +23 |
| I796 | 0.959 | |
| V796 | 0.980 | +21 |
| L795 | 0.989 | |
| I795 | 1008 | +19 |
| Y1631 | 0.914 | |
| H1631 | 0.859 | −55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magnapera, A.; Riccio, A.; Curcio, A.; Tramontozzi, C.; Piermatteo, L.; D’Anna, S.; Alcaro, S.; Alteri, C.; La Frazia, S.; Artese, A.; et al. Insights into the Currently Available Drugs and Investigational Compounds Against RSV with a Focus on Their Drug-Resistance Profiles. Viruses 2025, 17, 793. https://doi.org/10.3390/v17060793
Magnapera A, Riccio A, Curcio A, Tramontozzi C, Piermatteo L, D’Anna S, Alcaro S, Alteri C, La Frazia S, Artese A, et al. Insights into the Currently Available Drugs and Investigational Compounds Against RSV with a Focus on Their Drug-Resistance Profiles. Viruses. 2025; 17(6):793. https://doi.org/10.3390/v17060793
Chicago/Turabian StyleMagnapera, Alessia, Anna Riccio, Antonio Curcio, Caterina Tramontozzi, Lorenzo Piermatteo, Stefano D’Anna, Stefano Alcaro, Claudia Alteri, Simone La Frazia, Anna Artese, and et al. 2025. "Insights into the Currently Available Drugs and Investigational Compounds Against RSV with a Focus on Their Drug-Resistance Profiles" Viruses 17, no. 6: 793. https://doi.org/10.3390/v17060793
APA StyleMagnapera, A., Riccio, A., Curcio, A., Tramontozzi, C., Piermatteo, L., D’Anna, S., Alcaro, S., Alteri, C., La Frazia, S., Artese, A., Salpini, R., & Svicher, V. (2025). Insights into the Currently Available Drugs and Investigational Compounds Against RSV with a Focus on Their Drug-Resistance Profiles. Viruses, 17(6), 793. https://doi.org/10.3390/v17060793