
Wastewater-Based Surveillance of Human Adenoviruses in Italy: Quantification by Digital PCR and Molecular Typing via Nanopore Amplicon Sequencing
 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, Viral Concentration, and RNA Extraction
2.2. Quantification of HAdV by dPCR Assay
2.3. Nested PCR Assays and Sanger Sequencing
2.4. Next-Generation Sequencing
2.5. Statistical and Bioinformatic Analyzes
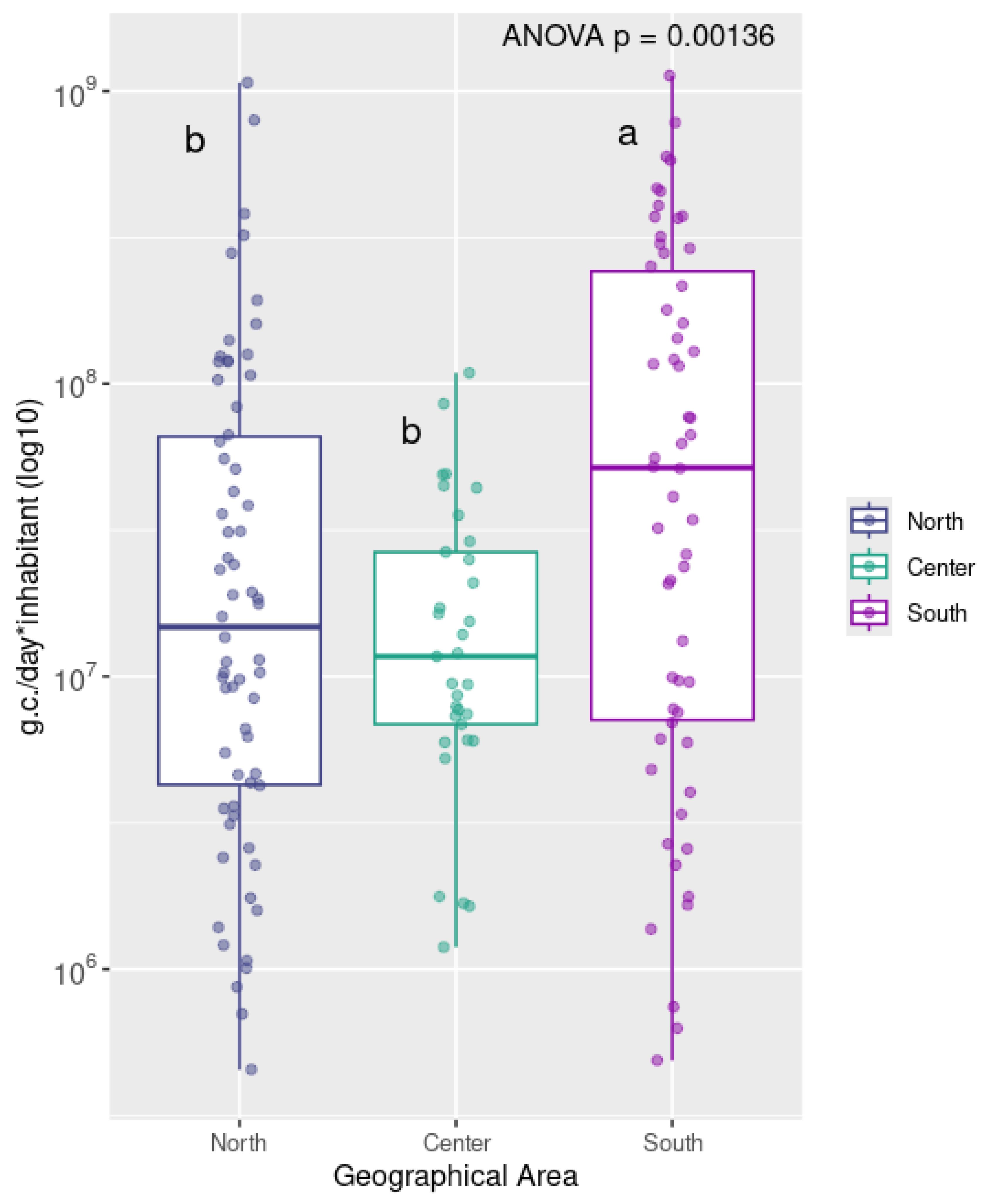
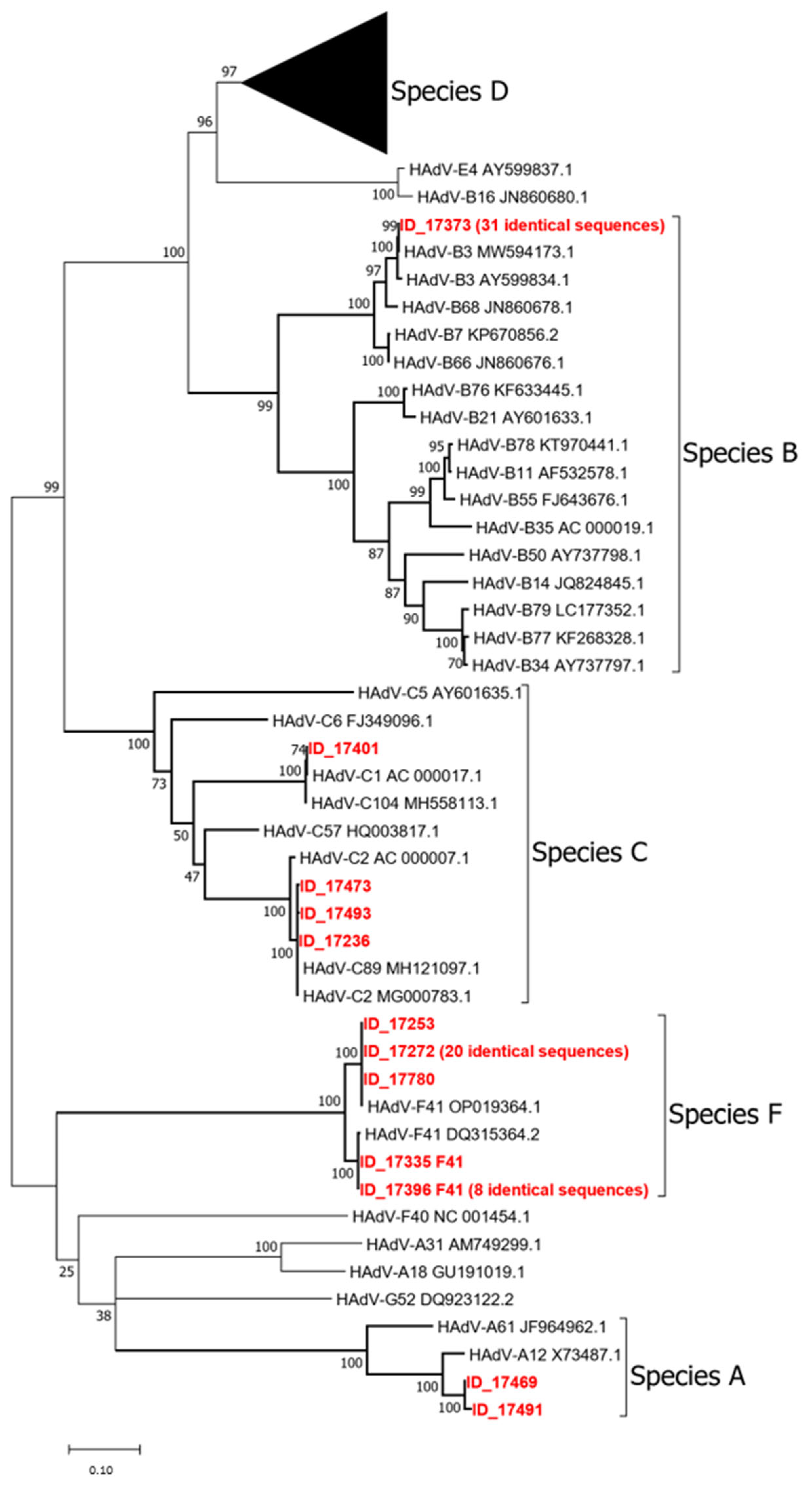
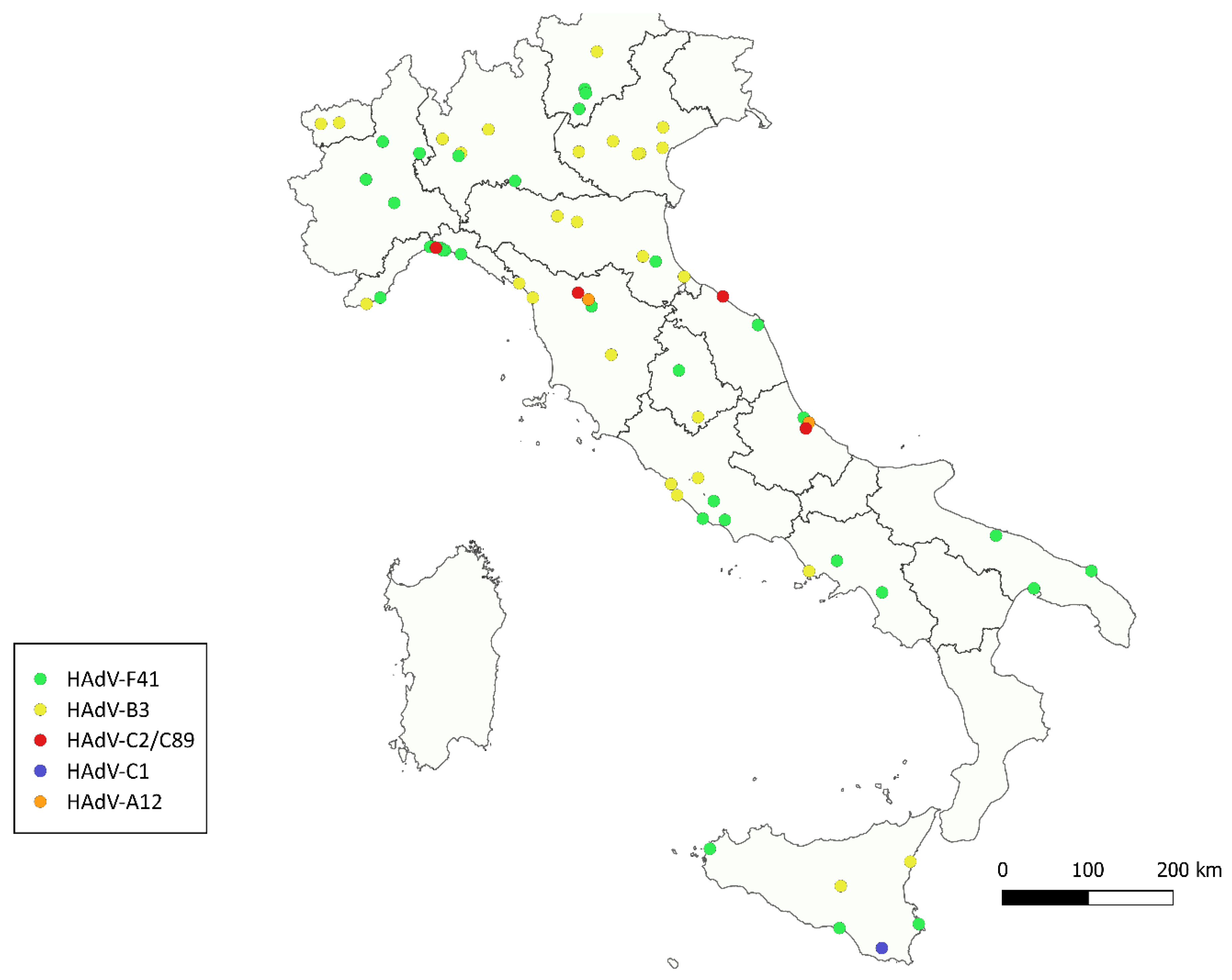
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J.; Echavarría, M.; Hess, M.; Jones, M.S.; Kaján, G.L.; Kajon, A.E.; Mittal, S.K.; et al. ICTV Virus Taxonomy Profile: Adenoviridae 2022. J. Gen. Virol. 2022, 103, 001721. [Google Scholar] [CrossRef]
- La Rosa, G.; Suffredini, E. Adenovirus. In Handbook of Foodborne Diseases; CRC Press: Boca Raton, FL, USA, 2018; pp. 13–24. [Google Scholar] [CrossRef]
- Kajon, A.E. Adenovirus infections: New insights for the clinical laboratory. J. Clin. Microbiol. 2024, 62, e0083622. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control (CDC). Outbreak of pharyngoconjunctival fever at a summer camp—North Carolina, 1991. MMWR Morb. Mortal. Wkly. Rep. 1992, 41, 342–344. [Google Scholar]
- Lessa, F.C.; Gould, P.L.; Pascoe, N.; Erdman, D.D.; Lu, X.; Bunning, M.L.; Marconi, V.C.; Lott, L.; Widdowson, M.A.; Anderson, L.J.; et al. Health care transmission of a newly emergent adenovirus serotype in health care personnel at a military hospital in Texas, 2007. J. Infect. Dis. 2009, 200, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.P.; Marquez, J.L.; Nord, J.; Stamper, A.R.; Edwards, E.A.; Valentini, N.; Frank, C.J.; Washer, L.L.; Ernst, R.D.; Park, J.I.; et al. Detection of a Human Adenovirus Outbreak, Including Some Critical Infections, Using Multipathogen Testing at a Large University, September 2022–January 2023. Open Forum Infect. Dis. 2024, 11, ofae192. [Google Scholar] [CrossRef]
- WHO. Adenoviruses: Background Document for the WHO Guidelines for Drinking Water Quality and the WHO Guidelines on Sanitation and Health; Drinking-water and sanitation related pathogens series; World Health Organization: Geneva, Switzerland, 2025. [Google Scholar] [CrossRef]
- Enriquez, C.E.; Hurst, C.J.; Gerba, C.P. Survival of the enteric adenoviruses 40 and 41 in tap, sea, and waste water. Water Res. 1995, 29, 2548–2553. [Google Scholar] [CrossRef]
- Jiang, S.C. Human Adenoviruses in Water: Occurrence and Health Implications: A Critical Review. Environ. Sci. Technol. 2006, 40, 7132–7140. [Google Scholar] [CrossRef]
- Takuissu, G.R.; Kenmoe, S.; Ebogo-Belobo, J.T.; Kengne-Ndé, C.; Mbaga, D.S.; Bowo-Ngandji, A.; Ndzie, J.L.O.; Kenfack-Momo, R.; Tchatchouang, S.; Kenfack-Zanguim, J.; et al. Exploring adenovirus in water environments: A systematic review and meta-analysis. Int. J. Environ. Health Res. 2024, 34, 2504–2516. [Google Scholar] [CrossRef]
- Mena, K.D.; Gerba, C.P. Waterborne adenovirus. Rev. Environ. Contam. Toxicol. 2009, 198, 133–167. [Google Scholar] [CrossRef]
- Bonadonna, L.; La Rosa, G. A Review and Update on Waterborne Viral Diseases Associated with Swimming Pools. Int. J. Environ. Res. Public Health 2019, 16, 166. [Google Scholar] [CrossRef]
- Iaconelli, M.; Muscillo, M.; Della Libera, S.; Fratini, M.; Meucci, L.; De Ceglia, M.; Giacosa, D.; La Rosa, G. One-year Surveillance of Human Enteric Viruses in Raw and Treated Wastewaters, Downstream River Waters, and Drinking Waters. Food Environ. Virol. 2017, 9, 79–88. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Brandtner, D.; Mancini, P.; Veneri, C.; Ferraro, G.B.; Bonadonna, L.; Lucentini, L.; Suffredini, E. Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology. Water 2021, 13, 2503. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, J.; Xiao, A.; Gu, X.; Lee, W.L.; Armas, F.; Alm, E.J. SARS-CoV-2 titers inwastewater are higher than expected from clinically confirmed cases. mSystems 2020, 5, e00614–e00620. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Pourshaban, M.; Iaconelli, M.; Muscillo, M. Quantitative real-time PCR of enteric viruses in influent and effluent samples from wastewater treatment plants in Italy. Ann. Dell’istituto Super. Sanita 2010, 46, 266–273. [Google Scholar] [CrossRef]
- Lu, X.; Erdman, D.D. Molecular typing of human adenoviruses by PCR and sequencing of a partial region of the hexon gene. Arch. Virol. 2006, 151, 1587–1602. [Google Scholar] [CrossRef]
- Adefisoye, M.A.; Nwodo, U.U.; Green, E.; Okoh, A.I. Quantitative PCR Detection and Characterisation of Human Adenovirus, Rotavirus and Hepatitis A Virus in Discharged Effluents of Two Wastewater Treatment Facilities in the Eastern Cape, South Africa. Food Environ. Virol. 2016, 8, 262–274. [Google Scholar] [CrossRef]
- Osuolale, O.; Okoh, A. Incidence of human adenoviruses and Hepatitis A virus in the final effluent of selected wastewater treatment plants in Eastern Cape Province, South Africa. Virol. J. 2015, 12, 98. [Google Scholar] [CrossRef]
- Ibrahim, C.; Hassen, A.; Pothier, P.; Mejri, S.; Hammami, S. Molecular detection and genotypic characterization of enteric adenoviruses in a hospital wastewater. Environ. Sci. Pollut. Res. 2018, 25, 10977–10987. [Google Scholar] [CrossRef]
- Elmahdy, E.M.; Ahmed, N.I.; Shaheen, M.N.F.; Mohamed, E.-C.B.; Loutfy, S.A. Molecular detection of human adenovirus in urban wastewater in Egypt and among children suffering from acute gastroenteritis. J. Water Health 2019, 17, 287–294. [Google Scholar] [CrossRef]
- Carducci, A.; Battistini, R.; Rovini, E.; Verani, M. Viral Removal by Wastewater Treatment: Monitoring of Indicators and Pathogens. Food Environ. Virol. 2009, 1, 85–91. [Google Scholar] [CrossRef]
- Iaconelli, M.; Valdazo-González, B.; Equestre, M.; Ciccaglione, A.; Marcantonio, C.; Della Libera, S.; La Rosa, G. Molecular characterization of human adenoviruses in urban wastewaters using next generation and Sanger sequencing. Water Res. 2017, 121, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, L.; Renteria, S.C.U.; Ceriotti, F.; Ammoni, E.; Galli, C.; Seiti, A.; Castiglioni, S.; Cereda, D.; Binda, S.; Pariani, E. Wastewater Surveillance Captured an Increase in Adenovirus Circulation in Milan (Italy) during the First Quarter of 2022. Viruses 2022, 14, 2351. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Kim, J.; Lewis, G. Evaluation of methodology for detection of human adenoviruses in wastewater, drinking water, stream water and recreational waters. J. Appl. Microbiol. 2010, 108, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.-T.; Phanikumar, M.S.; Xagoraraki, I.; Rose, J.B. Quantitative Detection of Human Adenoviruses in Wastewater and Combined Sewer Overflows Influencing a Michigan River. Appl. Environ. Microbiol. 2010, 76, 715–723. [Google Scholar] [CrossRef]
- Fumian, T.M.; Vieira, C.B.; Leite, J.P.G.; Miagostovich, M.P. Assessment of burden of virus agents in an urban sewage treatment plant in Rio de Janeiro, Brazil. J. Water Health 2013, 11, 110–119. [Google Scholar] [CrossRef]
- Rodrigues, M.T.; Henzel, A.; Staggemeier, R.; de Quevedo, D.M.; Rigotto, C.; Heinzelmann, L.; Nascimento, C.A.D.; Spilki, F.R. Human adenovirus spread, rainfalls, and the occurrence of gastroenteritis cases in a Brazilian basin. Environ. Monit. Assess. 2015, 187, 720. [Google Scholar] [CrossRef]
- O’Brien, E.; Nakyazze, J.; Wu, H.; Kiwanuka, N.; Cunningham, W.; Kaneene, J.B.; Xagoraraki, I. Viral diversity and abundance in polluted waters in Kampala, Uganda. Water Res. 2017, 127, 41–49. [Google Scholar] [CrossRef]
- Rames, E.; Roiko, A.; Stratton, H.; Macdonald, J. DNA Heat Treatment for Improving qPCR Analysis of Human Adenovirus in Wastewater. Food Environ. Virol. 2017, 9, 354–357. [Google Scholar] [CrossRef]
- Ogorzaly, L.; Walczak, C.; Galloux, M.; Etienne, S.; Gassilloud, B.; Cauchie, H.M. Human Adenovirus Diversity in Water Samples Using a Next-Generation Amplicon Sequencing Approach. Food Environ. Virol. 2015, 7, 112–121. [Google Scholar] [CrossRef]
- Khales, P.; Razizadeh, M.H.; Ghorbani, S.; Moattari, A.; Sarvari, J.; Saadati, H.; Sayyahfar, S.; Salavatiha, Z.; Hasanabad, M.H.; Poortahmasebi, V.; et al. Human adenoviruses in children with gastroenteritis: A systematic review and meta-analysis. BMC Infect. Dis. 2024, 24, 478. [Google Scholar] [CrossRef]
- Nascimento, L.G.D.; Fialho, A.M.; Andrade, J.d.S.R.d.; de Assis, R.M.S.; Fumian, T.M. Human enteric adenovirus F40/41 as a major cause of acute gastroenteritis in children in Brazil, 2018 to 2020. Sci. Rep. 2022, 12, 11220. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.A.; Gonzalez, G.; Reynolds, L.J.; Bennett, C.; Campbell, C.; Nolan, T.M.; Byrne, A.; Fennema, S.; Holohan, N.; Kuntamukkula, S.R.; et al. Adeno-Associated Virus 2 and Human Adenovirus F41 in Wastewater during Outbreak of Severe Acute Hepatitis in Children, Ireland. Emerg. Infect. Dis. 2023, 29, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Lambisia, A.W.; Makori, T.O.; Mutunga, M.; Cheruiyot, R.; Murunga, N.; Quick, J.; Githinji, G.; Nokes, D.J.; Houldcroft, C.J.; Agoti, C.N. Genomic epidemiology of human adenovirus F40 and F41 in coastal Kenya: A retrospective hospital-based surveillance study (2013–2022). Virus Evol. 2023, 9, vead023. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Hiroi, S.; Hirai, Y.; Kaida, A. Prevalence of Human Adenovirus Type 3 Associated with Pharyngoconjunctival Fever in Children in Osaka, Japan during and after the COVID-19 Pandemic. Jpn. J. Infect. Dis. 2024, 77, 292–295. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Wu, K.; Wang, C.; Liu, W.; Zhu, Y.; Ye, L.; Li, X.; Zhou, R.; Zhu, B.; et al. An Outbreak of Human Adenovirus Infection Among Children Post COVID-19 Pandemic in Southern China. J. Med. Virol. 2024, 96, e70139. [Google Scholar] [CrossRef]
- Giardina, F.A.; Pellegrinelli, L.; Novazzi, F.; Vian, E.; Biscaro, V.; Russo, C.; Ranno, S.; Pagani, E.; Masi, E.; Tiberio, C.; et al. Epidemiological impact of human adenovirus as causative agent of respiratory infections: An Italian multicentre retrospective study, 2022–2023. J. Infect. Chemother. 2024, 30, 1097–1103. [Google Scholar] [CrossRef]
- Payne, S.B.; Grilli, E.A.; Smith, A.J.; Hoskins, T.W. Investigation of an outbreak of adenovirus type 3 infection in a boys’ boarding school. Epidemiol. Infect. 1984, 93, 277–283. [Google Scholar] [CrossRef]
- Harley, D.; Harrower, B.; Lyon, M.; Dick, A. A primary school outbreak of pharyngoconjunctival fever caused by adenovirus type 3. Commun. Dis. Intell. 2001, 25, 9–12. [Google Scholar]
- Tessier, T.M.; Dodge, M.J.; MacNeil, K.M.; Evans, A.M.; Prusinkiewicz, M.A.; Mymryk, J.S. Almost famous: Human adenoviruses (and what they have taught us about cancer). Tumour Virus Res. 2021, 12, 200225. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lee, S.K.; Ko, D.-H.; Hyun, J.; Kim, H.-S.; Song, W.; Kim, H.S. Associations of Adenovirus Genotypes in Korean Acute Gastroenteritis Patients with Respiratory Symptoms and Intussusception. BioMed Res. Int. 2017, 2017, 1602054. [Google Scholar] [CrossRef]
- Portes, S.A.R.; Volotão, E.d.M.; Rocha, M.S.; Rebelo, M.C.; Xavier, M.d.P.T.P.; de Assis, R.M.; Rose, T.L.; Miagostovich, M.P.; Leite, J.P.G.; Carvalho-Costa, F.A. A non-enteric adenovirus A12 gastroenteritis outbreak in Rio de Janeiro, Brazil. Mem. Do Inst. Oswaldo Cruz 2016, 111, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Huzly, D.; Ganzenmueller, T.; Beck, R.; Schulz, T.F.; Heim, A. A human adenovirus species B subtype 21a associated with severe pneumonia. J. Infect. 2014, 69, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Pfortmueller, C.A.; Barbani, M.T.; Schefold, J.C.; Hage, E.; Heim, A.; Zimmerli, S. Severe acute respiratory distress syndrome (ARDS) induced by human adenovirus B21: Report on 2 cases and literature review. J. Crit. Care 2019, 51, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Hoppe, E.; Mugisha, L.; Petrzelkova, K.; Akoua-Koffi, C.; Couacy-Hymann, E.; Anoh, A.E.; Mossoun, A.; Schubert, G.; Wiersma, L.; et al. High prevalence and diversity of species D adenoviruses (HAdV-D) in human populations of four Sub-Saharan countries. Virol. J. 2014, 11, 25. [Google Scholar] [CrossRef]
- Kurskaya, O.G.; Prokopyeva, E.A.; Dubovitskiy, N.A.; Solomatina, M.V.; Sobolev, I.A.; Derko, A.A.; Nokhova, A.R.; Anoshina, A.V.; Leonova, N.V.; Simkina, O.A.; et al. Genetic Diversity of the Human Adenovirus C Isolated from Hospitalized Children in Russia (2019–2022). Viruses 2024, 16, 386. [Google Scholar] [CrossRef]
- Garnett, C.T.; Talekar, G.; Mahr, J.A.; Huang, W.; Zhang, Y.; Ornelles, D.A.; Gooding, L.R. Latent Species C Adenoviruses in Human Tonsil Tissues. J. Virol. 2009, 83, 2417–2428. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Assay | PCR ID | Primer and Probe ID | Nucleotide Sequence (5′-3′) | Orientation | Amplicon Size (bp) | Reference |
|---|---|---|---|---|---|---|
| dPCR | 494 | 1080 | CWTACATGCACATCKCSGG | + | 69 | [16] |
| 1081 | CRCGGGCRAAYTGCACCAG | - | ||||
| 1082 | FAM-CCGGGCTCAGGTACTCCGAGGCGTCCT-TAMRA | - | ||||
| Nested PCR | 582 | 1551 | TICTTTGACATICGIGGIGTICTIGA | + | 764 to 896 | [17] |
| 1553 | CTGTCIACIGCCTGRTTCCACA | - | ||||
| 583 | 1554 | GGYCCYAGYTTYAARCCCTAYTC | + | 688 to 821 | ||
| 1555 | GGTTCTGTCICCCAGAGARTCIAGCA | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veneri, C.; Bonanno Ferraro, G.; Congiu, D.; Franco, A.; Brandtner, D.; Mancini, P.; Iaconelli, M.; The SARI Network; Lucentini, L.; Suffredini, E.; et al. Wastewater-Based Surveillance of Human Adenoviruses in Italy: Quantification by Digital PCR and Molecular Typing via Nanopore Amplicon Sequencing. Viruses 2025, 17, 791. https://doi.org/10.3390/v17060791
Veneri C, Bonanno Ferraro G, Congiu D, Franco A, Brandtner D, Mancini P, Iaconelli M, The SARI Network, Lucentini L, Suffredini E, et al. Wastewater-Based Surveillance of Human Adenoviruses in Italy: Quantification by Digital PCR and Molecular Typing via Nanopore Amplicon Sequencing. Viruses. 2025; 17(6):791. https://doi.org/10.3390/v17060791
Chicago/Turabian StyleVeneri, Carolina, G. Bonanno Ferraro, D. Congiu, A. Franco, D. Brandtner, P. Mancini, M. Iaconelli, The SARI Network, L. Lucentini, E. Suffredini, and et al. 2025. "Wastewater-Based Surveillance of Human Adenoviruses in Italy: Quantification by Digital PCR and Molecular Typing via Nanopore Amplicon Sequencing" Viruses 17, no. 6: 791. https://doi.org/10.3390/v17060791
APA StyleVeneri, C., Bonanno Ferraro, G., Congiu, D., Franco, A., Brandtner, D., Mancini, P., Iaconelli, M., The SARI Network, Lucentini, L., Suffredini, E., & La Rosa, G. (2025). Wastewater-Based Surveillance of Human Adenoviruses in Italy: Quantification by Digital PCR and Molecular Typing via Nanopore Amplicon Sequencing. Viruses, 17(6), 791. https://doi.org/10.3390/v17060791