
Combined Use of RT-qPCR and NGS for Identification and Surveillance of SARS-CoV-2 Variants of Concern in Residual Clinical Laboratory Samples in Miami-Dade County, Florida
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. NGS/ARTIC SARS-CoV-2 Sequencing Method
2.3. Bioinformatics
2.4. RT-qPCR Target Selection and Primer/Probe Design
2.5. In Silico Validation of Targeted RT-qPCR Assays
2.6. In Vitro Validation of Targeted RT-qPCR Assays
3. Results
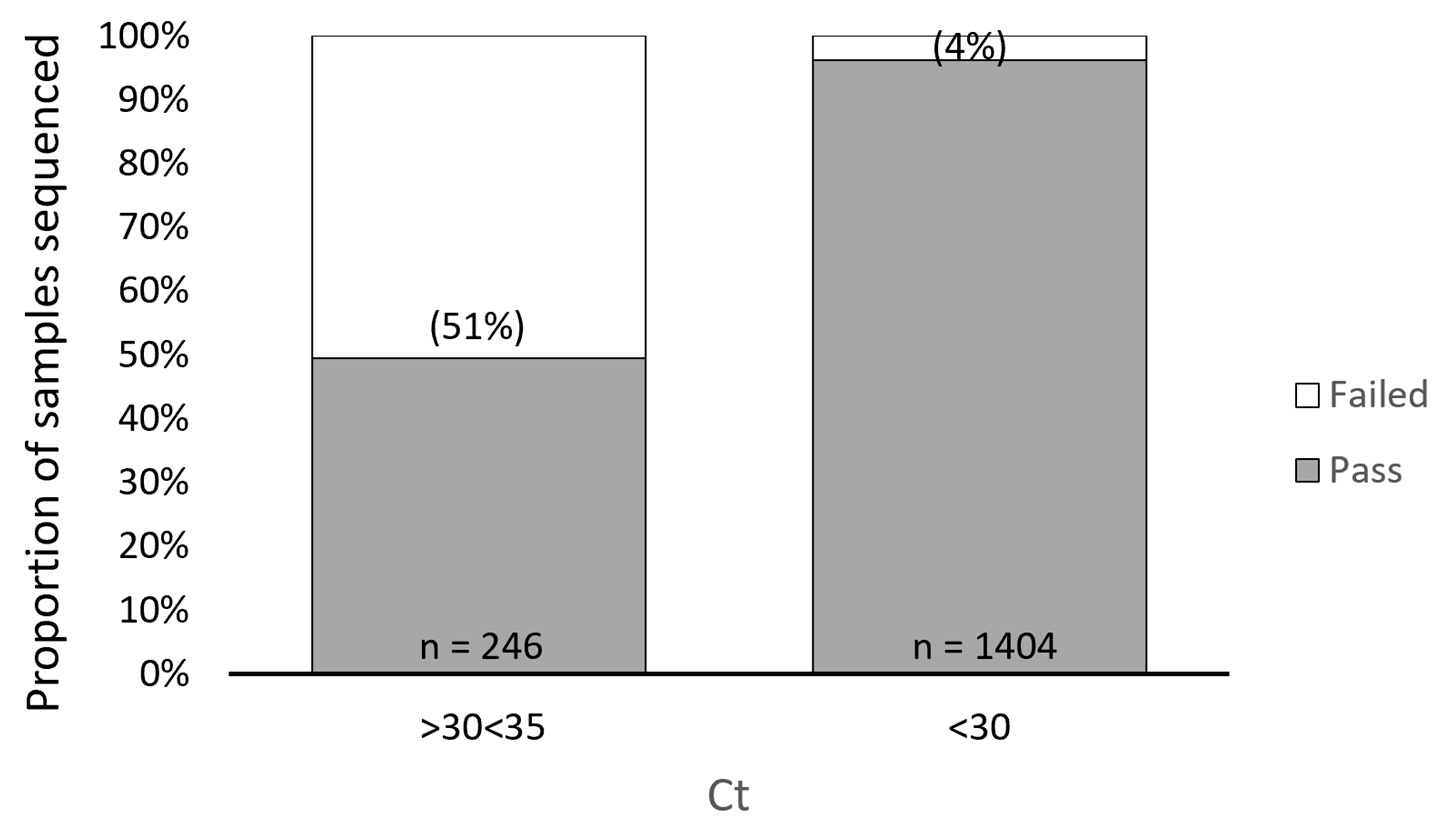
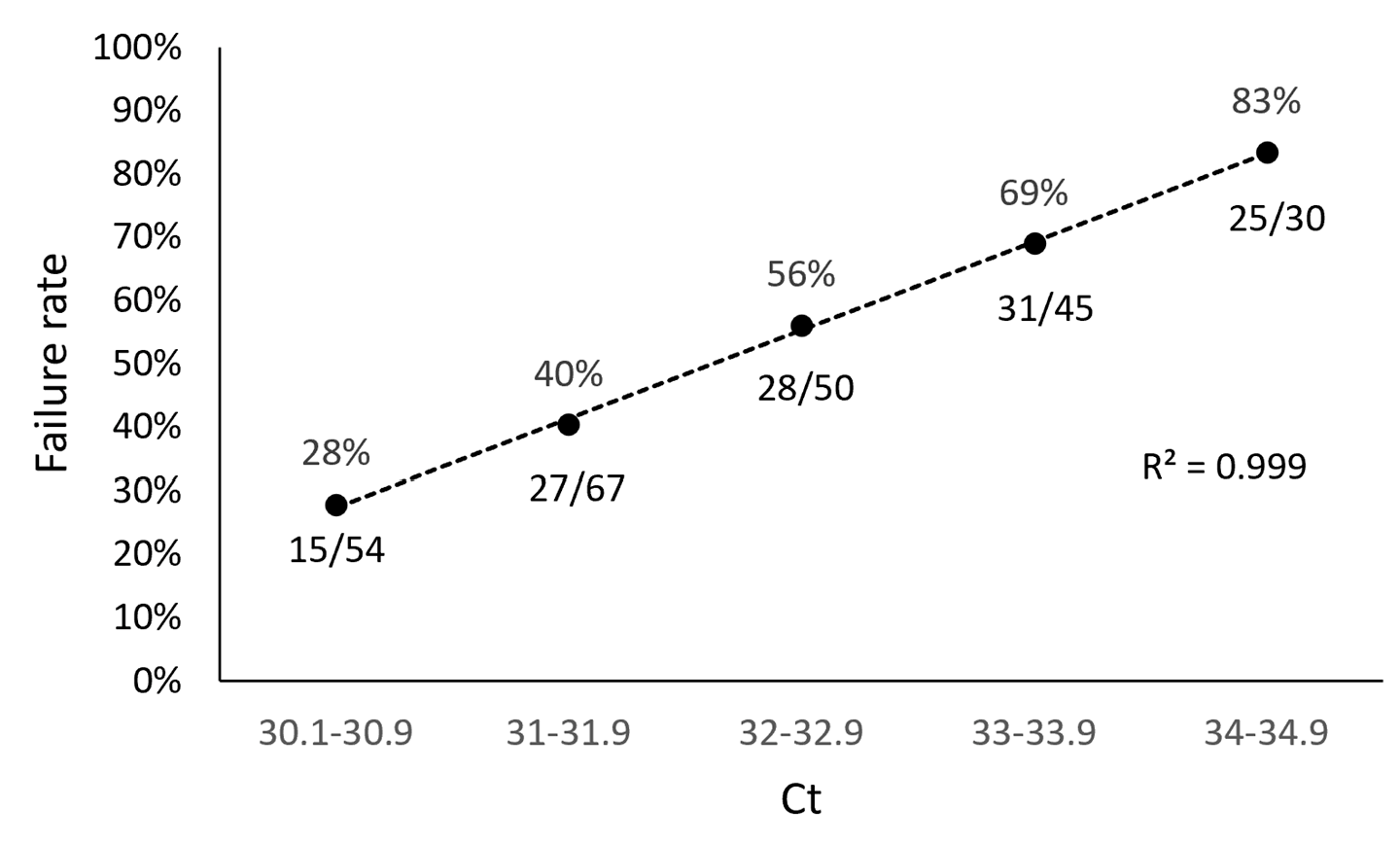
3.1. ARTIC SARS-CoV-2 Sequencing Method Performance Is Directly Related to Sample Ct Values
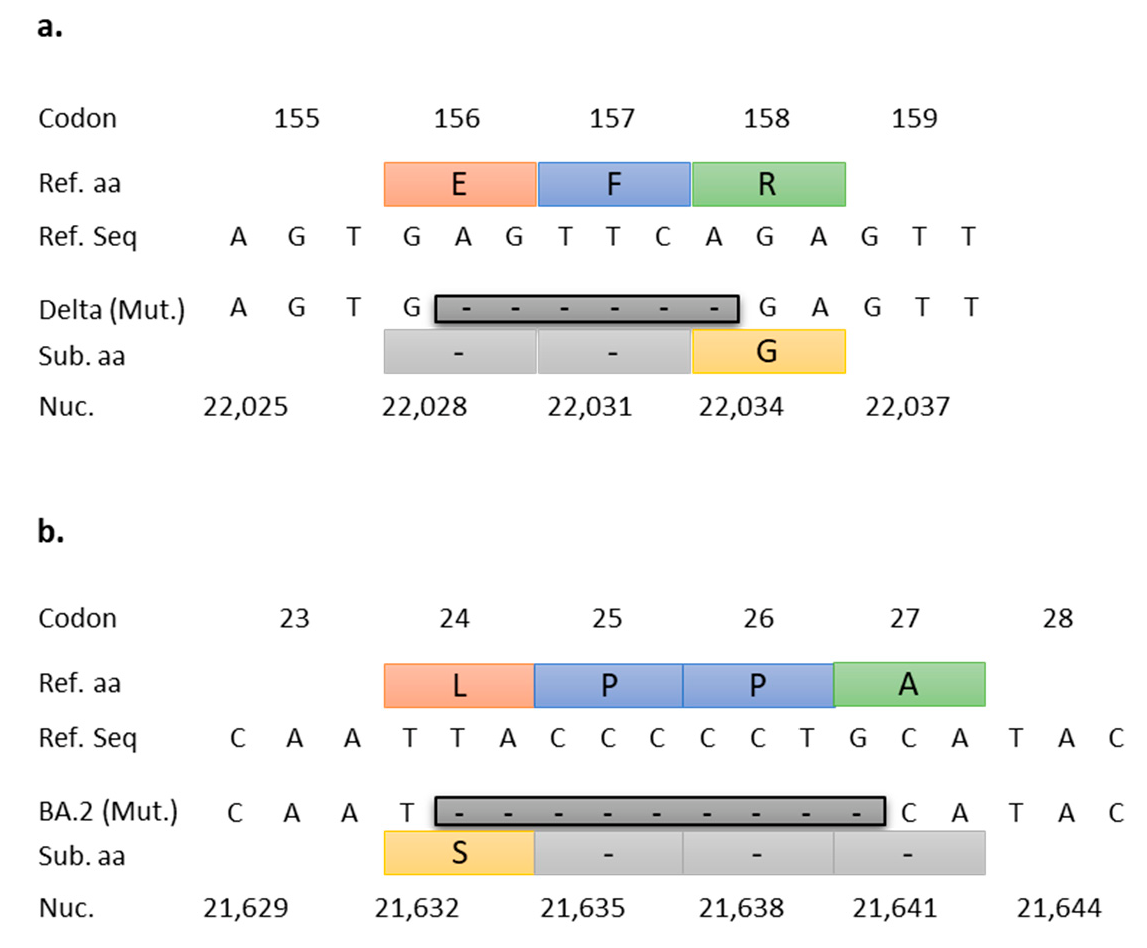
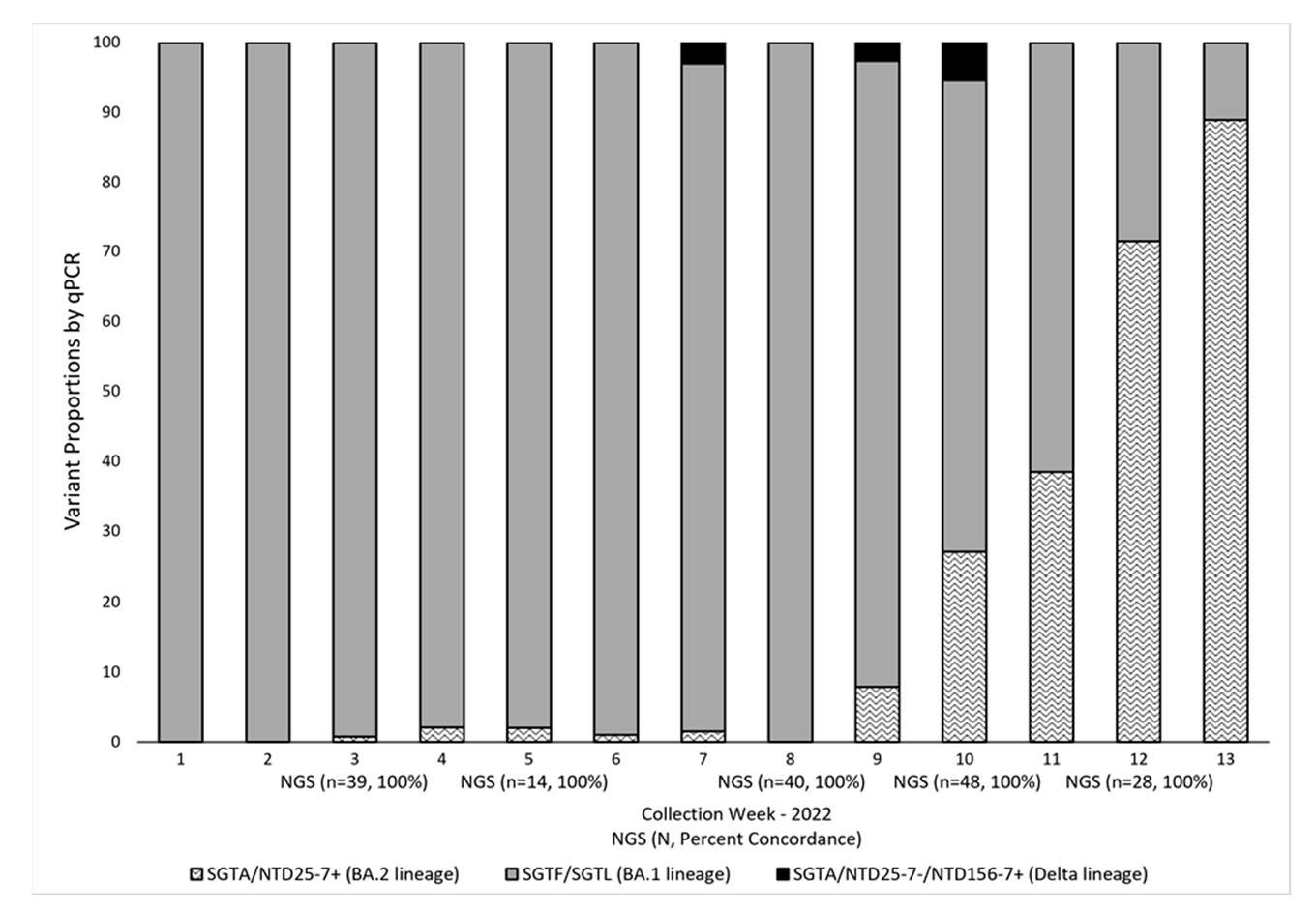
3.2. Targeted RT-qPCR for Detection and Surveillance of VOCs Delta and Omicron
3.3. In Silico Validation Results
3.4. In Vitro Validation Results
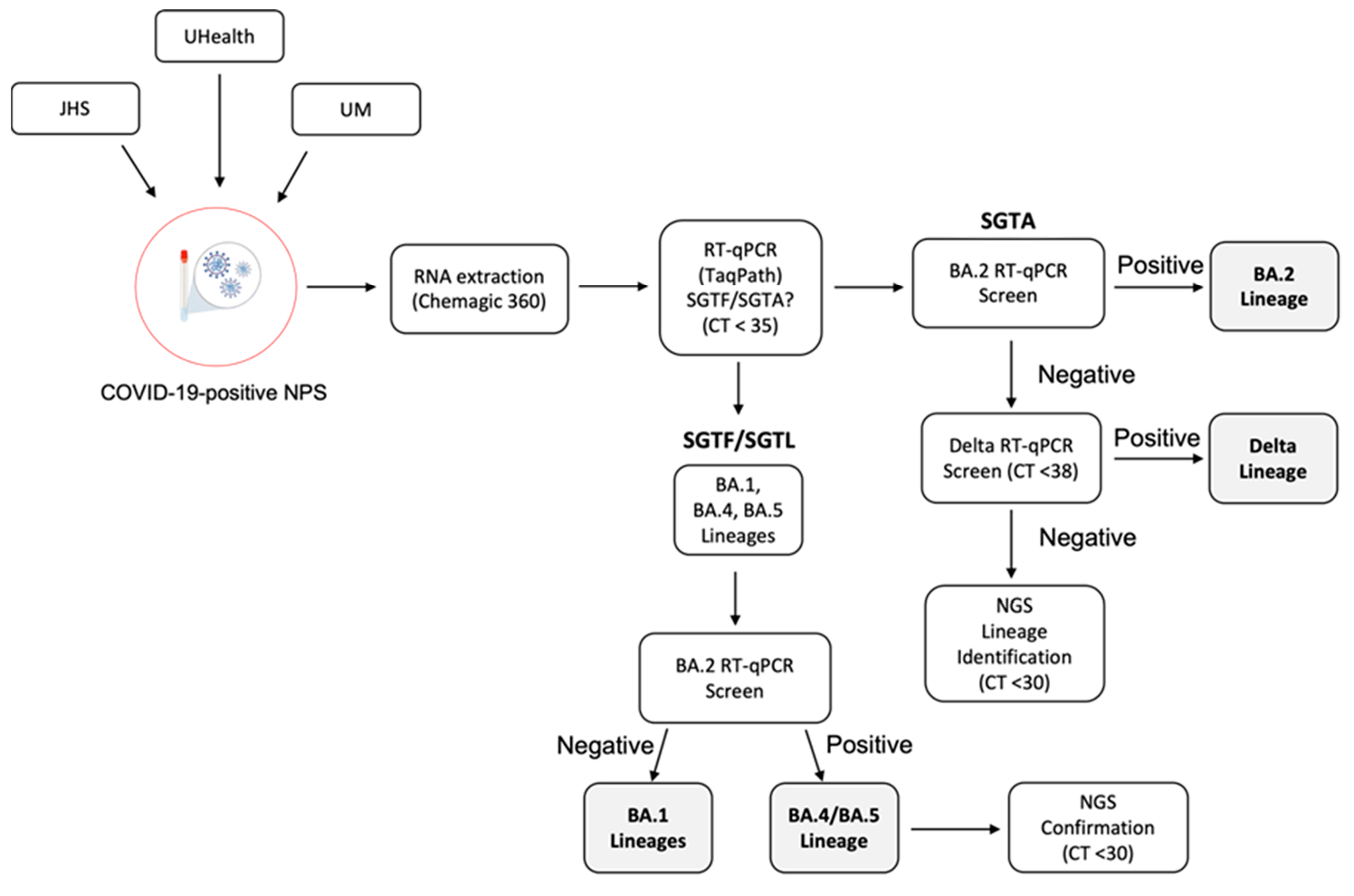
3.5. SARS-CoV-2 VOC Surveillance Workflow
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Health Organization (WHO) Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 26 February 2021).
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 26 February 2021).
- ECDC. SARS-CoV-2 Variants of Concern as of 24 February 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 26 February 2022).
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 22 August 2021).
- Liu, Y.; Rocklöv, J. The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus. J. Travel Med. 2021, 28, taab124. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Bolze, A.; Cirulli, E.T.; Luo, S.; White, S.; Wyman, D.; Rossi, A.D.; Machado, H.; Cassens, T.; Jacobs, S.; Schiabor Barrett, K.M.; et al. SARS-CoV-2 variant Delta rapidly displaced variant Alpha in the United States and led to higher viral loads. medRxiv 2021. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Liu, L.; Iketani, S.; Guo, Y.; Chan, J.F.; Wang, M.; Luo, Y.; Chu, H.; Huang, Y.; Nair, M.S.; Yu, J.; et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rocklöv, J. The effective reproduction number for the omicron SARS-CoV-2 variant of concern is several times higher than Delta. J. Travel Med. 2022, 29, taac037. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Stella, A.O.; Walker, G.; Akerman, A.; Milogiannakis, V.; Brilot, F.; Amatayakul-Chantler, S.; Roth, N.; Coppola, G.; Schofield, P.; et al. SARS-CoV-2 Omicron: Evasion of potent humoral responses and resistance to clinical immunotherapeutics relative to viral variants of concern. medRxiv 2021. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Chan, J.F.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; Chik, K.K.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, C.; Mayer, C.K.; Claassen, M.; Maponga, T.; Burgers, W.A.; Keeton, R.; Riou, C.; Sutherland, A.D.; Suliman, T.; Shaw, M.L.; et al. Breakthrough infections with SARS-CoV-2 omicron despite mRNA vaccine booster dose. Lancet 2022, 399, 625–626. [Google Scholar] [CrossRef] [PubMed]
- CDC. COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker/#variant-proportions (accessed on 22 August 2021).
- Nasir, J.A.; Kozak, R.A.; Aftanas, P.; Raphenya, A.R.; Smith, K.M.; Maguire, F.; Maan, H.; Alruwaili, M.; Banerjee, A.; Mbareche, H.; et al. A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. Viruses 2020, 12, 895. [Google Scholar] [CrossRef]
- Baker, D.J.; Aydin, A.; Le-Viet, T.; Kay, G.L.; Rudder, S.; de Oliveira Martins, L.; Tedim, A.P.; Kolyva, A.; Diaz, M.; Alikhan, N.F.; et al. CoronaHiT: High-throughput sequencing of SARS-CoV-2 genomes. Genome Med. 2021, 13, 21. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- GISAID. SARS-CoV-2 Lineage Variant Summary: Primer Monitor Tool. Available online: https://primer-monitor.neb.com/lineages (accessed on 3 June 2021).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef]
- Thomas, E.; Delabat, S.; Carattini, Y.L.; Andrews, D.M. SARS-CoV-2 and Variant Diagnostic Testing Approaches in the United States. Viruses 2021, 13, 2492. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Delabat, S.; Andrews, D.M. Diagnostic Testing for SARS-CoV-2 Infection. Curr. Hepatol. Rep. 2021, 20, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Destras, G.; Gaymard, A.; Stefic, K.; Marlet, J.; Eymieux, S.; Regue, H.; Semanas, Q.; d’Aubarede, C.; Billaud, G.; et al. Two-step strategy for the identification of SARS-CoV-2 variant of concern 202012/01 and other variants with spike deletion H69-V70, France, August to December 2020. Euro Surveill. 2021, 26, 2100008. [Google Scholar] [CrossRef]
- Borges, V.; Sousa, C.; Menezes, L.; Gonçalves, A.M.; Picão, M.; Almeida, J.P.; Vieita, M.; Santos, R.; Silva, A.R.; Costa, M.; et al. Tracking SARS-CoV-2 lineage B.1.1.7 dissemination: Insights from nationwide spike gene target failure (SGTF) and spike gene late detection (SGTL) data, Portugal, week 49 2020 to week 3 2021. Euro Surveill. 2021, 26, 2100130. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef]
- Bedotto, M.; Fournier, P.E.; Houhamdi, L.; Levasseur, A.; Delerce, J.; Pinault, L.; Padane, A.; Chamieh, A.; Tissot-Dupont, H.; Brouqui, P.; et al. Implementation of an in-house real-time reverse transcription-PCR assay for the rapid detection of the SARS-CoV-2 Marseille-4 variant. J. Clin. Virol. 2021, 139, 104814. [Google Scholar] [CrossRef]
- Peterson, S.W.; Lidder, R.; Daigle, J.; Wonitowy, Q.; Dueck, C.; Nagasawa, A.; Mulvey, M.R.; Mangat, C.S. RT-qPCR detection of SARS-CoV-2 mutations S 69-70 del, S N501Y and N D3L associated with variants of concern in Canadian wastewater samples. Sci. Total Environ. 2022, 810, 151283. [Google Scholar] [CrossRef]
- Gand, M.; Vanneste, K.; Thomas, I.; Van Gucht, S.; Capron, A.; Herman, P.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Deepening of In Silico Evaluation of SARS-CoV-2 Detection RT-qPCR Assays in the Context of New Variants. Genes 2021, 12, 565. [Google Scholar] [CrossRef]
- Yaniv, K.; Ozer, E.; Shagan, M.; Lakkakula, S.; Plotkin, N.; Bhandarkar, N.S.; Kushmaro, A. Direct RT-qPCR assay for SARS-CoV-2 variants of concern (Alpha, B.1.1.7 and Beta, B.1.351) detection and quantification in wastewater. Environ. Res. 2021, 201, 111653. [Google Scholar] [CrossRef] [PubMed]
- Utah DoH ARTIC/Illumina Bioinformatic Workflow. Available online: https://github.com/CDCgov/SARS-CoV-2_Sequencing/tree/master/protocols/BFX-UT_ARTIC_Illumina (accessed on 29 January 2021).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS-CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, e00647-20. [Google Scholar] [CrossRef]
- Rahman, M.S.; Islam, M.R.; Alam, A.S.M.R.; Islam, I.; Hoque, M.N.; Akter, S.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Evolutionary dynamics of SARS-CoV-2 nucleocapsid protein and its consequences. J. Med. Virol. 2021, 93, 2177–2195. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/Probe | Sequence 5′-3′ | |
|---|---|---|
| Delta (NTD156-7) | Forward Primer | TGGGTGTTTATTACCACAA |
| Reverse Primer | GGCTGAGAGACATATTCAAA | |
| Probe | FAM-ATGGAAAGT/ZEN/GGAGTTTATTCTAGT-3IABkFQ | |
| Omicron BA.2, BA.4, BA.5 (NTD25-7) | Forward Primer | TTTATTGCCACTAGTCTCTAGTCAG |
| Reverse Primer | GGTAATAAACACCACGTGTGAAAG | |
| Probe | Cy5-AGAACTCAA/TAO/TCATACACTAATT-3IAbRQSp |
| NTD156-7 Forward Primer | Mutated Sequences | NTD156-7 Reverse Primer | Mutated Sequences | NTD156-7 Probe | Mutated Sequences | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ref Base | # | % | Ref Base | # | % | Ref Base | # | % | |
| 5′ | T | 48 | 0.004% | G | 40 | 0.003% | A | 196 | 0.015% |
| G | 3 | 0.000% | G | 722 | 0.055% | T | 187 | 0.014% | |
| G | 4 | 0.000% | C | 638 | 0.049% | G | 1011 | 0.077% | |
| G | 17,428 | 1.335% | T | 68 | 0.005% | G | 16 | 0.001% | |
| T | 0 | 0.000% | G | 346 | 0.027% | A | 35 | 0.003% | |
| G | 2 | 0.000% | A | 512 | 0.039% | A | 10 | 0.001% | |
| T | 28 | 0.002% | G | 77 | 0.006% | A | 31 | 0.002% | |
| T | 30 | 0.002% | A | 455 | 0.035% | G | 126 | 0.010% | |
| T | 15 | 0.001% | G | 45 | 0.003% | T | 251 | 0.019% | |
| A | 152 | 0.012% | A | 19 | 0.001% | G | 274 | 0.021% | |
| T | 76 | 0.006% | C | 142 | 0.011% | G | 57 | 0.004% | |
| T | 15 | 0.001% | A | 142 | 0.011% | A | 60 | 0.005% | |
| A | 0 | 0.000% | T | 4 | 0.000% | G | 22 | 0.002% | |
| C | 3 | 0.000% | A | 156 | 0.012% | T | 26 | 0.002% | |
| C | 3 | 0.000% | T | 36 | 0.003% | T | 35 | 0.003% | |
| A | 2 | 0.000% | T | 7 | 0.001% | T | 34 | 0.003% | |
| C | 3 | 0.000% | C | 12 | 0.001% | A | 3 | 0.000% | |
| A | 2 | 0.000% | A | 10 | 0.001% | T | 60 | 0.005% | |
| A | 29 | 0.002% | A | 181 | 0.014% | T | 12 | 0.001% | |
| A | 447 | 0.034% | C | 54 | 0.004% | ||||
| T | 18 | 0.001% | |||||||
| A | 20 | 0.002% | |||||||
| G | 149 | 0.011% | |||||||
| 3′ | T | 29 | 0.002% | ||||||
| NTD156-7 Oligos | Sequence 5′-3′ | % Sequences with any mutation | % Sequences with mutation in last 5 bases | ||||||
| Forward Primer | TGGGTGTTTATTACCACAA | 1.367 | 0.003 | ||||||
| Reverse Primer | GGCTGAGAGACATATTCAAA | 0.311 | 0.050 | ||||||
| Probe | ATGGAAAGTGGAGTTTATTCTAGT | 0.210 | 0.020 | ||||||
| NTD25-7 Forward Primer | Mutated Sequences | NTD25-7 Reverse Primer | Mutated Sequences | NTD25-7 Probe | Mutated Sequences | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ref Base | # | % | Ref Base | # | % | Ref Base | # | % | |
| 5′ | T | 18 | 0.008% | G | 24 | 0.011% | A | 0 | 0.000% |
| T | 12 | 0.005% | G | 385 | 0.174% | G | 15 | 0.007% | |
| T | 6 | 0.003% | T | 19 | 0.009% | A | 0 | 0.000% | |
| A | 32 | 0.014% | A | 12 | 0.005% | A | 0 | 0.000% | |
| T | 27 | 0.012% | A | 81 | 0.037% | C | 3 | 0.001% | |
| T | 4 | 0.002% | T | 284 | 0.129% | T | 34 | 0.015% | |
| G | 69 | 0.031% | A | 281 | 0.127% | C | 1 | 0.000% | |
| C | 18 | 0.008% | A | 9 | 0.004% | A | 0 | 0.000% | |
| C | 31 | 0.014% | A | 6 | 0.003% | A | 1 | 0.000% | |
| A | 28 | 0.013% | C | 926 | 0.420% | T | 45 | 0.020% | |
| C | 48 | 0.022% | A | 8 | 0.004% | C | 2 | 0.001% | |
| T | 3 | 0.001% | C | 8 | 0.004% | A | 1 | 0.000% | |
| A | 8 | 0.004% | C | 10 | 0.005% | T | 3 | 0.001% | |
| G | 31 | 0.014% | A | 9 | 0.004% | A | 4 | 0.002% | |
| T | 18 | 0.008% | C | 14 | 0.006% | C | 14 | 0.006% | |
| C | 3672 | 1.664% | G | 5 | 0.002% | A | 0 | 0.000% | |
| T | 17 | 0.008% | T | 15 | 0.007% | C | 8 | 0.004% | |
| C | 480 | 0.217% | G | 134 | 0.061% | T | 12 | 0.005% | |
| T | 38 | 0.017% | T | 66 | 0.030% | A | 0 | 0.000% | |
| A | 31 | 0.014% | G | 337 | 0.153% | A | 1 | 0.000% | |
| G | 242 | 0.110% | A | 135 | 0.061% | T | 12 | 0.005% | |
| T | 33 | 0.015% | A | 9 | 0.004% | T | 0 | 0.000% | |
| C | 30 | 0.014% | A | 13 | 0.006% | ||||
| A | 52 | 0.024% | G | 2 | 0.001% | ||||
| 3′ | G | 285 | 0.129% | ||||||
| NTD25-7 Oligos | Sequence 5′-3′ | % Sequences with any mutation | % Sequences with mutation in last 5 bases | ||||||
| Forward Primer | TTTATTGCCACTAGTCTCTAGTCAG | 2.371 | 0.291 | ||||||
| Reverse Primer | GGTAATAAACACCACGTGTGAAAG | 1.265 | 0.225 | ||||||
| Probe | AGAACTCAATCATACACTAATT | 0.071 | 0.011 | ||||||
| Variant (NGS) | n | NTD156-7 Result | Mean Ct | Percent Agreement | |
|---|---|---|---|---|---|
| Positive | Negative | ||||
| B.1.617.2 (Delta) | 9 | 9 | 0 | 28.60 | 100% |
| AY.2 (Delta) | 1 | 1 | 0 | 25.20 | 100% |
| AY.3 (Delta) | 3 | 3 | 0 | 28.90 | 100% |
| AY.3.1 (Delta) | 2 | 2 | 0 | 31.05 | 100% |
| AY.4 (Delta) | 1 | 1 | 0 | 26.94 | 100% |
| AY.5 (Delta) | 1 | 1 | 0 | 37.73 | 100% |
| AY.12 (Delta) | 1 | 1 | 0 | 26.77 | 100% |
| AY.24 (Delta) | 1 | 1 | 0 | 20.19 | 100% |
| AY.25 (Delta) | 5 | 5 | 0 | 27.86 | 100% |
| B.1 | 1 | 0 | 1 | na | 100% |
| B.1.1.7 (Alpha) | 6 | 0 | 6 | na | 100% |
| B.1.427 (Epsilon) | 2 | 0 | 2 | na | 100% |
| B.1.429 (Epsilon) | 2 | 0 | 2 | na | 100% |
| B.1.526 (Iota) | 3 | 0 | 3 | na | 100% |
| B.1.621 (Mu) | 2 | 0 | 2 | na | 100% |
| B.1.621.1 (Mu) | 2 | 0 | 2 | na | 100% |
| B.1.623 | 1 | 0 | 1 | na | 100% |
| B.1.628 | 1 | 0 | 1 | na | 100% |
| C.37 (Lambda) | 3 | 0 | 3 | na | 100% |
| P.1 (Gamma) | 3 | 0 | 3 | na | 100% |
| Total | 50 | 24 | 26 | ||
| Variant (NGS) | n | NTD25-7 Results | Mean Ct | Percent Agreement | |
|---|---|---|---|---|---|
| Positive | Negative | ||||
| BA.2 (Omicron) | 51 | 51 | 0 | 27.2 | 100% |
| BA.2.3 | 2 | 2 | 0 | 20.1 | 100% |
| BA.1 (Omicron) | 9 | 0 | 9 | na | 100% |
| BA.1.1 (Omicron) | 17 | 0 | 17 | na | 100% |
| AY.3 (Delta) | 2 | 0 | 2 | na | 100% |
| AY.47 (Delta) | 1 | 0 | 1 | na | 100% |
| Total | 82 | 53 | 29 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carattini, Y.L.; Griswold, A.; Williams, S.; Valiathan, R.; Zhou, Y.; Shukla, B.; Abbo, L.M.; Parra, K.; Jorda, M.; Nimer, S.D.; et al. Combined Use of RT-qPCR and NGS for Identification and Surveillance of SARS-CoV-2 Variants of Concern in Residual Clinical Laboratory Samples in Miami-Dade County, Florida. Viruses 2023, 15, 593. https://doi.org/10.3390/v15030593
Carattini YL, Griswold A, Williams S, Valiathan R, Zhou Y, Shukla B, Abbo LM, Parra K, Jorda M, Nimer SD, et al. Combined Use of RT-qPCR and NGS for Identification and Surveillance of SARS-CoV-2 Variants of Concern in Residual Clinical Laboratory Samples in Miami-Dade County, Florida. Viruses. 2023; 15(3):593. https://doi.org/10.3390/v15030593
Chicago/Turabian StyleCarattini, Yamina L., Anthony Griswold, Sion Williams, Ranjini Valiathan, Yi Zhou, Bhavarth Shukla, Lilian M. Abbo, Katiuska Parra, Merce Jorda, Stephen D. Nimer, and et al. 2023. "Combined Use of RT-qPCR and NGS for Identification and Surveillance of SARS-CoV-2 Variants of Concern in Residual Clinical Laboratory Samples in Miami-Dade County, Florida" Viruses 15, no. 3: 593. https://doi.org/10.3390/v15030593
APA StyleCarattini, Y. L., Griswold, A., Williams, S., Valiathan, R., Zhou, Y., Shukla, B., Abbo, L. M., Parra, K., Jorda, M., Nimer, S. D., Sologon, C., Gallegos, H. R., Weiss, R. E., Ferreira, T., Memon, A., Paige, P. G., Thomas, E., & Andrews, D. M. (2023). Combined Use of RT-qPCR and NGS for Identification and Surveillance of SARS-CoV-2 Variants of Concern in Residual Clinical Laboratory Samples in Miami-Dade County, Florida. Viruses, 15(3), 593. https://doi.org/10.3390/v15030593