
A Phylogeographic Analysis of Porcine Parvovirus 1 in Africa
 , , , , , and add
Show full author list
, , , , , and add
Show full author list
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Swine Samples
2.2. Sequence Analysis
2.3. Viral Population Dynamics and Phylogeography
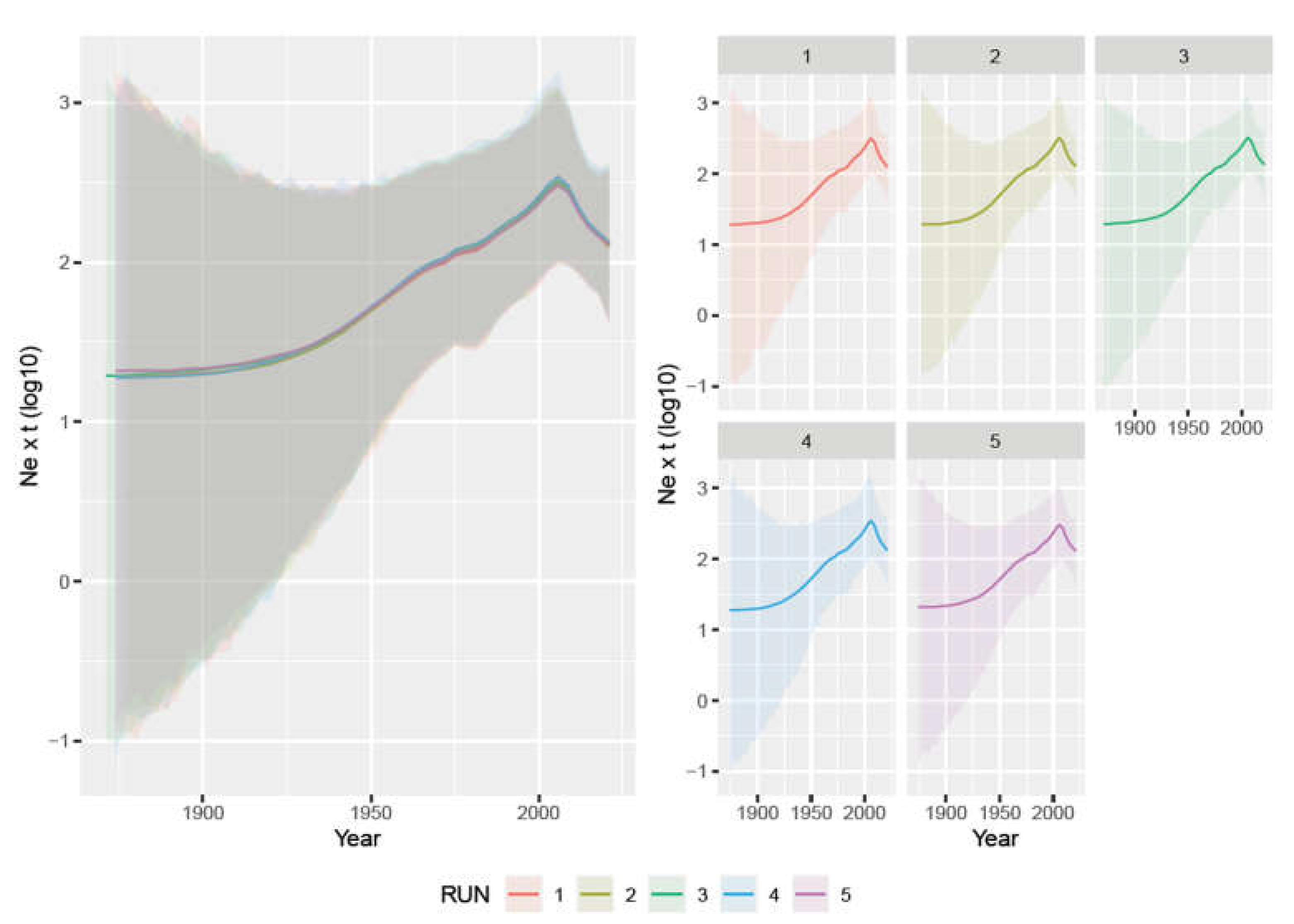
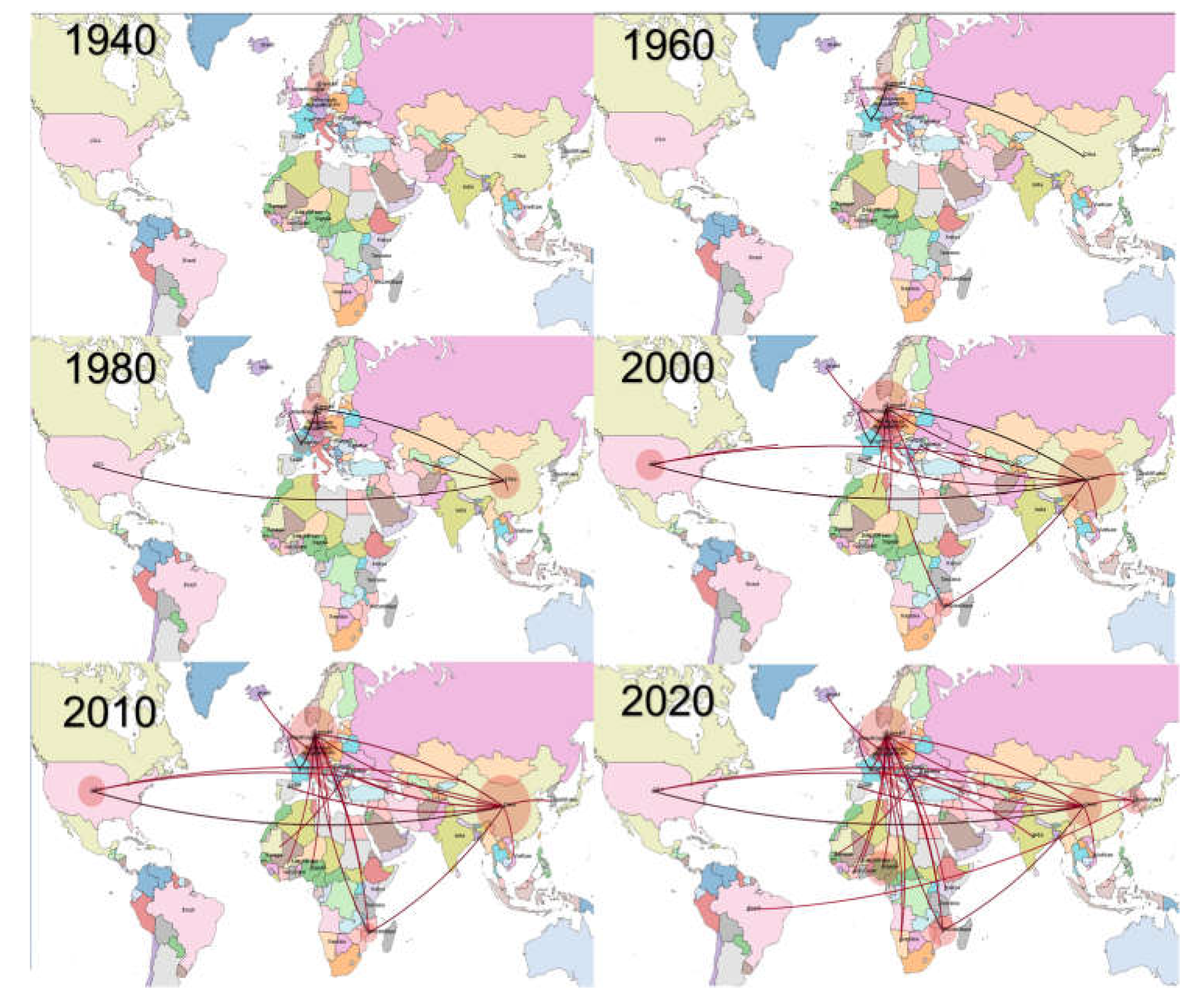
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Streck, A.F.; Truyen, U. Porcine Parvovirus. Curr. Issues Mol. Biol. 2020, 37, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, I.; Olasz, F.; Cságola, A.; Tijssen, P.; Zádori, Z. Biology of Porcine Parvovirus (Ungulate Parvovirus 1). Viruses 2017, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Parke, C.R.; Burgess, G.W. An Economic Assessment of Porcine Parvovirus Vaccination. Aust. Vet. J. 1993, 70, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.T.; Kim, R.Y.; Nguyen, V.G.; Chung, H.C.; Park, B.K. Perspectives on the Evolution of Porcine Parvovirus. Viruses 2017, 9, 196. [Google Scholar] [CrossRef]
- Vereecke, N.; Kvisgaard, L.K.; Baele, G.; Boone, C.; Kunze, M.; Larsen, L.E.; Theuns, S.; Nauwynck, H. Molecular Epidemiology of Porcine Parvovirus Type 1 (PPV1) and the Reactivity of Vaccine-Induced Antisera against Historical and Current PPV1 Strains. Virus Evol. 2022, 8, veac053. [Google Scholar] [CrossRef]
- Zimmermann, P.; Ritzmann, M.; Selbitz, H.J.; Heinritzi, K.; Truyen, U. VP1 Sequences of German Porcine Parvovirus Isolates Define Two Genetic Lineages. J. Gen. Virol. 2006, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Dán, Á.; Tombácz, K.; Lorincz, M.; Kiss, T.; Becskei, Z.; Spînu, M.; Tuboly, T.; Cságola, A. Phylogeny and Evolutionary Genetics of Porcine Parvovirus in Wild Boars. Infect. Genet. Evol. 2012, 12, 1163–1171. [Google Scholar] [CrossRef]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular Epidemiology Evolution of Porcine Parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef]
- Streck, A.; Bonatto, S.L.; Homeier, T.; Souza, C.K.; Gonçalves, K.R.; Gava, D.; Canal, C.W.; Truyen, U. High Rate of Viral Evolution in the Capsid Protein of Porcine Parvovirus. J. Gen. Virol. 2011, 92, 2628–2636. [Google Scholar] [CrossRef]
- Bergeron, J.; Hébert, B.; Tijssen, P. Genome Organization of the Kresse Strain of Porcine Parvovirus: Identification of the Allotropic Determinant and Comparison with Those of NADL-2 and Field Isolates. J. Virol. 1996, 70, 2508–2515. [Google Scholar] [CrossRef]
- Zeeuw, E.J.L.; Leinecker, N.; Herwig, V.; Selbitz, H.J.; Truyen, U. Study of the Virulence and Cross-Neutralization Capability of Recent Porcine Parvovirus Field Isolates and Vaccine Viruses in Experimentally Infected Pregnant Gilts. J. Gen. Virol. 2007, 88, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Jóźwik, A.; Manteutel, J.; Selbitz, H.J.; Truyen, U. Vaccination against Porcine Parvovirus Protects against Disease, but Does Not Prevent Infection and Virus Shedding after Challenge Infection with a Heterologous Virus Strain. J. Gen. Virol. 2009, 90, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Molini, U.; Franzo, G.; Gous, L.; Moller, S.; Hemberger, Y.M.; Chiwome, B.; Marruchella, G.; Khaiseb, S.; Cattoli, G.; Dundon, W.G. Three Different Genotypes of Porcine Circovirus 2 (PCV-2) Identified in Pigs and Warthogs in Namibia. Arch. Virol. 2021, 166, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- Luka, P.D.; Adedeji, A.J.; Jambol, A.R.; Ifende, I.V.; Luka, H.G.; Choji, N.D.; Weka, R.; Settypalli, T.B.K.; Achenbach, J.E.; Cattoli, G.; et al. Coinfections of African Swine Fever Virus, Porcine Circovirus 2 and 3, and Porcine Parvovirus 1 in Swine in Nigeria. Arch. Virol. 2022, 167, 2715–2722. [Google Scholar] [CrossRef]
- Molini, U.; Franzo, G.; Settypalli, T.B.K.; Hemberger, M.Y.; Khaiseb, S.; Cattoli, G.; Dundon, W.G.; Lamien, C.E. Viral Co-Infections of Warthogs in Namibia with African Swine Fever Virus and Porcine Parvovirus 1. Animals 2022, 12, 1697. [Google Scholar] [CrossRef]
- Molini, U.; Coetzee, L.M.; Hemberger, M.Y.; Khaiseb, S.; Cattoli, G.; Dundon, W.G. Evidence Indicating Transmission of Porcine Parvovirus 1 between Warthogs and Domestic Pigs in Namibia. Vet. Res. Commun. 2022. [Google Scholar] [CrossRef]
- Standley, K. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. (Outlines Version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Ginestet, C. Ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees With Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Segalés, J.; Kekarainen, T.; Cortey, M. The Natural History of Porcine Circovirus Type 2: From an Inoffensive Virus to a Devastating Swine Disease? Vet. Microbiol. 2013, 165, 13–20. [Google Scholar] [CrossRef]
- Franzo, G.; Faustini, G.; Legnardi, M.; Cecchinato, M.; Drigo, M.; Tucciarone, C.M. Phylodynamic and Phylogeographic Reconstruction of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in Europe: Patterns and Determinants. Transbound. Emerg. Dis. 2022, 69, e2175–e2184. [Google Scholar] [CrossRef]
- A Profile of the South African Pork Market Value Chain 2020. Available online: https://dalrrd.gov.za (accessed on 10 January 2023).
- Stein, P.; Uddhammar, E. China in Africa: The Role of Trade, Investments, and Loans Amidst Shifting Geopolitical Ambitions. ORF Occasional Paper 2021. Available online: https://policycommons.net/artifacts/1808834/china-in-africa/2543755/ (accessed on 10 January 2023).
- Franzo, G.; Settypalli, T.B.K.; Agusi, E.R.; Meseko, C.; Minoungou, G.; Ouoba, B.L.; Habibata, Z.L.; Wade, A.; de Barros, J.L.; Tshilenge, C.G.; et al. Porcine Circovirus-2 in Africa: Identification of Continent-Specific Clusters and Evidence of Independent Viral Introductions from Europe, North America and Asia. Transbound. Emerg. Dis. 2021, 69, e1142–e1152. [Google Scholar] [CrossRef] [PubMed]
- Tegegne, D.; Tsegaye, G.; Aman, S.; Faustini, G.; Franzo, G. Molecular Epidemiology and Genetic Characterization of PCV2 Circulating in Wild Boars in Southwestern Ethiopia. J. Trop. Med. 2022, 2022, 5185247. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Zerbo, H.L.; Ouoba, B.L.; Dji-Tombo, A.D.; Kindo, M.G.; Sawadogo, R.; Chang’a, J.; Bitanyi, S.; Kamigwe, A.; Mayenga, C.; et al. A Phylogeographic Analysis of Porcine Parvovirus 1 in Africa. Viruses 2023, 15, 207. https://doi.org/10.3390/v15010207
Franzo G, Zerbo HL, Ouoba BL, Dji-Tombo AD, Kindo MG, Sawadogo R, Chang’a J, Bitanyi S, Kamigwe A, Mayenga C, et al. A Phylogeographic Analysis of Porcine Parvovirus 1 in Africa. Viruses. 2023; 15(1):207. https://doi.org/10.3390/v15010207
Chicago/Turabian StyleFranzo, Giovanni, Habibata Lamouni Zerbo, Bruno Lalidia Ouoba, Adama Drabo Dji-Tombo, Marietou Guitti Kindo, Rasablaga Sawadogo, Jelly Chang’a, Stella Bitanyi, Aloyce Kamigwe, Charles Mayenga, and et al. 2023. "A Phylogeographic Analysis of Porcine Parvovirus 1 in Africa" Viruses 15, no. 1: 207. https://doi.org/10.3390/v15010207
APA StyleFranzo, G., Zerbo, H. L., Ouoba, B. L., Dji-Tombo, A. D., Kindo, M. G., Sawadogo, R., Chang’a, J., Bitanyi, S., Kamigwe, A., Mayenga, C., Lo, M. M., Ndiaye, M., Ba, A., Diop, G. L., Anahory, I. V., Mapaco, L. P., Achá, S. J., Kouakou, V. K., Couacy-Hymann, E., ... Dundon, W. G. (2023). A Phylogeographic Analysis of Porcine Parvovirus 1 in Africa. Viruses, 15(1), 207. https://doi.org/10.3390/v15010207