
Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Definitions—Outbreaks, Cases, Clusters, and Hotspots
2.2. Sample Selection and RNA Extraction
2.3. IAV-End-RT-PCR
2.4. Next-Generation Sequencing and Consensus Generation
2.5. Phylogenetic and Phylogeographic Analysis
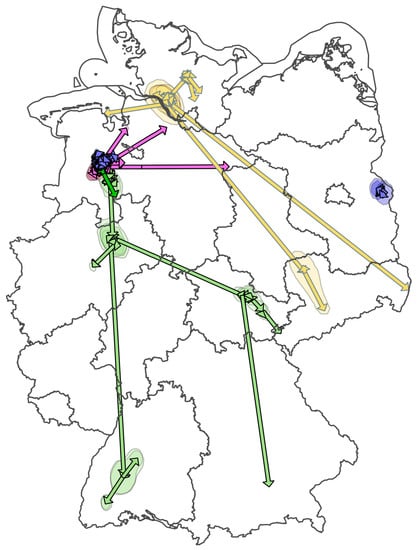
3. Results
3.1. Background Information
3.2. Direct Wild Bird Introductions and Spread—H5N8 Ger-11-16 and Ger-12-16.1
3.2.1. H5N8 Ger-11-16
Epidemiological Data
Phylogenetic Analysis of Ger-11-16
Wild Bird-Mediated Transmissions of Ger-11-16
Farm-to-Farm Transmissions within Ger-11-16
Circulation Period of Ger-11-16
3.2.2. H5N8 Ger-12-16.1
Epidemiological Data, Genetic Distinction, and Phylogenetic Analysis
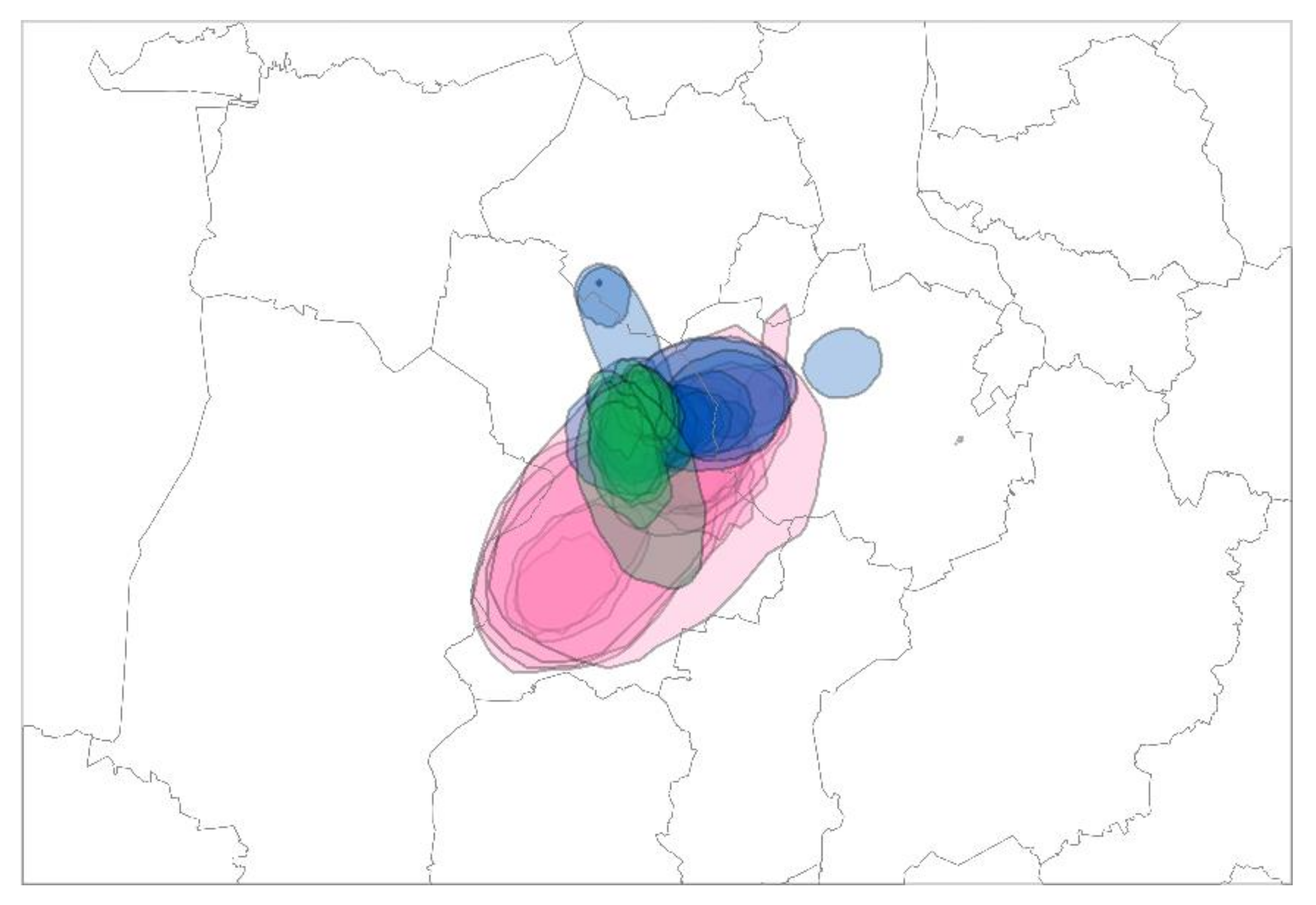
3.3. Cluster I and II: Cluster Development and Modification by Genetic Drift—H5N8 Ger-10-16.2
3.3.1. Cluster I—“Brandenburg (BB)”
3.3.2. Cluster II—“Cloppenburg/Oldenburg (CLOL)”
3.4. Cluster III: Genetic Modification by Reassortment in Poultry Holdings—H5N5 Ger-12-16-N5.1 and Ger-12-16-N5.2
3.4.1. Epidemiological Data
3.4.2. Genetic Modification by Reassortment
3.5. Cluster IV, V, and VI: Cluster Development and Distant Spread—H5N8 Ger-10-20-N8
Epidemiological Data and Time-Scaled Phylogeny Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pohlmann, A.; Starick, E.; Harder, T.; Grund, C.; Höper, D.; Globig, A.; Staubach, C.; Dietze, K.; Strebelow, G.; Ulrich, R.G.; et al. Outbreaks among Wild Birds and Domestic Poultry Caused by Reassorted Influenza A(H5N8) Clade 2.3.4.4 Viruses, Germany, 2016. Emerg. Infect. Dis. 2017, 23, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef]
- Fusaro, A.; Monne, I.; Mulatti, P.; Zecchin, B.; Bonfanti, L.; Ormelli, S.; Milani, A.; Cecchettin, K.; Lemey, P.; Moreno, A.; et al. Genetic Diversity of Highly Pathogenic Avian Influenza A(H5N8/H5N5) Viruses in Italy, 2016–2017. Emerg. Infect. Dis. 2017, 23, 1543–1547. [Google Scholar] [CrossRef]
- Meier, S.; Hussy, D.; Hofmann, M.; Renzullo, S.; Vogler, B.; Sigrist, B.; Hoop, R.; Albini, S. Outbreak of Highly Pathogenic Avian Influenza H5N8 in November 2016 in Wild Birds in Switzerland. Schweiz. Arch. Tierheilkd. 2017, 159, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, P.; Brouwer, A.; Venkatesh, D.; Duncan, D.; Dovas, C.I.; Georgiades, G.; Monne, I.; Fusaro, A.; Dan, A.; Smietanka, K.; et al. Comparison of 2016-17 and Previous Epizootics of Highly Pathogenic Avian Influenza H5 Guangdong Lineage in Europe. Emerg. Infect. Dis. 2018, 24, 2270–2283. [Google Scholar] [CrossRef] [PubMed]
- Guinat, C.; Nicolas, G.; Vergne, T.; Bronner, A.; Durand, B.; Courcoul, A.; Gilbert, M.; Guerin, J.L.; Paul, M.C. Spatio-temporal patterns of highly pathogenic avian influenza virus subtype H5N8 spread, France, 2016 to 2017. Euro Surveill 2018, 23, 1700791. [Google Scholar] [CrossRef]
- Pohlmann, A.; Starick, E.; Grund, C.; Höper, D.; Strebelow, G.; Globig, A.; Staubach, C.; Conraths, F.J.; Mettenleiter, T.C.; Harder, T.; et al. Swarm incursions of reassortants of highly pathogenic avian influenza virus strains H5N8 and H5N5, clade 2.3.4.4b, Germany, winter 2016/17. Sci. Rep. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; Hoffmann, D.; Grund, C.; Koethe, S.; Hussy, D.; Meier, S.M.; King, J.; Schinkoethe, J.; Ulrich, R.; Harder, T.; et al. Genetic Characterization and Zoonotic Potential of Highly Pathogenic Avian Influenza Virus A(H5N6/H5N5), Germany, 2017-2018. Emerg. Infect. Dis. 2019, 25, 1973–1976. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef]
- King, J.; Harder, T.; Globig, A.; Stacker, L.; Gunther, A.; Grund, C.; Beer, M.; Pohlmann, A. Highly pathogenic avian influenza virus incursions of subtype H5N8, H5N5, H5N1, H5N4, and H5N3 in Germany during 2020-21. Virus Evol. 2022, 8, veac035. [Google Scholar] [CrossRef]
- Verhagen, J.H.; van der Jeugd, H.P.; Nolet, B.A.; Slaterus, R.; Kharitonov, S.P.; de Vries, P.P.; Vuong, O.; Majoor, F.; Kuiken, T.; Fouchier, R.A. Wild bird surveillance around outbreaks of highly pathogenic avian influenza A(H5N8) virus in the Netherlands, 2014, within the context of global flyways. Euro Surveill. 2015, 20, 21–32. [Google Scholar] [CrossRef] [PubMed]
- El-Shesheny, R.; Barman, S.; Feeroz, M.M.; Hasan, M.K.; Jones-Engel, L.; Franks, J.; Turner, J.; Seiler, P.; Walker, D.; Friedman, K.; et al. Genesis of Influenza A(H5N8) Viruses. Emerg. Infect. Dis. 2017, 23, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Poen, M.J.; Venkatesh, D.; Bestebroer, T.M.; Vuong, O.; Scheuer, R.D.; Oude Munnink, B.B.; de Meulder, D.; Richard, M.; Kuiken, T.; Koopmans, M.P.G.; et al. Co-circulation of genetically distinct highly pathogenic avian influenza A clade 2.3.4.4 (H5N6) viruses in wild waterfowl and poultry in Europe and East Asia, 2017–2018. Virus. Evol. 2019, 5, vez004. [Google Scholar] [CrossRef]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R.; Kuiken, T. Changing Role of Wild Birds in the Epidemiology of Avian Influenza A Viruses. Adv. Virus Res. 2018, 100, 279–307. [Google Scholar] [CrossRef] [PubMed]
- Ku, K.B.; Park, E.H.; Yum, J.; Kim, J.A.; Oh, S.K.; Seo, S.H. Highly pathogenic avian influenza A(H5N8) virus from waterfowl, South Korea, 2014. Emerg. Infect. Dis. 2014, 20, 1587–1588. [Google Scholar] [CrossRef]
- Zhou, L.C.; Liu, J.; Pei, E.L.; Xue, W.J.; Lyu, J.M.; Cai, Y.T.; Wu, D.; Wu, W.; Liu, Y.Y.; Jin, H.Y.; et al. Novel Avian Influenza A(H5N8) Viruses in Migratory Birds, China, 2013–2014. Emerg. Infect. Dis. 2016, 22, 1121–1123. [Google Scholar] [CrossRef]
- Lee, D.H.; Bertran, K.; Kwon, J.H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef]
- Harder, T.C.; Maurer-Stroh, S.; Pohlmann, A.; Starick, E.; Höreth-Böntgen, D.; Albrecht, K.; Pannwitz, G.; Teifke, J.; Gunalan, V.; Lee, R.T.; et al. Influenza A(H5N8) Virus Similar to Strain in Korea Causing Highly Pathogenic Avian Influenza in Germany. Emerg. Infect. Dis. 2015, 21, 860–863. [Google Scholar] [CrossRef]
- Lee, D.H.; Bahl, J.; Torchetti, M.K.; Killian, M.L.; Ip, H.S.; DeLiberto, T.J.; Swayne, D.E. Highly Pathogenic Avian Influenza Viruses and Generation of Novel Reassortants, United States, 2014-2015. Emerg. Infect. Dis. 2016, 22, 1283–1285. [Google Scholar] [CrossRef]
- Lee, D.H.; Sharshov, K.; Swayne, D.E.; Kurskaya, O.; Sobolev, I.; Kabilov, M.; Alekseev, A.; Irza, V.; Shestopalov, A. Novel Reassortant Clade 2.3.4.4 Avian Influenza A(H5N8) Virus in Wild Aquatic Birds, Russia, 2016. Emerg. Infect. Dis. 2017, 23, 359–360. [Google Scholar] [CrossRef]
- Li, M.; Liu, H.; Bi, Y.; Sun, J.; Wong, G.; Liu, D.; Li, L.; Liu, J.; Chen, Q.; Wang, H.; et al. Highly Pathogenic Avian Influenza A(H5N8) Virus in Wild Migratory Birds, Qinghai Lake, China. Emerg. Infect. Dis. 2017, 23, 637–641. [Google Scholar] [CrossRef]
- Chen, J.; Liang, B.; Hu, J.; Liu, H.; Sun, J.; Li, M.; Chen, Q.; He, Y.; Liu, D. Circulation, Evolution and Transmission of H5N8 virus, 2016–2018. J. Infect. 2019, 79, 363–372. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Multiple Reassorted Viruses as Cause of Highly Pathogenic Avian Influenza A(H5N8) Virus Epidemic, the Netherlands, 2016. Emerg. Infect. Dis. 2017, 23, 1974–1981. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, V.; Goncharova, N.; Susloparov, I.; Kolosova, N.; Gudymo, A.; Svyatchenko, S.; Danilenko, A.; Durymanov, A.; Gavrilova, E.; Maksyutov, R.; et al. Isolation and characterization of H5Nx highly pathogenic avian influenza viruses of clade 2.3.4.4 in Russia. Virology 2018, 525, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Majo, N.; Sanchez-Gonzalez, R.; Vergara-Alert, J. Emergence and spread of highly pathogenic avian influenza A(H5N8) in Europe in 2016-2017. Transbound. Emerg. Dis. 2018, 65, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenstrom, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Cattoli, G.; Fusaro, A.; Monne, I.; Capua, I. H5N1 Virus Evolution in Europe-An Updated Overview. Viruses 2009, 1, 1351–1363. [Google Scholar] [CrossRef]
- Takekawa, J.Y.; Newman, S.H.; Xiao, X.; Prosser, D.J.; Spragens, K.A.; Palm, E.C.; Yan, B.; Li, T.; Lei, F.; Zhao, D.; et al. Migration of waterfowl in the East Asian flyway and spatial relationship to HPAI H5N1 outbreaks. Avian Dis. 2010, 54, 466–476. [Google Scholar] [CrossRef]
- Saito, T.; Tanikawa, T.; Uchida, Y.; Takemae, N.; Kanehira, K.; Tsunekuni, R. Intracontinental and intercontinental dissemination of Asian H5 highly pathogenic avian influenza virus (clade 2.3.4.4) in the winter of 2014-2015. Rev. Med. Virol. 2015, 25, 388–405. [Google Scholar] [CrossRef]
- Ip, H.S.; Dusek, R.J.; Bodenstein, B.; Torchetti, M.K.; DeBruyn, P.; Mansfield, K.G.; DeLiberto, T.; Sleeman, J.M. High Rates of Detection of Clade 2.3.4.4 Highly Pathogenic Avian Influenza H5 Viruses in Wild Birds in the Pacific Northwest During the Winter of 2014-15. Avian Dis. 2016, 60, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92, e00433-18. [Google Scholar] [CrossRef]
- Poen, M.J.; Bestebroer, T.M.; Vuong, O.; Scheuer, R.D.; van der Jeugd, H.P.; Kleyheeg, E.; Eggink, D.; Lexmond, P.; van den Brand, J.M.A.; Begeman, L.; et al. Local amplification of highly pathogenic avian influenza H5N8 viruses in wild birds in the Netherlands, 2016 to 2017. Euro Surveill. 2018, 23, 17–00449. [Google Scholar] [CrossRef]
- Lycett, S.J.; Pohlmann, A.; Staubach, C.; Caliendo, V.; Woolhouse, M.; Beer, M.; Kuiken, T.; Global Consortium for H5N8 and Related Influenza Viruses. Genesis and spread of multiple reassortants during the 2016/2017 H5 avian influenza epidemic in Eurasia. Proc. Natl. Acad. Sci. USA 2020, 117, 20814–20825. [Google Scholar] [CrossRef]
- Mulatti, P.; Fusaro, A.; Scolamacchia, F.; Zecchin, B.; Azzolini, A.; Zamperin, G.; Terregino, C.; Cunial, G.; Monne, I.; Marangon, S. Integration of genetic and epidemiological data to infer H5N8 HPAI virus transmission dynamics during the 2016–2017 epidemic in Italy. Sci. Rep. 2018, 8, 18037. [Google Scholar] [CrossRef]
- Swieton, E.; Smietanka, K. Phylogenetic and molecular analysis of highly pathogenic avian influenza H5N8 and H5N5 viruses detected in Poland in 2016–2017. Transbound. Emerg. Dis. 2018, 65, 1664–1670. [Google Scholar] [CrossRef]
- Swieton, E.; Fusaro, A.; Shittu, I.; Niemczuk, K.; Zecchin, B.; Joannis, T.; Bonfante, F.; Smietanka, K.; Terregino, C. Sub-Saharan Africa and Eurasia Ancestry of Reassortant Highly Pathogenic Avian Influenza A(H5N8) Virus, Europe, December 2019. Emerg. Infect. Dis. 2020, 26, 1557–1561. [Google Scholar] [CrossRef]
- Naguib, M.M.; Graaf, A.; Fortin, A.; Luttermann, C.; Wernery, U.; Amarin, N.; Hussein, H.A.; Sultan, H.; Al Adhadh, B.; Hassan, M.K.; et al. Novel real-time PCR-based patho- and phylotyping of potentially zoonotic avian influenza A subtype H5 viruses at risk of incursion into Europe in 2017. Euro Surveill. 2017, 22, 30435. [Google Scholar] [CrossRef]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Harder, T.; Beer, M.; Pohlmann, A. Rapid multiplex MinION nanopore sequencing workflow for Influenza A viruses. BMC Infect Dis. 2020, 20, 648. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef]
- King, J.; Schulze, C.; Engelhardt, A.; Hlinak, A.; Lennermann, S.L.; Rigbers, K.; Skuballa, J.; Staubach, C.; Mettenleiter, T.C.; Harder, T.; et al. Novel HPAIV H5N8 Reassortant (Clade 2.3.4.4b) Detected in Germany. Viruses 2020, 12, 281. [Google Scholar] [CrossRef]
- King, J.; Harder, T.; Conraths, F.J.; Beer, M.; Pohlmann, A. The genetics of highly pathogenic avian influenza viruses of subtype H5 in Germany, 2006–2020. Transbound. Emerg. Dis. 2021, 68, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Bergervoet, S.A.; Ho, C.K.Y.; Heutink, R.; Bossers, A.; Beerens, N. Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events. Viruses 2019, 11, 501. [Google Scholar] [CrossRef]
- Denzin, N.; Bolling, M.; Pohlmann, A.; King, J.; Globig, A.; Conraths, F.J. Investigation into a Superspreading Event of the German 2020–2021 Avian Influenza Epidemic. Pathogens 2022, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Wilking, H.; Ziller, M.; Staubach, C.; Globig, A.; Harder, T.C.; Conraths, F.J. Chances and limitations of wild bird monitoring for the avian influenza virus H5N1—Detection of pathogens highly mobile in time and space. PLoS ONE 2009, 4, e6639. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beerens, N.; Heutink, R.; Pritz-Verschuren, S.; Germeraad, E.A.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Genetic relationship between poultry and wild bird viruses during the highly pathogenic avian influenza H5N6 epidemic in the Netherlands, 2017–2018. Transbound. Emerg. Dis. 2019, 66, 1370–1378. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reassortant | Subtype | First Date | Last Date | Total | Poultry | Wild |
|---|---|---|---|---|---|---|
| Ger-11-16-N8 | H5N8 | 7 November 2016 | 22 August 2017 | 34 | 25 | 9 |
| Ger-12-16-N8.1 | H5N8 | 1 January 2017 | 24 February 2017 | 8 | 2 | 6 |
| Ger-12-16-N8.2 | H5N8 | 13 December 2016 | 9 May 2017 | 71 | 61 | 10 |
| Ger-12-16-N5.1 | H5N5 | 13 December 2016 | 23 January 2017 | 7 | 2 | 5 |
| Ger-12-16-N5.2 | H5N5 | 22 January 2017 | 9 March 2017 | 22 | 15 | 7 |
| Ger-10-20-N8 | H5N8 | 26 October 2020 | 20 July 2021 | 161 | 122 | 39 |
| Ger-02-21-N8 | H5N8 | 26 February 2021 | 10 March 2021 | 3 | 3 | 0 |
| Ger-03-21-N8 | H5N8 | 2 March 2021 | 2 March 2021 | 1 | 1 | 0 |
| Ger-10-20-N5 | H5N5 | 26 October 2020 | 9 November 2020 | 2 | 1 | 1 |
| Ger-12-20-N3 | H5N3 | 14 December 2020 | 26 January 2021 | 5 | 0 | 5 |
| Ger-02-21-N4 | H5N4 | 17 February 2021 | 17 February 2021 | 2 | 0 | 2 |
| Ger-02-21-N1 | H5N1 | 17 February 2021 | 17 July 2021 | 9 | 4 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, J.; Staubach, C.; Lüder, C.; Koethe, S.; Günther, A.; Stacker, L.; Rubbenstroth, D.; Dietze, K.; Grund, C.; Conraths, F.J.; et al. Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany. Viruses 2022, 14, 1849. https://doi.org/10.3390/v14091849
King J, Staubach C, Lüder C, Koethe S, Günther A, Stacker L, Rubbenstroth D, Dietze K, Grund C, Conraths FJ, et al. Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany. Viruses. 2022; 14(9):1849. https://doi.org/10.3390/v14091849
Chicago/Turabian StyleKing, Jacqueline, Christoph Staubach, Christiane Lüder, Susanne Koethe, Anne Günther, Lina Stacker, Dennis Rubbenstroth, Klaas Dietze, Christian Grund, Franz J. Conraths, and et al. 2022. "Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany" Viruses 14, no. 9: 1849. https://doi.org/10.3390/v14091849
APA StyleKing, J., Staubach, C., Lüder, C., Koethe, S., Günther, A., Stacker, L., Rubbenstroth, D., Dietze, K., Grund, C., Conraths, F. J., Harder, T., Beer, M., & Pohlmann, A. (2022). Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany. Viruses, 14(9), 1849. https://doi.org/10.3390/v14091849