Abstract
Clostridioides difficile causes antibiotic-induced diarrhoea and pseudomembranous colitis in humans and animals. Current conventional treatment relies solely on antibiotics, but C. difficile infection (CDI) cases remain persistently high with concomitant increased recurrence often due to the emergence of antibiotic-resistant strains. Antibiotics used in treatment also induce gut microbial imbalance; therefore, novel therapeutics with improved target specificity are being investigated. Bacteriophages (phages) kill bacteria with precision, hence are alternative therapeutics for the targeted eradication of the pathogen. Here, we review current progress in C. difficile phage research. We discuss tested strategies of isolating C. difficile phages directly, and via enrichment methods from various sample types and through antibiotic induction to mediate prophage release. We also summarise phenotypic phage data that reveal their morphological, genetic diversity, and various ways they impact their host physiology and pathogenicity during infection and lysogeny. Furthermore, we describe the therapeutic development of phages through efficacy testing in different in vitro, ex vivo and in vivo infection models. We also discuss genetic modification of phages to prevent horizontal gene transfer and improve lysis efficacy and formulation to enhance stability and delivery of the phages. The goal of this review is to provide a more in-depth understanding of C. difficile phages and theoretical and practical knowledge on pre-clinical, therapeutic evaluation of the safety and effectiveness of phage therapy for CDI.
1. Scope of Current Review and Introduction to Clostridioides difficile Infection
There have been a number of reviews on Clostridioides difficile bacteriophages (phages) which summarise the mechanistic aspects that underpin our understanding and application of these phages [1,2,3,4]. In this review, we complement existing reviews by covering the body of data that has been largely gathered from our research and by others on a range of models developed to test the efficacy of C. difficile phages. We also emphasise the applied aspects of phages for phage product development.
C. difficile is a Gram-positive bacterium first isolated by Ivan Hall and Elizabeth O’Toole from the intestinal tract of infants where it was regarded as a commensal [5]. C. difficile infection (CDI) was later linked to antibiotic use and described as the cause of pseudomembranous colitis and nosocomial diarrhoea [6,7]. CDI is mediated by virulence factors located on a 19.6 kb pathogenicity locus (PaLoc), and the key toxins A and B are encoded by genes tcdA and tcdB, respectively [8,9]. Both toxins are cytotoxic, proinflammatory and cause disruption of tight junctions in human intestinal epithelial cells, resulting in fluid accumulation and damage to the large intestine. Expression of toxins A and B are controlled by the tcdR and tcdC genes, also located on the PaLoc [10,11]. Some strains, including the NAP1/027 epidemic strain, produce a third toxin, called the C. difficile binary toxin, which is located on the CdtLoc and may contribute to increased toxin production and disease severity [12,13]. The final toxin regulatory gene, tcdE, intercalates between toxins A and B, and is suspected of promoting the lysis of the cytoplasmic membrane and the release of the toxins from the cells [14]. Other virulence factors associated with C. difficile are linked to adhesion (such as pili, flagella, surface-layer proteins and physiological features), hydrolytic enzyme production, sporulation, biofilm production and cell wall glycopolymers [15,16,17,18,19,20].
Since the discovery of C. difficile, research has focused on virulence, pathogenicity and epidemiology of the bacterium to improve our understanding of CDI [21,22]. Despite advances made in the fields of antibiotic stewardship, infection control measures and surveillance policies, CDI remains a global health problem in the healthcare system [23,24,25,26,27]. The number of reported cases, recurrences and deaths is persistently high in many parts of the world [23,28]. In the UK, there are approximately twenty-two cases per 100,000 patient-bed days, in the EU, there are twenty-six infections in every 100,000 patient-bed days, and in the USA, there are one hundred and fifteen cases per 100,000 patient-bed days [16,23,24,29,30].
Strikingly, the infection has an associated ~45% recurrence rate and ~40% death rate [23,31]. The consequences of the infection are far reaching, affecting patient care and quality of life, and causing high economic costs; for example, in France, CDI’s annual costs are ~EUR 15 million [32]. These data highlight that CDI treatment and management strategies are insufficient, and that there is an unmet urgent need for alternative treatments to effectively treat the infection [4,22,31].
CDI is currently treated with the antibiotics fidaxomicin, vancomycin, and metronidazole [33,34]. New antibiotics such as tinidazole, rifaximin, rifalazil, and bezlotoxumab are currently being investigated and have some promise in treating CDI [31,35]. Antibiotics are, of course, essential, but bacteria are notorious for inducing gut dysbiosis, which triggers the outgrowth and colonisation of C. difficile and other pathogenic Enterobacteriaceae; hence, they are particularly problematic for CDI [36]. To overcome the low efficacy due to the development of antibiotic resistance and harmful side effects of antibiotic use in CDI, other therapies such as probiotics, immunotherapies, traditional and recombinant vaccines, monoclonal antibodies, faecal microbiota transfer, endolysins, and phage (a virus of bacteria) therapies are being developed as supplements or adjuncts to antibiotics [4,22,37].
Being natural and abundant organisms, phages are generally easy to isolate and characterise compared to the time and effort involved in developing many other therapies. Pertinent to CDI, phages have great advantages, as they can lyse bacteria with great precision to ensure the effective removal of the pathogen whilst maintaining other gut commensals, thus preventing dysbiosis [4]. Also, in the presence of susceptible bacterial strains, phages will continue to replicate and provide a continual supply of infective viral particles in the gut [38,39,40]. Importantly, phages are self-limiting, and thus are eliminated when the targeted bacteria have been cleared [41].
C. difficile can produce biofilms, which are aggregates of bacteria that adhere to surfaces, secrete protective extracellular polymeric substances, and can significantly impede antibiotic efficacy [42,43,44]. However, phages can effectively prevent the formation of C. difficile biofilms in vitro and penetrate mature biofilms to remove bacteria, which can potentially enable other therapeutics to access bacterial targets [45]. Clearly, these suitable properties of phages make them attractive and appropriate for CDI treatment, and this need has triggered several studies on their isolation and efficacy testing in infection models. We review and discuss the methods used to isolate and characterise the phages and the models we have developed and tested to assess phage lysis efficacy. We also discuss various ways C. difficile phages can be engineered and formulated to improve lysis, stability and therapeutic efficacy in humans and animals.
2. C. difficile Phage Isolation and Characterisation
Phages are the most abundant biological entity on earth. They are widespread throughout all environments, and their presence is closely linked with their host bacteria. C. difficile phages were first isolated in the 1980s and initially used as a bacterial typing tool. However, their efficiency for typing was restricted as they could only infect a limited number of C. difficile strains, and also due to the fastidious nature of C. difficile itself [46,47]. These characteristics of C. difficile has also negatively impacted therapeutic research conducted on its associated phages [48]. However, the rapid rise in CDI incidence and severity due to antibiotic failures has triggered renewed interest in therapeutic phage development [2,4,49,50,51,52,53,54]. In this section, we focus on the different methods used for C. difficile phage isolation, identification and therapeutic application.
2.1. Direct Isolation of Phages from Environmental and Clinical Samples
To begin with, we discuss direct screening of patient and animal faecal and environmental samples for the presence of infective C. difficile phages. For therapeutic purposes, strictly lytic phages, which infect and lyse target bacteria, are preferable to lysogenic phages that have the potential to integrate within the bacterial host chromosome. Lysogenic (or temperate) phages may cause horizontal gene transfer of genes associated with antimicrobial resistance (AMR) and other virulence factors associated with CDI [2,49,55]. The cycle the phage follows is identified via sequencing, and in genomes of lysogenic phages, genes associated with lysogeny, such as repressor and integrase genes, are found which are not present in lytic phages. Thus, attempts have been made by several research groups to screen for virulent lytic C. difficile phages.
Metagenomic studies have revealed that the human gut has over 1000 species of bacteria and associated phages [56,57]. Furthermore, the high diversity and richness of phages in faecal samples of healthy humans is speculated to be the catalyst for the success of faecal microbiota transplantation [58,59]. However, despite clear evidence of diverse phages in healthy individuals and patients, strictly lytic C. difficile phages have not been observed. Also, directly isolating phages from human faecal samples, or indeed isolating strictly lytic phages from any environment, has been unsuccessful [60].
Whilst the reason for the lack of isolation of strictly virulent phages remains unknown, it may largely be linked to adaptation strategies, where C. difficile phages have evolved to exist alongside their hosts through lysogeny to enhance their survival in harsh environmental conditions. This is further supported as all characterised C. difficile genomes encode multiple prophages, an active clustered regularly interspaced short palindromic repeat (CRISPR) targeting phages, and the phages themselves encode CRISPR arrays that target additional phages [61,62,63,64]. This obligate interconnectedness with their hosts potentially limited the evolution, and thus existence, of strictly lytic C. difficile phages. Alternatively, C. difficile lytic phages may exist but are not amenable to existing isolation procedures for this organism.
To increase the possibility of isolating strictly lytic C. difficile phages, environmental and clinical samples have been enriched [60,64,65,66,67,68]. The enrichment method involves incubating environmental and clinical samples in liquid media inoculated with susceptible bacterial hosts to enable amplification of effective phages [65,66,69,70]. However, this approach may limit the diversity of prospective phages observed due to bias towards the strain(s) included [70]. The enrichment media may also be supplemented with antibiotics to select for C. difficile growth and proliferation, reduce competition by other bacterial species and to allow optimum amplification of phages to occur [64,67]. Salts (MgCl2 and/or CaCl2) can also be added to the enrichment mix to enhance the stability and attachment of the putative phages present in the samples to the bacterial hosts [66,68]. However, studies enriching faecal samples have only observed C difficile phages in ~10% of samples examined and none from sewage [66], despite examining large numbers of a wide variety of samples, including those from healthy humans, inflammatory bowel disease patients and from healthy pigs, as well as pig caecal contents and slurries [60,65]. Sources from which C. difficile phages have been isolated include soil, sediment and estuarine samples, but all of these locations may be associated with human activities and, hence, could suggest why phages were isolated [64,67].
2.2. Isolation of C. difficile Phages through Prophage Induction
We stated in the previous section that C. difficile phages can be isolated from clinical and environmental samples through enrichment procedures. However, all difficile phages isolated to date are lysogenic and encode integrases in their genomes; this is despite their clear-plaque morphology, often broad host range (they can infect multiple C. difficile strains) and lysis ability determined using various in vitro, ex vivo and in vivo model systems [53,71,72,73,74]. Clearly, the presence of lysogeny-associated genes in the genomes of C. difficile phages indicates that they are temperate despite them behaving in a lytic manner. Furthermore, in some cases, lysogens have been isolated from the interaction between the phages and their bacterial hosts, signifying that the integrases are active [53,75].
In cases where strictly lytic phages that target species such as C. difficile cannot be found, it is pragmatic to isolate phages that infect pathogenic strains of interest and to assess their therapeutic potential. There is still lots to learn about phage lifestyles even if all integrases are active. There is a possibility that the integrases observed in some C. difficile phage genomes are only active within a subset of specific strains rather than the strains being examined. Certainly, our work has shown that in some cases, despite subjecting the strains to very high concentrations of phages, lysogens were not formed [53]. Therefore, if the phages are effective, research can be conducted to assess the risks associated with using temperate phages in their native state for therapeutic purposes. If temperate phage genes such as integrases pose a risk of horizontal gene transfer and will therefore fail to meet the regulatory standard, then the phage could potentially be genetically modified to delete all temperate associated genes [76].
To provide a solution to the problem of C. difficile phage isolation, we developed a method to induce prophages (lysogenic phages) from C. difficile strains. We previously hypothesised that prophage induction from environmental C. difficile strains might be effective to isolate therapeutically relevant phages that can lyse clinically relevant C. difficile strains [77,78]. To do this, bacterial strains are treated with sub-lethal concentrations of various DNA-damaging agents to mediate prophage release (Table 1). In E. coli, this exposure was shown to trigger the recA pathway and the SOS response, which resulted in the cleavage of prophage(s) from the host chromosome [79]. The released phages are recovered by centrifuging and filtering the cultures. The lytic activity of induced phages can be confirmed by spot tests or plaque assay techniques [80,81].

Table 1.
List of fully characterised, publicly available C. difficile phage genomes, their sources and methods of isolation.
Two DNA-damaging agents, mitomycin C (0.3–5 ug/mL final concentrations) and irradiation with UV light (302 nm wavelength), are commonly used to induce prophages in C. difficile. Mitomycin C is an alkylating agent that initiates DNA damage by causing mispairing of bases, DNA strand damage or cross-linking of complementary strands as shown in E. coli [79,82]. Although mitomycin C is widely used, norfloxacin (a fluoroquinolone) was found to enhance prophage induction, especially in strains not susceptible to induction by mitomycin C [78,83]. This may be attributed to the mechanism of norfloxacin action, which inactivates the DNA gyrase and topoisomerase IV, causing the disruption of DNA supercoiling that leads to damage [84]. There are no standardised procedures that guide the selection of the prophage-inducing agent in C. difficile, but studies have shown that the use of diverse agents on one strain could maximise prophage release and yield [78]. Regardless of the inducing agent used, to maximise yield, prophage induction has been carried out at different growth phases of the bacterial broth culture [78,83]. Although lysis and reduction of bacterial growth are generally considered to be good indicators of prophage release, we have also observed that the treated bacterial cultures often continued to grow or remained stationary despite phage release [78].
3. Diversity of C. difficile Phages
3.1. Morphological Diversity of C. difficile Phages
Isolated C. difficile phages to date belong to the Caudovirales family, which is the order of tailed phages (Table 1) [1,2,4,85,86]. Over the past decade, the phage taxonomy has been updated, and currently there is a new order called Tubulavirales along with ten new families [87]. However, as published data on C. difficile phages was based on the previous taxonomy, we will refer to these phages using the old taxonomy scheme for consistency.
There are thirty-five C. difficile phage genomes publicly available to date and all have dsDNA genomes. Twenty-six of the phages were classified as myoviruses, eight were siphoviruses and one is a phage tail-like protein (Table 1). The isolated myoviruses have been further sub-classified based on their tail lengths, which are the medium-tailed and short-tailed myoviruses [85]. No podovirus that targets C. difficile has been isolated [1]. The phage tail-like particles, also referred to as bacteriocins (or diffocins), lack a capsid and are widely isolated from various C. difficile strains either alone or simultaneously in addition to other phage morphologies [78,83]. Although the particles have been shown to have bactericidal ability, they were not able to replicate effectively to form plaques [83].
3.2. Genomic Diversity
Due to the highly diverse nature of phage genomes, there are no generalised conserved genes to characterise them as seen in the bacteria using the 16S rRNA gene [77,78]. However, specific C. difficile phage genes have been identified that could be used as molecular markers to examine diversity [77,78]. The major capsid protein is relatively conserved and has been used to identify C. difficile prophages in situ [78]. However, this marker is limited as it is too diverse and not recognisable in some phages, such as in phage phiCD27. Similarly, the minor capsid protein, gp20, is also too diverse to facilitate alignment and primer assertions [78]. Due to these limitations, the holin gene, which is identifiable in all phages, has been used to assess genetic diversity within C. difficile phages [77]. Although the holin is also limited due to its conserved nature, it is useful in distinguishing between siphoviruses and myoviruses that infect C. difficile.
The diversity of C. difficile phage genes and modularity within their genomes were described recently [1]. However, to both contextualise and understand the genetic relationships and genetic diversity within C. difficile and all Clostridial phages, we have applied our PhageClouds concept [88]. PhageClouds is a computational database, and the concept was developed for better visualisation and understanding of the relationships between phages that target any bacterium of choice [88]. This approach is based on creating phage genomic networks from whole genome similarities and thereby overcomes the limitations imposed by only examining one conserved gene. PhageClouds allows us to identify phages that are most closely genetically related to each other, here represented as particular clusters or clouds (Figure 1, Table S1). Where any phages share DNA, they group together, and we will see that the clouds are connected through those genetic similarities. On the other hand, different clouds of phages which are not connected do not have any DNA similarities.
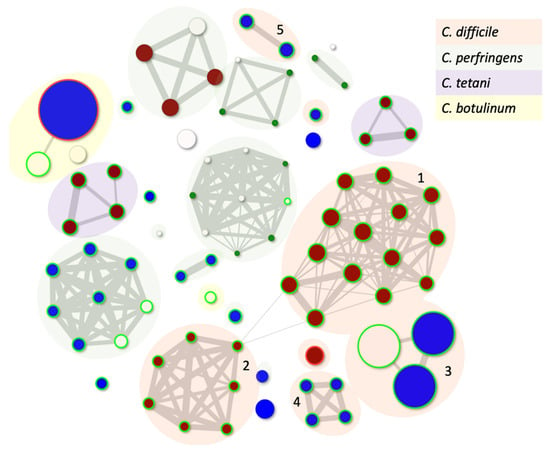
Figure 1.
Genetic relationships between all known Clostridial phages that can either independently plaque on bacterial hosts or can be induced from their host strain. The size of the dot is related to the genome size, and the colour is reflective of the morphology with red being a myovirus, blue siphovirus and white where the taxonomy is unknown. The green circle suggests the phages are temperate using Phage Leads, and the red circle shows that the phage carries an antibiotic resistance gene. Clouds numbered 1–5 represent clusters of C. difficile phages (Table S1). Phages included in the analysis are listed in Table S1.
Figure 1 shows the relationship between all known Clostridial phages. It is clear that there is no genetic relationship between the phages that infect C. difficile and other Clostridial species, as they form different clouds. There are five distinct groupings/clouds of C. difficile phages, although the major and largest two, clouds 1-2 (containing twenty-one and nine phages, respectively), are clearly connected. Interestingly, these clouds represent the myoviruses that target C. difficile (Table S1). The third cloud contains a group of eight relatively newly described related siphoviruses with genomes that are much larger than most C. difficile phages, approximately 133 kb (Clostridioides phage LIBA-2945, Clostridioides phage LIBA-6276, Clostridium phage phiCD211) [89,90]. These phages have not yet been shown to propagate using the lytic life cycle but can be induced from the genomes of their hosts and have intriguingly long tails [90]. The fourth cloud consists of the remaining C. difficile siphoviruses (Clostridium_phage_phiCD6356, Clostridium_phage_phiCD24-1, Clostridium_phage_phiCDKH01, Clostridium_phage CPD2), which are clearly genetically distinct from each other, and from all other phages sequenced to date, and thus appear on this figure as pairs or singletons.
4. Phage Mechanics of Infection
4.1. Impact of Tail Fibres on Attachment and Host Selection
The first stage of a successful phage infection is attachment to specific receptors on the bacterial host cell as shown in Figure 2 [88]. Phage binding occurs through several interactions between the receptor binding proteins (RBPs) via two stages. The first stage involves the phage tail fibres reversibly attaching to a receptor on the surface of the bacterial cell, and the second involves the irreversible attachment to the same receptor or a different receptor. The phage then injects its genetic material into the host cells [91]. Extensive research has focused on unraveling these mechanisms in Gram-negative bacteria, in particular E. coli phages, as both the organism and its phages are easier to mutate and handle in the laboratory [92]. In comparison, Gram-positive bacteria such as C. difficile are more difficult to work with in the laboratory, largely due to their fastidious nature and cell wall composition, which make them difficult to manipulate. However as there is growing interest in studying the mechanism of infection of phages targeting Gram-positive bacteria, methods to understand this interaction are being developed, and the phage receptors in Bacillus subtilis, Lactococcus lactis and Staphylococcus aureus have been identified through mutational studies [93,94,95,96]. Whilst for C. difficile phages the mechanisms of action by which phages infect their hosts are unknown, the methods developed for Gram-positive bacteria may be applied.
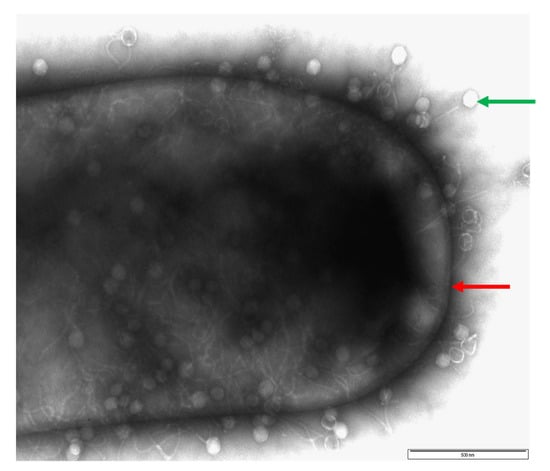
Figure 2.
Image of a CD105LC1 C. difficile cell (red arrow) surrounded by phage phiCDHS1 (green arrow), which have attached to the surface of the bacterial cell wall. Exponentially growing bacteria cultures were mixed with phage at a ratio of 1:100 (bacteria to phage ratio) and allowed to attach for 15 min. Afterward, the phage/bacterial cultures were fixed with 2% glutaraldehyde and mounted on glow-discharged pioloform/carbon-coated copper grids for 5 min. After being washed with water, samples were stained with uranyl acetate, air-dried and examined using a JEOL 1220 electron microscope at 80 kV voltage. Bar is approximately 500 nm.
However, our research group has been making progress in understanding phage–host interactions, and we investigated adsorption of C. difficile phages both to strains they do and strains they do not infect. Our study included three myoviruses (phiCDHM1, phiCDHM3 and phiCDHM6), and we identified phages phiCDHM1 and phiCDHM3 bound by ~75% to strains they infect and by less than 30% to strains they do not infect. However, phage phiCDHM6 adsorbed to all strains by ~30% regardless of whether or not it could infect the strain, despite the tail-fibre proteins of phiCDHM3 and phiCDHM6 sharing 100% homology at the amino-acid level. Thus, phage adsorption is phage–host specific, and our study provided insights into phage infection [97]. Currently, the phage receptors on C. difficile are unknown, however, we speculate that C. difficile phages could attach to the surface layer (S-layer) proteins on C. difficile cells, as phage tail-like bacteriocins were shown to use S-layer proteins as their receptors [94].
4.2. Phage Host Range
Successful phage binding of virulent phages leads to infection and the lysis of bacteria to release progeny. The range of available bacterial species or strains a phage can lyse is known as its host range, and phages that lyse multiple strains from different subgroups of the same bacterial species are more clinically useful for therapy [98]. In comparison, some phages have narrow host ranges and can only lyse one strain from a single subgroup [99]. To maximise efficacy, phage cocktails can be used which include a diverse set of phages which target different strains and thus can improve overall lysis efficiency [100]. To further improve phage efficacy and specificity, the RBPs and phage tail-fibre proteins could be genetically engineered, as they are involved in phage specificity. Resultant modified phages can therefore recognise, attach and lyse a broader set of host targets, and the method has been successfully shown in Pseudomonas aeruginosa and Acinetobacter baumannii phages [101].
4.3. Impact of Phage Infection on C. difficile Physiology
Analysis of phage genomes has highlighted C. difficile phage-encoded genes that can mediate transcriptional regulations in the bacterium. For example, C. difficile phage phiCDHM1 encodes the agr system, consisting of a cassette of genes (agrA, agrB, agrC and agrD) with the ability to modulate how the bacterium interacts with the environment. These genes impact bacterial motility, biofilm formation, defence, toxicity, replication, metabolism, sporulation, stress response and quorum sensing [3,74,102]. In addition, phage phiCD119 has been shown to modulate toxin production after lysogenisation [75].
To further understand the impact of phage infection on C. difficile, we recently conducted a transcriptional study investigating infection of phage phiCDHS-1 on C. difficile strain R20291 to determine which genes are expressed during infection. The analysis revealed that 10–20% of the bacterial host genes are differentially expressed during infection [103]. The majority of these genes were downregulated at the early stage of the phage life cycle, which includes genes responsible for metabolism and DNA replication [103]. A similar study of R20291 infection by phage CD38-2 showed that genes associated with transcriptional regulators and phosphotransferase system subunits involved in glucose, fructose, and glucitol/sorbitol uptake and metabolism were differentially expressed in the host. Other differentially expressed host genes were linked to phase variation regulated by the conserved phase-variable cell-wall protein [104]. Also, genes responsible for lysis–lysogeny decision were expressed at an early infection stage of C. difficile phage infection [103,105]. Furthermore, various genes related to pathogenicity, such as toxin production and regulation, anti-phage systems, bacterial sporulation and adhesion, were all regulated during phage infection [103,105]. Interestingly, though phage infection resulted in bacterial resistance and lysogeny development, the clones produced were less virulent, further supporting the use of C. difficile phage for therapeutic purposes, as will be discussed in the next section [103].
5. Therapeutic Application of C. difficile Phage Models of C. difficile Phage Therapy
Infection leading to lysis is the key phage asset, which can be harnessed for phage therapy and has been studied using in vitro, in vivo and ex vivo models [47,52,53,100,106]. In this section, we discuss the different models developed and used to study the lytic activity of C. difficile phages.
5.1. Culture-Based Assays
Several in vitro studies have been conducted to ascertain the efficacy of C. difficile phages to either kill or reduce bacteria using both host-range and virulence assays [45,52,53,106]. Host-range analysis is typically conducted by applying specific volumes of high-titre phage stocks on confluent cultures of C. difficile in semi-solid media, and the same phage is tested on multiple clinically relevant C. difficile strains which represent different ribotypes. Host range analysis has identified C. difficile phages to have narrow-to-broad host ranges, often lysing several ribotypes [53,64,65]. We used this method to screen the host range of our seven phages against 80 strains, representing 21 clinically relevant ribotypes from humans. We identified phiCDHM4 as having the narrowest host range, and lysed single representative strains from each of four ribotypes. In comparison, phages phiCDHM3, phiCDHS-1, and phiCDHM5 had broad host ranges and infected 20–31 strains representing 10–12 ribotypes. However, the results of host-range analysis showed complimentary coverage could be achieved by combining the phages as cocktails; for example, phage cocktail phiCDHM1+2+5+6 combined is able to lyse 18 ribotypes and 62 of the strains tested [53].
In addition to host-range analysis, killing or virulence assays are used to determine which phages or phage cocktail combinations are efficient at lysing target strains [50]. This method involves growing the target strain to an exponential stage and then infecting it with phage(s) and monitoring bacterial growth over a set time, typically 24 h. We used this method to identify optimal phage combinations for lysis of C. difficile, and we tested two-, three- and four-phage cocktails [53]. We found the three-phage cocktail, phiCDHM2+5+6, caused a 6 log10 reduction in bacterial counts over 24 h, whilst the four-phage cocktail, phiCDHM1+2+5+6, was more efficient and lysed the same culture within 3 h (0 log10) [53]. In addition, with the four-phage cocktail there was no regrowth of C. difficile over 24 h. As the four-phage cocktail was more efficacious, it could be a potential candidate for future phage clinical trials.
5.2. Biofilm Model
As discussed in the introduction, C. difficile strains can aggregate in complex biofilms in vitro, and these structures complicate therapeutic deployment of antibiotics and act as reservoirs for recurrent CDI [44,45,107,108,109,110]. Unlike antibiotics, data from our study showed that C. difficile phages can inhibit biofilm formation by penetrating and lysing established biofilms, which leads to a decrease in bacterial viability and biomass [45]. Furthermore, the four-phage cocktail we have developed, phiCDHM1+2+5+6, was more effective than using single phages at tackling biofilms and could be an assuring therapeutic option for controlling C. difficile biofilms [45].
5.3. Epithelial Cell Tissue Model
Human cell lines are informative ex vivo tools to study phage/bacterial interactions and therapeutic efficacy. We examined the interaction of the C. difficile 027 strain with phage phiCDHS-1 in the presence of two human epithelial cell lines [111]. The cell lines Caco-2 and HT-29 were selected, as both have previously been used to study the pathogenesis of C. difficile [112,113,114,115,116]. The data from the study revealed that pre-treatment of cell cultures with phiCDHS-1 one hour prior to introducing C. difficile significantly reduced C. difficile counts from 8 log10 to 3 log10 CFU/mL within eight hours. In comparison, by introducing phiCDHS-1 and C. difficile simultaneously to the epithelial cells, C. difficile counts were reduced from 8 log10 to 4 log10 CFU/mL within the same treatment time. There was established evidence that the phage was able to adsorb to the epithelial cells, which may have contributed to the effectiveness of the prophylactic treatment [111].
5.4. Batch Fermentation Model
In vitro gut and batch fermentation models simulate the human gut microbiome and have been developed as a useful tool to study the gut microbiome response to anti-infectives, including phages [52,106,117,118,119]. The main goal in using the fermentation model is to culture a complex intestinal microbiota to study microbial modulation and metabolism under controlled environmental conditions. This approach is both time-efficient and cost-effective compared to animal models [52,106,118,119,120].
One study used an in vitro batch fermentation model spiked with faecal material from a single donor [106,119]. Single-phage treatment with phiCD27 led to a substantial decrease in vegetative C. difficile cells numbers, low toxin level detection and no detrimental impact on human gut commensals [106,119]. Building on this model, we tested the effectiveness of our optimised four-phage cocktail, phiCDHM1+2+5+6, to clear C. difficile in a fermentation vessel inoculated with combined faecal slurries from four individuals from different ethnicity and age groups [52]. The phage cocktail efficiently cleared C. difficile from the model within 24 h, and C. difficile was not recovered. Phage prophylactic treatment was more effective than remedial treatment, consistent with previous data [52,106,119]. Encouragingly, in addition to preserving the gut commensals, phage treatment enhanced the colonisation of specific commensals, further strengthening their use in preventing dysbiosis and CDI [52].
5.5. Galleria Mellonella Infection Model
The use of G. mellonella as a bacterial infection model has risen in popularity within the last decade, ranging from simple survival assays to multifaceted experiments. The larval stage of the greater wax moth is used as a favorable ethical, financial and experimental ease model compared to other models [121,122]. G. mellonella is predominately used as a screening model to assess virulence of a particular bacterium or gene. The survival outcome and melanization of larvae during infection provide a macro-view of host–infection outcome, whilst changes in larval gene expression and proteomic responses have been measured to provide a more precise insight into infection [51,123,124]. The larval response to bacterial infection and toxic substances is similar to other commonly used cell lines and models [125]. Some caveats still exist, however, as G. mellonella larvae only possess an innate immune response, which, although sharing similarities to the mammalian humoral and cellular responses, lacks the complexity of mammalian-based models [126,127]. The lack of adaptive immune response, however, can be useful to study solely for the interactions between pathogen-innate immunity.
Before using this model to explore CDI phage therapy, we first established colonisation of the G. mellonella larvae with C. difficile using oral inoculation rather than the hemolymph for better reproducibility. Having established this model, we then tested the efficacy of phage cocktail phiCDHM1+2+5+6 to reduce C. difficile colonisation and improve the survival of challenged larvae. Three phage treatment regimens were tested: prophylactic (phage inoculation prior to bacterial infection); concurrent (simultaneous bacterial and phage infection); and remedial (phage treatment after bacterial infection). Prophylactic phage treatment was the most effective treatment, and 100% of larvae survived after 60 h. In comparison, there was a 0% survival rate of larvae infected only with C. difficile. Phage treatment also reduced C. difficile counts to 2 log10 CFU/larva, whilst in larva infected only with C. difficile, counts were 8 log10 CFU/larva [45]. This observation of prophylactic treatment being the most effective concurred with the biofilm data and with the cell tissue culture assays [45,111].
We were able to further refine the G. mellonella CDI model by measuring insect stress genes as biomarkers to detect and monitor disease progression and recuperation during phage therapy in the insects [51,103]. This approach allowed an increased resolution into determining the phage cocktail efficacy and other potential antimicrobial agents [51,123]. Such advancements in the development of the G. mellonella infection model provide an attractive alternative to more conventional approaches to studying CDI and might provide a valuable tool to track infection for other pathogens.
5.6. Hamster Infection Model
The study of phage therapy within in vitro models usually provides the preliminary groundwork in pre-clinical studies, allowing more experimental control and traceability without raising ethical complications. However, the linearity of such parameters limits the complexity or representability of assessment when compared to an in vivo model, where a systemic approach provides additional dimensions of interaction, such as an immune system, microbiota or even confounding variables [128]. Hamster models (especially Golden Syrian hamsters) have been the predominant choice to study CDI, sharing similarities with antibiotic-induced susceptibility and clinical manifestations observed in humans [129,130]. As a result, they are ideal candidates for phage therapy studies and efficacy testing of different cocktails.
The first reported phage therapy in a CDI-induced in vivo study was conducted by Ramesh et al. where hamsters were subjected to clindamycin-induced CDI and treated with various doses of phage CD140 [100]. Untreated hamsters were susceptible to CDI within 72 h, displaying diarrhoea and haemorrhagic and fluid-filled ceca, while all but one phage-treated hamster survived [100]. C. difficile was recovered from all culled hamsters and, interestingly, the strain recovered from them was resistant to phage CD140. The emphasis on the fleeting protection of phages were highlighted, as CD140 was not recovered from the cecal contents from the hamsters fourteen days after phage therapy. Additionally, through clindamycin-induced C. difficile reinfection fourteen days after phage therapy, all hamsters succumbed to C. difficile rechallenge of the same strain, further enforcing the temporal nature of phage therapy. The ability for hamsters to pick up environmental phages was observed, as 50% of the control group exhibited phage recovery, which was attributed to colonisation through phage-contaminated environments previously used to house phage-treated hamsters [100]. As a by-product of their study into in vivo phage lysogenisation with PCR, Govind et al. demonstrated an increased survival rate of phage-treated hamsters of 5 days, whereas the non-treated controls died within 48 h [75].
The assessment of phage therapy for CDI in vivo is still in its infancy; the use of phage cocktails in the hamster CDI model has only been reported from our laboratory. Different combinations of C. difficile phages were analysed first in vitro, and the five most promising combinations were assessed in the clindamycin-induced hamster colitis for up to five days [53]. The role of phage treatment in C. difficile colonisation was assessed through bacterial recovery from luminal and tissue samples. The most effective treatment was determined to be the four-phage combination (phiCDHM1+2+5+6), which was able to reduce recovered bacterial load in the lumen samples by 4 log10 CFU/g and tissue samples by 2 log10 CFU/g. Furthermore, this combination of phage intervention promoted increased survivability in hamsters by approximately ~32 h compared to untreated controls [53].
6. Genetic Manipulation of C. difficile Phages
Over the last fifteen years, significant progress has been made in developing tools to mutate C. difficile [131]. Researchers have faced numerous hurdles, as gene transfer in C. difficile is less efficient, and developing stable mutants in C. difficile has been a consistent problem due to the lack of counter-selection markers [132]. However, tools are now available to produce stable mutations in C. difficile, which can be applied to mutate C. difficile phages in their lysogenic state, but no genetic tools are currently available to mutate lytic phages [2]. This section will describe the three main C. difficile genetic manipulation systems, the ClosTron system, Clostridium shuttle plasmids and CRISPR, which are all tools that can be used to genetically mutate C. difficile phages [133]. Of particular interest would be the ability to delete the lysogeny-associated genes in phages, which will likely pose a problem for their downstream therapeutic applications [53].
6.1. ClosTron System
The ClosTron system uses broad-host-range group II introns for directed mutagenesis within Clostridia. Group II introns are described as catalytic RNAs that can excise themselves from RNA transcripts and then insert themselves into a new target [134]. The mobility of group II introns provides a method for gene disruption, and the intron target specificity can be altered by changing the DNA sequence that encodes the section of the intron. The group II introns carry an open reading frame in which they have a multifunctional intron-encoded protein (IEP) [135]. Through the action of the IEP, the introns are able to self-catalytically splice out of the targeted RNA sequence in the host gene. The IEP synthesises the corresponding complementary DNA strand via activity of reverse transcriptase [134]. The ClosTron system also includes an integrated functional antibiotic resistance gene within the coding region of the group II intron element; thereby, the acquisition of antibiotic resistance is coupled with integration and helps to positively select for integration events [136,137].
The directed mutagenesis process involves four clear steps: step 1 is intron design; step 2 is plasmid construction, both of which are done using an easy-to-follow online tool (http://clostron.com (accessed on 28 October 2022)); step 3 is plasmid transfer via conjugation; and step 4 is integrant isolation, which is performed using standard methods [136]. The ClosTron system has been successfully used for directed mutagenesis in C. difficile and has helped to improve our knowledge on adhesion and virulence genes, and genes involved in dissemination of C. difficile [138,139,140]. Similarly, the system can be used to mutate and study temperate C. difficile phages.
6.2. Clostridium Shuttle Plasmids
Other tools developed for direct mutagenesis via allele exchange include a range of pMTL8000 Clostridium-Escherichia coli modular shuttle plasmids [141]. Allele exchange occurs when the native allele of DNA is exchanged with a new allele that contains a mutation by homologous recombination [142]. The shuttle plasmids in the pMTL8000 range are modular and all include a Gram-negative replicon, an antibiotic selection marker, a Gram-positive replicon, a conjugal transfer function and/or a multiple cloning site [130]. The plasmids contain a Gram-negative replicon, which allows all the cloning to be initially done in E. coli, i.e., genes of interest and flanking regions added to the plasmid in E. coli, as cloning directly into C. difficile is difficult. Then the plasmid can be transferred to Clostridium via conjugation [143]. However, it should be noted that DNA transfer to C. difficile occurs at low frequencies and varies significantly between C. difficile strains. Furthermore, the conjugation efficiency is dependent on the length of the flanking regions used and the media used for the conjugation process [142].
The pMTL8000 plasmids are replication-defective plasmids (pseudosuicide) and are used for allele exchange via a two-step recombination process. The first recombination event involves the integration of the plasmid into the target genome and is referred to as a single crossover event. Strains in which the first recombination event has occurred grow rapidly on selective media, and their colonies are larger in size, which makes them easier to identify [131]. The second recombination event is the plasmid excision from the genome, at which point cells can either revert to wild-type or generate mutants [142]; however, the frequency of the second recombination event occurring to generate double-crossover mutants is low and very laborious [144]. To overcome this problem, a counter selection marker (toxic under certain conditions), the codA gene of E. coli, has been identified and used successfully to generate double-crossover mutants at a higher frequency [132].
The shuttle plasmids were used by our group to tag C. difficile phages in their lysogenic state with luminescence luxAB genes (reporter phages) with the aim of developing a phage-based diagnostic test. To design the reporter phages, non-essential phage genes were replaced with the luxAB genes, which would then be expressed during phage infection. We found this method required extensive optimisation, and once the reporter phages had successfully been constructed, the luxAB genes were unstable within the phage genome and were lost during phage replication. We are further optimising the method to develop stable mutations in C. difficile phages [145].
6.3. CRISPR Technology
The CRISPR system is an RNA-mediated immune system in prokaryotic cells, and the type II CRISPR-Cas9 has been widely used to genetically modify both eukaryotic and prokaryotic cells. In the CRISPR-Cas9 system, the Cas9 is directed by guide RNA (gRNA) to the region of chromosomal DNA in which to make the desired mutation, which then subsequently leads to breakage of the double-stranded DNA [146]. The system allows generation of stable mutants on the host chromosome and has been used to mutate numerous lytic and lysogenic phages of important pathogens including Bacillus subtilis, Vibrio Cholerae and E. coli 0157:H7 [147,148,149,150]. To date, the system has not been used to mutate C. difficile phages, but in the past three years, CRISPR-Cas9 plasmids have been designed to further study genes in C. difficile, which could potentially be applied to its associated phages [151].
The methods described in this section can be applied to temperate C. difficile phages, but future tools need to focus on developing methods to mutate lytic phages. For example, the lytic T7 E. coli phage was mutated using CRISPR–Cas3 technology. Phages were first propagated on a E. coli strain that harboured a plasmid, with the donor DNA and the phage sequences that flank either side of the gene to be deleted. Phage recombinants were enriched by plating on another E. coli strain that contained three plasmids that encode for CRISPR-Cas3 activity and the spacer sequence, which is complementary to the target gene. Phage recombinants were isolated at a rate of approximately 40% [152]. A simpler method was used to engineer a Lactococcus lactis phage P2, whereby the plasmid encoding CRISPR-Cas9 and the plasmid-encoded donor DNA were added to one strain. The authors showed that the method could be used to introduce insertion, deletion and point mutation in several sites in the genome of phage P2 [153]. It may be difficult to use similar CRISPR-based methods to mutate C. difficile phages, as it is currently difficult to transfer just one plasmid to C. difficile. Furthermore, there are no data to support stability of two or three plasmids in the same strain. Therefore, moving forward, a plasmid that encodes for CRISPR activity and includes the donor DNA is needed.
7. Encapsulation and Formulation of Therapeutic C. difficile Phages
Isolation, characterisation and purification of phages form the initial stages in the roadmap for commercialisation of therapeutic phages. It is equally important to evaluate their compatibility with commercialisation processes, including formulation, scale-up, storage and delivery. Success stories are often hindered by the lack of consideration to the post-processes and therefore should be evaluated alongside the first stage [154,155].
Formulating phage for delivery involves many challenges, since conventional off-the-shelf solutions are not suitable. Active pharmaceutical ingredients (APIs) are compatible with a catalogue of formulation ingredients and have been studied for decades; in the case of phages, however, there is limited knowledge. The first article discussing phage formulation was published in 2002 using the model Salmonella-specific phage, Felix O1. The authors encapsulated the phage in alginate and chitosan particles for oral delivery [156]. Since then, there has been a steady rise in the number of publications addressing the challenges around phage encapsulation and formulation.
Phages are prone to environmental changes such as temperature, humidity, pH and mechanical shearing, which makes them challenging to encapsulate and protect for therapeutic applications [157,158,159,160,161,162,163]. Apart from formulating phages in compatible materials, the technique used for encapsulation can prove detrimental to the viability of phages. Hence, the choice of encapsulating technique plays a crucial role in the development of commercially viable phage products.
Spray-drying is a well-known industrial method for the encapsulation of API into dry powder form, which is easier to transport in comparison to liquids and gels. The method uses high temperatures (50–300 °C) to form aerosolized droplets which undergo evaporation, leaving fine particles. It has also been employed for the encapsulation of phages due to its ease of use and one-step process for obtaining a dry powder carrying phages [157,158,164,165,166]. In all instances, there has been reported losses of viability of phage ranging from 0.5–2 log10 PFU/g attributed to the high operating temperatures. However, phage loss can be limited by testing different encapsulation materials and adding sugars such as trehalose, which can protect phages during the drying process.
C. difficile phages were encapsulated in a methyl-methacrylate copolymer known as Eudragit S100, which is responsive to changes in pH [167]. Here, Eudragit was used in combination with alginate to produce composite microparticles using microfluidic technology, which enables precise control over size and monodispersity of the microparticles without the need for shear or high temperature. The microparticles were able to protect the phage from pH 2 (SGF: simulated gastric fluid) for a period of 3 h. A total loss of 1 log10 PFU/mL was observed during this period, resulting in the final phage titre of 5 log10 PFU/mL. The results demonstrated that the combination of the formulation and the technique employed to produce microparticles helped minimise the loss of phage post SGF exposure. Encapsulating phages using this method will enable successful transit of the phage to the site of infection in the lower GI tract, where C. difficile colonises in humans [167].
In addition to the above encapsulation techniques, there are a plethora of combinations which can be tested to find the optimal method and material for phage encapsulation. A more advanced method which offers further benefits in protecting phages and ensuring a site-specific release is lipid nanoparticles [168]. These can further be functionalised and combined with polymeric materials to achieve a desired release profile. Further work is still required in this field; however, with the continued success of the encapsulation of mRNA and other biologics, it gives hope to the future of phage encapsulation.
8. Conclusions
Research on C. difficile phages has progressed significantly over the past decade, from phage isolation, to sequencing and understanding phage-host interactions. In addition, robust in vitro, ex vivo and in vivo models have been developed to test efficacy of phages, and the collective data highlights that phages are effective against C. difficile. The barrier we face is that all isolated phages are temperate, and thus with current regulation may not be ideal for therapy. However, with the progression of genetic tools, we will be able to mutate or delete undesirable genes and progress the therapeutic use of phages against C. difficile.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/article/10.3390/v14122772/s1, Table S1: Phages included in the phage cloud analysis shown in Figure 1.
Author Contributions
J.Y.N. and M.R.J.C. structured the manuscript; J.Y.N., A.M.T., S.J.R., J.S., G.K.V., A.S.A.D., J.K.J.C. and T.S.-P. contributed to drafting various sections of the manuscript; J.Y.N., A.M.T. and M.R.J.C. coordinated the sections together to produce the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Biotechnology and Biological Sciences Research Council (BBSRC), grant number RM38G0140 awarded to M.R.J.C.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank Natalie Allcock and Ania Straatman-Iwanowska (Electron Microscopy facility, Core Biotechnology Services, University of Leicester, Leicester, United Kingdom) for producing the electron image of Figure 2.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heuler, J.; Fortier, L.C.; Sun, X. Clostridioides difficile phage biology and application. FEMS Microbiol. Rev. 2021, 45, fuab012. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Clokie, M.R.J. Clostridium difficile phages: Still difficult? Front. Microbiol. 2014, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. Bacteriophage behavioral ecology: How phages alter their bacterial host’s habits. Bacteriophage 2014, 4, e29866. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Uematsu, S. Phage therapy for Clostridioides difficile infection. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.C.; O’Toole, E. Intestinal flora in new-born infants. Am. J. Dis. Child. 1935, 49, 390–402. [Google Scholar] [CrossRef]
- Cohen, L.E.; McNeill, C.J.; Wells, R.F. Clindamycin-Associated Colitis. JAMA 1973, 223, 1379–1380. [Google Scholar] [CrossRef]
- Noren, T.; Tang-Feldman, Y.J.; Cohen, S.H.; Silva, J.; Olcen, P. Clindamycin resistant strains of Clostridium difficile isolated from cases of C. difficile associated diarrhea (CDAD) in a hospital in Sweden. Diagn. Microbiol. Infect. Dis. 2002, 42, 149–151. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Lacy, D.B. Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell. Infect. Microbiol. 2012, 2, 28. [Google Scholar] [CrossRef]
- Lyerly, D.M.; Lockwood, D.E.; Richardson, S.H.; Wilkins, T.D. Biological activities of toxins A and B of Clostridium difficile. Infect. Immun. 1982, 35, 1147–1150. [Google Scholar] [CrossRef]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef]
- Carter, G.P.; Lyras, D.; Allen, D.L.; Mackin, K.E.; Howarth, P.M.; O’Connor, J.R.; Rood, J.I. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J. Bacteriol. 2007, 189, 7290–7301. [Google Scholar] [CrossRef]
- Carter, G.P.; Douce, G.R.; Govind, R.; Howarth, P.M.; Mackin, K.E.; Spencer, J.; Buckley, A.M.; Antunes, A.; Kotsanas, D.; Jenkin, G.A.; et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 2011, 7, e1002317. [Google Scholar] [CrossRef]
- Rupnik, M.; Grabnar, M.; Geric, B. Binary toxin producing Clostridium difficile strains. Anaerobe 2003, 9, 289–294. [Google Scholar] [CrossRef]
- Govind, R.; Dupuy, B. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 2012, 8, e1002727. [Google Scholar] [CrossRef]
- Abt, M.C.; McKenney, P.T.; Pamer, E.G. Clostridium difficile colitis: Pathogenesis and host defence. Nat. Rev. Microbiol. 2016, 14, 609–620. [Google Scholar] [CrossRef]
- Wilcox, M.H.; Shetty, N.; Fawley, W.N.; Shemko, M.; Coen, P.; Birtles, A.; Cairns, M.; Curran, M.D.; Dodgson, K.J.; Green, S.M.; et al. Changing Epidemiology of Clostridium difficile Infection Following the Introduction of a National Ribotyping-Based Surveillance Scheme in England. Clin. Infect. Dis. 2012, 55, 1056–1063. [Google Scholar] [CrossRef]
- Vohra, P.; Poxton, I.R. Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology 2011, 157, 1343–1353. [Google Scholar] [CrossRef]
- Seddon, S.V.; Hemingway, I.; Borriello, S.P. Hydrolytic enzyme production by Clostridium difficile and its relationship to toxin production and virulence in the hamster model. J. Med. Microbiol. 1990, 31, 169–174. [Google Scholar] [CrossRef]
- Underwood, S.; Guan, S.; Vijayasubhash, V.; Baines, S.D.; Graham, L.; Lewis, R.J.; Wilcox, M.H.; Stephenson, K. Characterization of the Sporulation Initiation Pathway of Clostridium difficile and Its Role in Toxin Production. J. Bacteriol. 2009, 191, 7296–7305. [Google Scholar] [CrossRef]
- Baban, S.T.; Kuehne, S.A.; Barketi-Klai, A.; Cartman, S.T.; Kelly, M.L.; Hardie, K.R.; Kansau, I.; Collignon, A.; Minton, N.P. The Role of Flagella in Clostridium difficile Pathogenesis: Comparison between a Non-Epidemic and an Epidemic Strain. PLoS ONE 2013, 8, e73026. [Google Scholar] [CrossRef]
- Lyerly, D.M.; Krivan, H.C.; Wilkins, T.D. Clostridium difficile: Its disease and toxins. Clin. Microbiol. Rev. 1988, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Scutera, S.; Savoia, D. Novel avenues for Clostridium difficile infection drug discovery. Expert Opin. Drug Discov. 2013, 8, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile Infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Gould, C.V.; McDonald, L.C. Current Status of Clostridium difficile Infection Epidemiology. Clin. Infect. Dis. 2012, 55, S65–S70. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.B.; Hollenbeak, C.S. Clostridium difficile colitis: Factors associated with outcome and assessment of mortality at a national level. J. Gastrointest. Surg. 2011, 15, 1548–1555. [Google Scholar] [CrossRef]
- Shek, F.W.; Stacey, B.S.F.; Rendell, J.; Hellier, M.D.; Hanson, P.J.V. The rise of Clostridium difficile: The effect of length of stay, patient age and antibiotic use. J. Hosp. Infect. 2000, 45, 235–237. [Google Scholar] [CrossRef]
- Riley, T.V.; Codde, J.P.; Rouse, I.L. Increased length of hospital stay due to Clostridium difficile associated diarrhoea. Lancet 1995, 345, 455–456. [Google Scholar] [CrossRef]
- Tay, H.L.; Chow, A.; Ng, T.M.; Lye, D.C. Risk factors and treatment outcomes of severe Clostridioides difficile infection in Singapore. Sci. Rep. 2019, 9, 13440. [Google Scholar] [CrossRef]
- UKHSA. Clostridioides Difficile Infection Updated Guidance on Management and Treatment; UKHSA Publication gateway number GOV-12513; UKHSA: London, UK, 2022; p. 33.
- King, A.; Mullish, B.H.; Williams, H.R.T.; Aylin, P. Comparative epidemiology of Clostridium difficile infection: England and the USA. Int. J. Qual. Health Care 2017, 29, 785–791. [Google Scholar] [CrossRef]
- Petrosillo, N.; Granata, G.; Cataldo, M.A. Novel Antimicrobials for the Treatment of Clostridium difficile Infection. Front. Med. 2018, 5, 96. [Google Scholar] [CrossRef]
- Dinh, A.; Le Monnier, A.; Emery, C.; Alami, S.; Torreton, É.; Duburcq, A.; Barbier, F. Predictors and burden of hospital readmission with recurrent Clostridioides difficile infection: A French nation-wide inception cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1297–1305. [Google Scholar] [CrossRef]
- Kociolek, L.K.; Gerding, D.N. Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 150–160. [Google Scholar] [CrossRef]
- Stevens, V.W.; Nelson, R.E.; Schwab-Daugherty, E.M.; Khader, K.; Jones, M.M.; Brown, K.A.; Greene, T.; Croft, L.D.; Neuhauser, M.; Glassman, P.; et al. Comparative effectiveness of vancomycin and metronidazole for the prevention of recurrence and death in patients with Clostridium difficile infection. JAMA Intern. Med. 2017, 177, 546–553. [Google Scholar] [CrossRef]
- Alves, J.D.F.; Yamaguti, A.; de Mendonça, J.S.; de Melo Gamba, C.; Fonseca, C.L.; Paraskevopoulos, D.K.S.; de Paula, A.I.; Hosino, N.; Costa, S.F.; Guimarães, T. Metronidazole for Treatment of Clostridioides difficile Infections in Brazil: A Single-Center Experience and Risk Factors for Mortality. Antibiotics 2022, 11, 1162. [Google Scholar] [CrossRef]
- Giau, V.V.; Lee, H.; An, S.S.A.; Hulme, J. Recent advances in the treatment of C. difficile using biotherapeutic agents. Infect. Drug Resist. 2019, 12, 1597–1615. [Google Scholar] [CrossRef]
- Singh, T.; Bedi, P.; Bumrah, K.; Singh, J.; Rai, M.; Seelam, S. Updates in Treatment of Recurrent Clostridium difficile Infection. J. Clin. Med. Res. 2019, 11, 465–471. [Google Scholar] [CrossRef]
- Kutter, E.M.; Kuhl, S.J.; Abedon, S.T. Re-establishing a place for phage therapy in Western medicine. Future Microbiol. 2015, 10, 685–688. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Mangieri, N.; Picozzi, C.; Cocuzzi, R.; Foschino, R. Evaluation of a Potential Bacteriophage Cocktail for the Control of Shiga-Toxin Producing Escherichia coli in Food. Front. Microbiol. 2020, 11, 1801. [Google Scholar] [CrossRef]
- Cos, P.; Tote, K.; Horemans, T.; Maes, L. Biofilms: An Extra Hurdle for Effective Antimicrobial Therapy. Curr. Pharm. Des. 2010, 16, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Gebreyohannes, G.; Nyerere, A.; Bii, C.; Sbhatu, D.B. Challenges of intervention, treatment, and antibiotic resistance of biofilm-forming microorganisms. Heliyon 2019, 5, e02192. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.F.; Valiente, E.; Faulds-Pain, A.; Donahue, E.H.; Wren, B.W. Characterisation of Clostridium difficile Biofilm Formation, a Role for Spo0A. PLoS ONE 2012, 7, e50527. [Google Scholar] [CrossRef] [PubMed]
- Nale, J.Y.; Chutia, M.; Carr, P.; Hickenbotham, P.; Clokie, M.R.J. ‘Get in early’; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front. Microbiol. 2016, 7, 1383. [Google Scholar] [CrossRef] [PubMed]
- Sell, T.L.; Schaberg, D.R.; Fekety, F.R. Bacteriophage and bacteriocin typing scheme for Clostridium difficile. J. Clin. Microbiol. 1983, 17, 1148–1152. [Google Scholar] [CrossRef]
- Dei, R. Observations on phage-typing of Clostridium difficile: Preliminary evaluation of a phage panel. Eur. J. Epidemiol. 1989, 5, 351–354. [Google Scholar] [CrossRef]
- Phothichaisri, W.; Ounjai, P.; Phetruen, T.; Janvilisri, T.; Khunrae, P.; Singhakaew, S.; Wangroongsarb, P.; Chankhamhaengdecha, S. Characterization of Bacteriophages Infecting Clinical Isolates of Clostridium difficile. Front. Microbiol. 2018, 9, 1701. [Google Scholar] [CrossRef]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef]
- Luong, T.; Salabarria, A.C.; Roach, D.R. Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going? Clin. Ther. 2020, 42, 1659–1680. [Google Scholar] [CrossRef]
- Nale, J.Y.; Chutia, M.; Cheng, J.K.J.; Clokie, M.R.J. Refining the Galleria mellonella Model by Using Stress Marker Genes to Assess Clostridioides difficile Infection and Recuperation during Phage Therapy. Microorganisms 2020, 8, 1306. [Google Scholar] [CrossRef]
- Nale, J.; Redgwell, T.A.; Millard, A.; Clokie, M.R.J. Efficacy of an Optimised Bacteriophage Cocktail to Clear Clostridium difficile in a Batch Fermentation Model. Antibiotics 2018, 7, 13. [Google Scholar] [CrossRef]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 2020, 11, e00019-20. [Google Scholar] [CrossRef]
- Goh, S.; Hussain, H.; Chang, B.J.; Emmett, W.; Riley, T.V.; Mullany, P. Phage ϕC2 Mediates Transduction of Tn6215, Encoding Erythromycin Resistance, between Clostridium difficile Strains. mBio 2013, 4, e00840-13. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Townsend, E.M.; Kelly, L.; Muscatt, G.; Box, J.D.; Hargraves, N.; Lilley, D.; Jameson, E. The Human Gut Phageome: Origins and Roles in the Human Gut Microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 498. [Google Scholar] [CrossRef]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.L.; Chan, P.K.S.; Chan, M.C.W.; Wu, J.C.Y.; et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018, 67, 634–643. [Google Scholar]
- Horgan, M.; O’Sullivan, O.; Coffey, A.; Fitzgerald, G.F.; van Sinderen, D.; McAuliffe, O.; Ross, R.P. Genome analysis of the Clostridium difficile phage PhiCD6356, a temperate phage of the Siphoviridae family. Gene 2010, 462, 34–43. [Google Scholar] [CrossRef]
- He, M.; Sebaihia, M.; Lawley, T.D.; Stabler, R.A.; Dawson, L.F.; Martin, M.J.; Holt, K.E.; Seth-Smith, H.M.; Quail, M.A.; Rance, R.; et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. USA 2010, 107, 7527–7532. [Google Scholar] [CrossRef]
- Ramírez-Vargas, G.; Goh, S.; Rodríguez, C. The Novel Phages phiCD5763 and phiCD2955 Represent Two Groups of Big Plasmidial Siphoviridae Phages of Clostridium difficile. Front. Microbiol. 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Flores, C.O.; Lawley, T.D.; Clokie, M.R.J. Abundant and Diverse Clustered Regularly Interspaced Short Palindromic Repeat Spacers in Clostridium difficile Strains and Prophages Target Multiple Phage Types within This Pathogen. mBio 2014, 5, e01045-01013. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.J.; Barylski, J.; Hargreaves, K.R.; Millard, A.A.; Vinner, G.K.; Clokie, M.R. Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology. Viruses 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Riley, T.V.; Chang, B.J. Isolation and Characterization of Temperate Bacteriophages of Clostridium difficile. Appl. Environ. Microbiol. 2005, 71, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Meessen-Pinard, M.; Sekulovic, O.; Fortier, L.C. Evidence of In Vivo Prophage Induction during Clostridium difficile Infection. Appl. Environ. Microbiol. 2012, 78, 7662–7670. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Colvin, H.V.; Patel, K.V.; Clokie, J.; Clokie, M.R.J. Genetically diverse Clostridium difficile strains harbouring abundant prophages in an estuarine environment. Appl. Environ. Microbiol. 2013, 79, 6236–6243. [Google Scholar] [CrossRef]
- Whittle, M.J.; Bilverstone, T.W.; van Esveld, R.J.; Lücke, A.C.; Lister, M.M.; Kuehne, S.A.; Minton, N.P. A Novel Bacteriophage with Broad Host Range against Clostridioides difficile Ribotype 078 Supports SlpA as the Likely Phage Receptor. Microbiol. Spectr. 2022, 10, e0229521. [Google Scholar] [CrossRef]
- Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef]
- Goh, S.; Chang, B.J.; Riley, T.V. Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 2005, 54, 129–135. [Google Scholar] [CrossRef]
- Govind, R.; Vediyappan, G.; Rolfe, R.D.; Dupuy, B.; Fralick, J.A. Bacteriophage-Mediated Toxin Gene Regulation in Clostridium difficile. J. Virol. 2009, 83, 12037–12045. [Google Scholar] [CrossRef]
- Sekulovic, O.; Meessen-Pinard, M.; Fortier, L.C. Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J. Bacteriol. 2011, 193, 2726–2734. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. What Does the Talking?: Quorum Sensing Signalling Genes Discovered in a Bacteriophage Genome. PLoS ONE 2014, 9, e85131. [Google Scholar] [CrossRef]
- Govind, R.; Fralick, J.A.; Rolfe, R.D. In vivo lysogenization of a Clostridium difficile bacteriophage ΦCD119. Anaerobe 2011, 17, 125–129. [Google Scholar]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Shan, J.; Patel, P.V.; Hickenbotham, P.T.; Nale, J.Y.; Hargreaves, K.R.; Clokie, M.R.J. Prophage carriage and diversity within clinically relevant strains of Clostridium difficile. Appl. Environ. Microbiol. 2012, 78, 6027–6034. [Google Scholar] [CrossRef]
- Nale, J.Y.; Shan, J.; Hickenbotham, P.T.; Fawley, W.N.; Wilcox, M.H.; Clokie, M.R.J. Diverse Temperate Bacteriophage Carriage in Clostridium difficile 027 Strains. PLoS ONE 2012, 7, e37263. [Google Scholar] [CrossRef]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol. 2009, 501, 69–76. [Google Scholar] [CrossRef]
- Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages using the small drop plaque assay system. Methods Mol. Biol. 2009, 501, 81–85. [Google Scholar] [CrossRef]
- Paolo, U.G. Induction by Mitomycin C of recA Protein Synthesis in Bacteria and Spheroplast. J. Biol. Chem. 1982, 257, 14932–14936. [Google Scholar]
- Sangster, W.; Hegarty, J.P.; Stewart, D.B., Sr. Phage tail-like particles kill Clostridium difficile and represent an alternative to conventional antibiotics. Surgery 2015, 157, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Matsushiro, A.; Sato, K.; Miyamoto, H.; Yamamura, T.; Honda, T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J. Bacteriol. 1999, 181, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Clokie, J. A Taxonomic Review of Clostridium difficile Phages and Proposal of a Novel Genus, “Phimmp04likevirus”. Viruses 2015, 7, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Phage Classification and Characterization. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Methods in Molecular Biology™; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 127–140. [Google Scholar]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Pineros, G.; Millard, A.; Michniewski, S.; Scanlan, D.; Sirén, K.; Reyes, A.; Petersen, B.; Clokie, M.R.J.; Sicheritz-Pontén, T. From Trees to Clouds: PhageClouds for Fast Comparison of ∼640,000 Phage Genomic Sequences and Host-Centric Visualization Using Genomic Network Graphs. PHAGE 2021, 2, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Sekulovic, O.; Dupuy, B.; Soutourina, O.; Monot, M.; Fortier, L.C. High Prevalence and Genetic Diversity of Large phiCD211 (phiCDIF1296T)-Like Prophages in Clostridioides difficile. Appl. Environ. Microbiol. 2018, 84, e02164-02117. [Google Scholar] [CrossRef]
- Murillo, T.; Ramírez-Vargas, G.; Riedel, T.; Overmann, J.; Andersen, J.M.; Guzmán-Verri, C.; Chaves-Olarte, E.; Rodríguez, C. Two Groups of Cocirculating, Epidemic Clostridiodes difficile Strains Microdiversify through Different Mechanisms. Genome Biol. Evol. 2018, 10, 982–998. [Google Scholar] [CrossRef]
- Dowah, A.S.A.; Clokie, M.R.J. Review of the nature, diversity and structure of bacteriophage receptor binding proteins that target Gram-positive bacteria. Biophys. Rev. 2018, 10, 535–542. [Google Scholar] [CrossRef]
- Mahony, J.; van Sinderen, D. Gram-positive phage-host interactions. Front. Microbiol. 2015, 6, 61. [Google Scholar] [CrossRef]
- Mahony, J.; Cambillau, C.; van Sinderen, D. Host recognition by lactic acid bacterial phages. FEMS Microbiol. Rev. 2017, 41, S16–S26. [Google Scholar] [CrossRef]
- Li, X.; Koç, C.; Kühner, P.; Stierhof, Y.D.; Krismer, B.; Enright, M.C.; Penadés, J.R.; Wolz, C.; Stehle, T.; Cambillau, C.; et al. An essential role for the baseplate protein Gp45 in phage adsorption to Staphylococcus aureus. Sci. Rep. 2016, 6, 26455. [Google Scholar] [CrossRef]
- Habann, M.; Leiman, P.G.; Vandersteegen, K.; Van den Bossche, A.; Lavigne, R.; Shneider, M.M.; Bielmann, R.; Eugster, M.R.; Loessner, M.J.; Klumpp, J. Listeria phage A511, a model for the contractile tail machineries of SPO1-related bacteriophages. Mol. Microbiol. 2014, 92, 84–99. [Google Scholar] [CrossRef]
- São-José, C.; Baptista, C.; Santos, M.A. Bacillus subtilis operon encoding a membrane receptor for bacteriophage SPP1. J. Bacteriol. 2004, 186, 8337–8346. [Google Scholar] [CrossRef]
- Thanki, A.M.; Taylor-Joyce, G.; Dowah, A.; Nale, J.Y.; Malik, D.; Clokie, M.R.J. Unravelling the Links between Phage Adsorption and Successful Infection in Clostridium difficile. Viruses 2018, 10, 411. [Google Scholar] [CrossRef]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef]
- Dingle, K.E.; Elliott, B.; Robinson, E.; Griffiths, D.; Eyre, D.W.; Stoesser, N.; Vaughan, A.; Golubchik, T.; Fawley, W.N.; Wilcox, M.H.; et al. Evolutionary History of the Clostridium difficile Pathogenicity Locus. Genome Biol. Evol. 2014, 6, 36–52. [Google Scholar] [CrossRef]
- Ramesh, V.; Fralick, J.A.; Rolfe, R.D. Prevention of Clostridium difficile -induced ileocecitis with Bacteriophage. Anaerobe 1999, 5, 69–78. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiology and Molecular Biology Reviews 2016, 80, 523–543. [Google Scholar] [CrossRef]
- Fortier, L.-C. Bacteriophages Contribute to Shaping Clostridioides (Clostridium) difficile Species. Front. Microbiol. 2018, 9, 2033. [Google Scholar] [CrossRef]
- Nale, J.Y.; Al-Tayawi, T.S.; Heaphy, S.; Clokie, M.R.J. Impact of Phage CDHS-1 on the Transcription, Physiology and Pathogenicity of a Clostridioides difficile Ribotype 027 Strain, R20291. Viruses 2021, 13, 2262. [Google Scholar] [CrossRef] [PubMed]
- Sekulovic, O.; Fortier, L.C. Global transcriptional response of Clostridium difficile carrying the phiCD38-2 prophage. Appl. Environ. Microbiol. 2014, 81, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Y.; Dong, K.; Kuo, C.J.; Li, C.; Zhu, Y.Q.; Qin, J.; Li, Q.T.; Chang, Y.F.; Guo, X.; et al. Isolation and Characterization of the Novel Phage JD032 and Global Transcriptomic Response during JD032 Infection of Clostridioides difficile Ribotype 078. mSystems 2020, 5, e00017-20. [Google Scholar] [CrossRef] [PubMed]
- Meader, E.; Mayer, M.J.; Gasson, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe 2010, 16, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Normington, C.; Moura, I.B.; Bryant, J.A.; Ewin, D.J.; Clark, E.V.; Kettle, M.J.; Harris, H.C.; Spittal, W.; Davis, G.; Henn, M.R.; et al. Biofilms harbour Clostridioides difficile, serving as a reservoir for recurrent infection. npj Biofilms Microbiomes 2021, 7, 16. [Google Scholar] [CrossRef]
- Frost, L.R.; Cheng, J.K.J.; Unnikrishnan, M. Clostridioides difficile biofilms: A mechanism of persistence in the gut? PLoS Pathogens 2021, 17, e1009348. [Google Scholar] [CrossRef]
- Vuotto, C.; Donelli, G.; Buckley, A.; Chilton, C. Clostridium difficile Biofilm. Adv. Exp. Med. Biol. 2018, 1050, 97–115. [Google Scholar] [CrossRef]
- Dapa, T.; Unnikrishnan, M. Biofilm formation by Clostridium difficile. Gut Microbes 2013, 4, 397–402. [Google Scholar] [CrossRef]
- Shan, J.; Ramachandran, A.; Thanki, A.M.; Vukusic, F.B.I.; Barylski, J.; Clokie, M.R.J. Bacteriophages are more virulent to bacteria with human cells than they are in bacterial culture; insights from HT-29 cells. Sci. Rep. 2018, 8, 5091. [Google Scholar] [CrossRef]
- Drudy, D.; Donoghue, D.P.; Baird, A.; Fenelon, L.; Farrelly, C. Flow cytometric analysis of Clostridium difficile adherence to human intestinal epithelial cells. J. Med. Microbiol. 2001, 50, 526–534. [Google Scholar] [CrossRef]
- Anonye, B.O.; Hassall, J.; Patient, J.; Detamornrat, U.; Aladdad, A.M.; Schüller, S.; Rose, F.R.A.J.; Unnikrishnan, M. Probing Clostridium difficile Infection in Complex Human Gut Cellular Models. Front. Microbiol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Hoffmann, P.; Burmester, M.; Langeheine, M.; Brehm, R.; Empl, M.T.; Seeger, B.; Breves, G. Caco-2/HT29-MTX co-cultured cells as a model for studying physiological properties and toxin-induced effects on intestinal cells. PLoS ONE 2021, 16, e0257824. [Google Scholar] [CrossRef]
- Castro-Córdova, P.; Mora-Uribe, P.; Reyes-Ramírez, R.; Cofré-Araneda, G.; Orozco-Aguilar, J.; Brito-Silva, C.; Mendoza-León, M.J.; Kuehne, S.A.; Minton, N.P.; Pizarro-Guajardo, M.; et al. Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 2021, 12, 1140. [Google Scholar] [CrossRef]
- Cerquetti, M.; Serafino, A.; Sebastianelli, A.; Mastrantonio, P. Binding of Clostridium difficile to Caco-2 epithelial cell line and to extracellular matrix proteins. FEMS Immunol. Med. Microbiol. 2002, 32, 211–218. [Google Scholar] [CrossRef]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Comparison of oritavancin versus vancomycin as treatments for clindamycin-induced Clostridium difficile PCR ribotype 027 infection in a human gut model. J. Antimicrob. Chemother. 2008, 62, 1078–1085. [Google Scholar] [CrossRef]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 2009, 63, 520–525. [Google Scholar] [CrossRef]
- Meader, E.; Mayer, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Evaluation of bacteriophage therapy to control Clostridium difficile and toxin production in an in vitro human colon model system. Anaerobe 2013, 22, 25–30. [Google Scholar] [CrossRef]
- Freeman, J.; Baines, S.D.; Jabes, D.; Wilcox, M.H. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J. Antimicrob. Chemother. 2005, 56, 717–725. [Google Scholar] [CrossRef]
- Tsai, C.J.; Loh, J.M.; Proft, T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 2016, 7, 214–229. [Google Scholar] [CrossRef]
- Cutuli, M.A.; Petronio Petronio, G.; Vergalito, F.; Magnifico, I.; Pietrangelo, L.; Venditti, N.; Di Marco, R. Galleria mellonella as a consolidated in vivo model hosts: New developments in antibacterial strategies and novel drug testing. Virulence 2019, 10, 527–541. [Google Scholar] [CrossRef]
- Piatek, M.; Sheehan, G.; Kavanagh, K. Galleria mellonella: The Versatile Host for Drug Discovery, In Vivo Toxicity Testing and Characterising Host-Pathogen Interactions. Antibiotics 2021, 10, 1545. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, G.; Kavanagh, K. Proteomic Analysis of the Responses of Candida albicans during Infection of Galleria mellonella Larvae. J. Fungi 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Allegra, E.; Titball, R.W.; Carter, J.; Champion, O.L. Galleria mellonella larvae allow the discrimination of toxic and non-toxic chemicals. Chemosphere 2018, 198, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Browne, N.; Heelan, M.; Kavanagh, K. An analysis of the structural and functional similarities of insect hemocytes and mammalian phagocytes. Virulence 2013, 4, 597–603. [Google Scholar] [CrossRef]
- Kavanagh, K.; Reeves, E.P. Exploiting the potential of insects for in vivo pathogenicity testing of microbial pathogens. FEMS Microbiol. Rev. 2004, 28, 101–112. [Google Scholar] [CrossRef]
- Elzinga, J.; van der Oost, J.; de Vos, W.M.; Smidt, H. The Use of Defined Microbial Communities To Model Host-Microbe Interactions in the Human Gut. Microbiol. Mol. Biol. Rev. 2019, 83, e00054-18. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef]
- Best, E.L.; Freeman, J.; Wilcox, M.H. Models for the study of Clostridium difficile infection. Gut Microbes 2012, 3, 145–167. [Google Scholar] [CrossRef]
- Minton, N.P.; Ehsaan, M.; Humphreys, C.M.; Little, G.T.; Baker, J.; Henstra, A.M.; Liew, F.; Kelly, M.L.; Sheng, L.; Schwarz, K.; et al. A roadmap for gene system development in Clostridium. Anaerobe 2016, 41, 104–112. [Google Scholar] [CrossRef]
- Cartman, S.T.; Kelly, M.L.; Heeg, D.; Heap, J.T.; Minton, N.P. Precise Manipulation of the Clostridium difficile Chromosome Reveals a Lack of Association between the <i>tcdC</i> Genotype and Toxin Production. Appl. Environ. Microbiol. 2012, 78, 4683–4690. [Google Scholar] [CrossRef]
- Joseph, R.C.; Kim, N.M.; Sandoval, N.R. Recent Developments of the Synthetic Biology Toolkit for Clostridium. Front. Microbiol. 2018, 9, 154. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The ClosTron: A universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 2007, 70, 452–464. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Minton, N.P. ClosTron-mediated engineering of Clostridium. Bioengineered 2012, 3, 247–254. [Google Scholar] [CrossRef]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The ClosTron: Mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar] [CrossRef]
- Heap, J.T.; Cartman, S.T.; Kuehne, S.A.; Cooksley, C.; Minton, N.P. ClosTron-targeted mutagenesis. Methods Mol. Biol. 2010, 646, 165–182. [Google Scholar] [CrossRef]
- Zhu, D.; Bullock, J.; He, Y.; Sun, X. Cwp22, a novel peptidoglycan cross-linking enzyme, plays pleiotropic roles in Clostridioides difficile. Environ. Microbiol. 2019, 21, 3076–3090. [Google Scholar] [CrossRef]
- Gu, H.; Yang, Y.; Wang, M.; Chen, S.; Wang, H.; Li, S.; Ma, Y.; Wang, J. Novel Cysteine Desulfidase CdsB Involved in Releasing Cysteine Repression of Toxin Synthesis in Clostridium difficile. Front. Cell. Infect. Microbiol. 2017, 7, 531. [Google Scholar] [CrossRef]
- Permpoonpattana, P.; Phetcharaburanin, J.; Mikelsone, A.; Dembek, M.; Tan, S.; Brisson, M.C.; La Ragione, R.; Brisson, A.R.; Fairweather, N.; Hong, H.A.; et al. Functional characterization of Clostridium difficile spore coat proteins. J. Bacteriol. 2013, 195, 1492–1503. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Minton, N.P. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 2009, 78, 79–85. [Google Scholar] [CrossRef]
- Faulds-Pain, A.; Wren, B.W. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl. Environ. Microbiol. 2013, 79, 4768–4771. [Google Scholar] [CrossRef]
- Purdy, D.; O’Keeffe, T.A.; Elmore, M.; Herbert, M.; McLeod, A.; Bokori-Brown, M.; Ostrowski, A.; Minton, N.P. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol. Microbiol. 2002, 46, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.K.; Ehsaan, M.; Philip, S.; Collery, M.M.; Janoir, C.; Collignon, A.; Cartman, S.T.; Minton, N.P. Expanding the Repertoire of Gene Tools for Precise Manipulation of the Clostridium difficile Genome: Allelic Exchange Using pyrE Alleles. PLoS ONE 2013, 8, e56051. [Google Scholar] [CrossRef] [PubMed]
- Thanki, A.M. Development of a Phage-Based Diagnostic Test for the Identification of Clostridium difficile. Ph.D. Thesis, Loughborough University, Loughborough, UK, 2016. [Google Scholar]
- McAllister, K.N.; Bouillaut, L.; Kahn, J.N.; Self, W.T.; Sorg, J.A. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci. Rep. 2017, 7, 14672. [Google Scholar] [CrossRef] [PubMed]
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR–Cas Systems. Viruses 2018, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Dietrich, S.; Hoppert, M.; Hertel, R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses 2018, 10, 241. [Google Scholar] [CrossRef]
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional Analysis of Bacteriophage Immunity through a Type I-E CRISPR-Cas System in Vibrio cholerae and Its Application in Bacteriophage Genome Engineering. J. Bacteriol. 2016, 198, 578–590. [Google Scholar] [CrossRef]
- Hoshiga, F.; Yoshizaki, K.; Takao, N.; Miyanaga, K.; Tanji, Y. Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 2019, 366, fnz041. [Google Scholar] [CrossRef]
- Wang, S.; Hong, W.; Dong, S.; Zhang, Z.T.; Zhang, J.; Wang, L.; Wang, Y. Genome engineering of Clostridium difficile using the CRISPR-Cas9 system. Clin. Microbiol. Infect. 2018, 24, 1095–1099. [Google Scholar] [CrossRef]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Malik, D.J. Approaches for manufacture, formulation, targeted delivery and controlled release of phage-based therapeutics. Curr. Opin. Biotechnol. 2021, 68, 262–271. [Google Scholar] [CrossRef]
- Ma, Y.; Pacan, J.C.; Wang, Q.; Xu, Y.; Huang, X.; Korenevsky, A.; Sabour, P.M. Microencapsulation of bacteriophage felix O1 into chitosan-alginate microspheres for oral delivery. Appl. Environ. Microbiol. 2008, 74, 4799–4805. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Singh, A.; Vandersteegen, K.; Klumpp, J.; Lavigne, R.; Van den Mooter, G. Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections. Eur. J. Pharm. Biopharm. 2013, 84, 578–582. [Google Scholar] [CrossRef]
- Leung, S.S.Y.; Parumasivam, T.; Gao, F.G.; Carter, E.A.; Carrigy, N.B.; Vehring, R.; Finlay, W.H.; Morales, S.; Britton, W.J.; Kutter, E.; et al. Effects of storage conditions on the stability of spray dried, inhalable bacteriophage powders. Int. J. Pharm. 2017, 521, 141–149. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Meeus, J.; Lavigne, R.; Van den Mooter, G. Instability of bacteriophages in spray-dried trehalose powders is caused by crystallization of the matrix. Int. J. Pharm. 2014, 472, 202–205. [Google Scholar] [CrossRef]
- Philip, A.K.; Philip, B. Colon targeted drug delivery systems: A review on primary and novel approaches. Oman Med. J. 2010, 25, 79–87. [Google Scholar] [CrossRef]
- Dini, C.; Islan, G.A.; de Urraza, P.J.; Castro, G.R. Novel biopolymer matrices for microencapsulation of phages: Enhanced protection against acidity and protease activity. Macromol. Biosci. 2012, 12, 1200–1208. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.R.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Amidon, S.; Brown, J.E.; Dave, V.S. Colon-targeted oral drug delivery systems: Design trends and approaches. AAPS PharmSciTech 2015, 16, 731–741. [Google Scholar] [CrossRef]
- Stanford, K.; McAllister, T.A.; Niu, Y.D.; Stephens, T.P.; Mazzocco, A.; Waddell, T.E.; Johnson, R.P. Oral delivery systems for encapsulated bacteriophages targeted at Escherichia coli O157:H7 in feedlot cattle. J. Food Prot. 2010, 73, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Matinkhoo, S.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H.; Vehring, R. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 2011, 100, 5197–5205. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Rezaie-Yazdi, Z.; Leppanen, M.; Stapley, A.G.F.; Leaper, M.C.; Malik, D.J. Microencapsulation of Salmonella-Specific Bacteriophage Felix O1 Using Spray-Drying in a pH-Responsive Formulation and Direct Compression Tableting of Powders into a Solid Oral Dosage Form. Pharmaceuticals 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Vladisavljević, G.T.; Clokie, M.R.J.; Malik, D.J. Microencapsulation of Clostridium difficile specific bacteriophages using microfluidic glass capillary devices for colon delivery using pH triggered release. PLoS ONE 2017, 12, e0186239. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kumari, A.; Kumari Negi, A.; Galav, V.; Thakur, S.; Agrawal, M.; Sharma, V. Nanotechnology Based Approaches in Phage Therapy: Overcoming the Pharmacological Barriers. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).