
Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages

 ,
, 
 ,
,
Abstract
1. Scope of Current Review and Introduction to Clostridioides difficile Infection
2. C. difficile Phage Isolation and Characterisation
2.1. Direct Isolation of Phages from Environmental and Clinical Samples
2.2. Isolation of C. difficile Phages through Prophage Induction
3. Diversity of C. difficile Phages
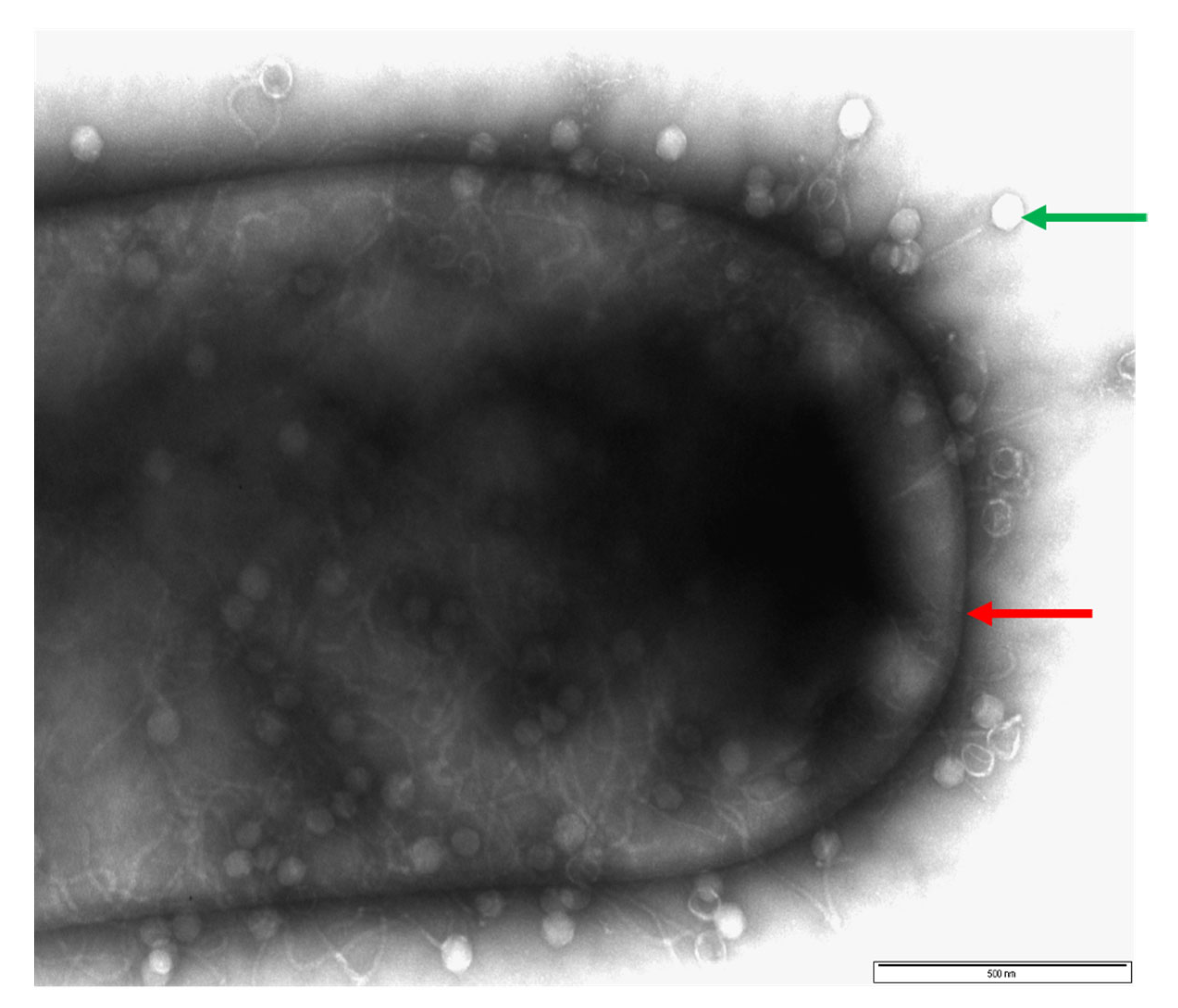
3.1. Morphological Diversity of C. difficile Phages
3.2. Genomic Diversity
4. Phage Mechanics of Infection
4.1. Impact of Tail Fibres on Attachment and Host Selection
4.2. Phage Host Range
4.3. Impact of Phage Infection on C. difficile Physiology
5. Therapeutic Application of C. difficile Phage Models of C. difficile Phage Therapy
5.1. Culture-Based Assays
5.2. Biofilm Model
5.3. Epithelial Cell Tissue Model
5.4. Batch Fermentation Model
5.5. Galleria Mellonella Infection Model
5.6. Hamster Infection Model
6. Genetic Manipulation of C. difficile Phages
6.1. ClosTron System
6.2. Clostridium Shuttle Plasmids
6.3. CRISPR Technology
7. Encapsulation and Formulation of Therapeutic C. difficile Phages
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Heuler, J.; Fortier, L.C.; Sun, X. Clostridioides difficile phage biology and application. FEMS Microbiol. Rev. 2021, 45, fuab012. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Clokie, M.R.J. Clostridium difficile phages: Still difficult? Front. Microbiol. 2014, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. Bacteriophage behavioral ecology: How phages alter their bacterial host’s habits. Bacteriophage 2014, 4, e29866. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Uematsu, S. Phage therapy for Clostridioides difficile infection. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.C.; O’Toole, E. Intestinal flora in new-born infants. Am. J. Dis. Child. 1935, 49, 390–402. [Google Scholar] [CrossRef]
- Cohen, L.E.; McNeill, C.J.; Wells, R.F. Clindamycin-Associated Colitis. JAMA 1973, 223, 1379–1380. [Google Scholar] [CrossRef]
- Noren, T.; Tang-Feldman, Y.J.; Cohen, S.H.; Silva, J.; Olcen, P. Clindamycin resistant strains of Clostridium difficile isolated from cases of C. difficile associated diarrhea (CDAD) in a hospital in Sweden. Diagn. Microbiol. Infect. Dis. 2002, 42, 149–151. [Google Scholar] [CrossRef]
- Pruitt, R.N.; Lacy, D.B. Toward a structural understanding of Clostridium difficile toxins A and B. Front. Cell. Infect. Microbiol. 2012, 2, 28. [Google Scholar] [CrossRef]
- Lyerly, D.M.; Lockwood, D.E.; Richardson, S.H.; Wilkins, T.D. Biological activities of toxins A and B of Clostridium difficile. Infect. Immun. 1982, 35, 1147–1150. [Google Scholar] [CrossRef]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef]
- Carter, G.P.; Lyras, D.; Allen, D.L.; Mackin, K.E.; Howarth, P.M.; O’Connor, J.R.; Rood, J.I. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J. Bacteriol. 2007, 189, 7290–7301. [Google Scholar] [CrossRef]
- Carter, G.P.; Douce, G.R.; Govind, R.; Howarth, P.M.; Mackin, K.E.; Spencer, J.; Buckley, A.M.; Antunes, A.; Kotsanas, D.; Jenkin, G.A.; et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 2011, 7, e1002317. [Google Scholar] [CrossRef]
- Rupnik, M.; Grabnar, M.; Geric, B. Binary toxin producing Clostridium difficile strains. Anaerobe 2003, 9, 289–294. [Google Scholar] [CrossRef]
- Govind, R.; Dupuy, B. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 2012, 8, e1002727. [Google Scholar] [CrossRef]
- Abt, M.C.; McKenney, P.T.; Pamer, E.G. Clostridium difficile colitis: Pathogenesis and host defence. Nat. Rev. Microbiol. 2016, 14, 609–620. [Google Scholar] [CrossRef]
- Wilcox, M.H.; Shetty, N.; Fawley, W.N.; Shemko, M.; Coen, P.; Birtles, A.; Cairns, M.; Curran, M.D.; Dodgson, K.J.; Green, S.M.; et al. Changing Epidemiology of Clostridium difficile Infection Following the Introduction of a National Ribotyping-Based Surveillance Scheme in England. Clin. Infect. Dis. 2012, 55, 1056–1063. [Google Scholar] [CrossRef]
- Vohra, P.; Poxton, I.R. Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology 2011, 157, 1343–1353. [Google Scholar] [CrossRef]
- Seddon, S.V.; Hemingway, I.; Borriello, S.P. Hydrolytic enzyme production by Clostridium difficile and its relationship to toxin production and virulence in the hamster model. J. Med. Microbiol. 1990, 31, 169–174. [Google Scholar] [CrossRef]
- Underwood, S.; Guan, S.; Vijayasubhash, V.; Baines, S.D.; Graham, L.; Lewis, R.J.; Wilcox, M.H.; Stephenson, K. Characterization of the Sporulation Initiation Pathway of Clostridium difficile and Its Role in Toxin Production. J. Bacteriol. 2009, 191, 7296–7305. [Google Scholar] [CrossRef]
- Baban, S.T.; Kuehne, S.A.; Barketi-Klai, A.; Cartman, S.T.; Kelly, M.L.; Hardie, K.R.; Kansau, I.; Collignon, A.; Minton, N.P. The Role of Flagella in Clostridium difficile Pathogenesis: Comparison between a Non-Epidemic and an Epidemic Strain. PLoS ONE 2013, 8, e73026. [Google Scholar] [CrossRef]
- Lyerly, D.M.; Krivan, H.C.; Wilkins, T.D. Clostridium difficile: Its disease and toxins. Clin. Microbiol. Rev. 1988, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Scutera, S.; Savoia, D. Novel avenues for Clostridium difficile infection drug discovery. Expert Opin. Drug Discov. 2013, 8, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile Infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Gould, C.V.; McDonald, L.C. Current Status of Clostridium difficile Infection Epidemiology. Clin. Infect. Dis. 2012, 55, S65–S70. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.B.; Hollenbeak, C.S. Clostridium difficile colitis: Factors associated with outcome and assessment of mortality at a national level. J. Gastrointest. Surg. 2011, 15, 1548–1555. [Google Scholar] [CrossRef]
- Shek, F.W.; Stacey, B.S.F.; Rendell, J.; Hellier, M.D.; Hanson, P.J.V. The rise of Clostridium difficile: The effect of length of stay, patient age and antibiotic use. J. Hosp. Infect. 2000, 45, 235–237. [Google Scholar] [CrossRef]
- Riley, T.V.; Codde, J.P.; Rouse, I.L. Increased length of hospital stay due to Clostridium difficile associated diarrhoea. Lancet 1995, 345, 455–456. [Google Scholar] [CrossRef]
- Tay, H.L.; Chow, A.; Ng, T.M.; Lye, D.C. Risk factors and treatment outcomes of severe Clostridioides difficile infection in Singapore. Sci. Rep. 2019, 9, 13440. [Google Scholar] [CrossRef]
- UKHSA. Clostridioides Difficile Infection Updated Guidance on Management and Treatment; UKHSA Publication gateway number GOV-12513; UKHSA: London, UK, 2022; p. 33.
- King, A.; Mullish, B.H.; Williams, H.R.T.; Aylin, P. Comparative epidemiology of Clostridium difficile infection: England and the USA. Int. J. Qual. Health Care 2017, 29, 785–791. [Google Scholar] [CrossRef]
- Petrosillo, N.; Granata, G.; Cataldo, M.A. Novel Antimicrobials for the Treatment of Clostridium difficile Infection. Front. Med. 2018, 5, 96. [Google Scholar] [CrossRef]
- Dinh, A.; Le Monnier, A.; Emery, C.; Alami, S.; Torreton, É.; Duburcq, A.; Barbier, F. Predictors and burden of hospital readmission with recurrent Clostridioides difficile infection: A French nation-wide inception cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1297–1305. [Google Scholar] [CrossRef]
- Kociolek, L.K.; Gerding, D.N. Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 150–160. [Google Scholar] [CrossRef]
- Stevens, V.W.; Nelson, R.E.; Schwab-Daugherty, E.M.; Khader, K.; Jones, M.M.; Brown, K.A.; Greene, T.; Croft, L.D.; Neuhauser, M.; Glassman, P.; et al. Comparative effectiveness of vancomycin and metronidazole for the prevention of recurrence and death in patients with Clostridium difficile infection. JAMA Intern. Med. 2017, 177, 546–553. [Google Scholar] [CrossRef]
- Alves, J.D.F.; Yamaguti, A.; de Mendonça, J.S.; de Melo Gamba, C.; Fonseca, C.L.; Paraskevopoulos, D.K.S.; de Paula, A.I.; Hosino, N.; Costa, S.F.; Guimarães, T. Metronidazole for Treatment of Clostridioides difficile Infections in Brazil: A Single-Center Experience and Risk Factors for Mortality. Antibiotics 2022, 11, 1162. [Google Scholar] [CrossRef]
- Giau, V.V.; Lee, H.; An, S.S.A.; Hulme, J. Recent advances in the treatment of C. difficile using biotherapeutic agents. Infect. Drug Resist. 2019, 12, 1597–1615. [Google Scholar] [CrossRef]
- Singh, T.; Bedi, P.; Bumrah, K.; Singh, J.; Rai, M.; Seelam, S. Updates in Treatment of Recurrent Clostridium difficile Infection. J. Clin. Med. Res. 2019, 11, 465–471. [Google Scholar] [CrossRef]
- Kutter, E.M.; Kuhl, S.J.; Abedon, S.T. Re-establishing a place for phage therapy in Western medicine. Future Microbiol. 2015, 10, 685–688. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Mangieri, N.; Picozzi, C.; Cocuzzi, R.; Foschino, R. Evaluation of a Potential Bacteriophage Cocktail for the Control of Shiga-Toxin Producing Escherichia coli in Food. Front. Microbiol. 2020, 11, 1801. [Google Scholar] [CrossRef]
- Cos, P.; Tote, K.; Horemans, T.; Maes, L. Biofilms: An Extra Hurdle for Effective Antimicrobial Therapy. Curr. Pharm. Des. 2010, 16, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Gebreyohannes, G.; Nyerere, A.; Bii, C.; Sbhatu, D.B. Challenges of intervention, treatment, and antibiotic resistance of biofilm-forming microorganisms. Heliyon 2019, 5, e02192. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.F.; Valiente, E.; Faulds-Pain, A.; Donahue, E.H.; Wren, B.W. Characterisation of Clostridium difficile Biofilm Formation, a Role for Spo0A. PLoS ONE 2012, 7, e50527. [Google Scholar] [CrossRef] [PubMed]
- Nale, J.Y.; Chutia, M.; Carr, P.; Hickenbotham, P.; Clokie, M.R.J. ‘Get in early’; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front. Microbiol. 2016, 7, 1383. [Google Scholar] [CrossRef] [PubMed]
- Sell, T.L.; Schaberg, D.R.; Fekety, F.R. Bacteriophage and bacteriocin typing scheme for Clostridium difficile. J. Clin. Microbiol. 1983, 17, 1148–1152. [Google Scholar] [CrossRef]
- Dei, R. Observations on phage-typing of Clostridium difficile: Preliminary evaluation of a phage panel. Eur. J. Epidemiol. 1989, 5, 351–354. [Google Scholar] [CrossRef]
- Phothichaisri, W.; Ounjai, P.; Phetruen, T.; Janvilisri, T.; Khunrae, P.; Singhakaew, S.; Wangroongsarb, P.; Chankhamhaengdecha, S. Characterization of Bacteriophages Infecting Clinical Isolates of Clostridium difficile. Front. Microbiol. 2018, 9, 1701. [Google Scholar] [CrossRef]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef]
- Luong, T.; Salabarria, A.C.; Roach, D.R. Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going? Clin. Ther. 2020, 42, 1659–1680. [Google Scholar] [CrossRef]
- Nale, J.Y.; Chutia, M.; Cheng, J.K.J.; Clokie, M.R.J. Refining the Galleria mellonella Model by Using Stress Marker Genes to Assess Clostridioides difficile Infection and Recuperation during Phage Therapy. Microorganisms 2020, 8, 1306. [Google Scholar] [CrossRef]
- Nale, J.; Redgwell, T.A.; Millard, A.; Clokie, M.R.J. Efficacy of an Optimised Bacteriophage Cocktail to Clear Clostridium difficile in a Batch Fermentation Model. Antibiotics 2018, 7, 13. [Google Scholar] [CrossRef]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage Combinations Significantly Reduce Clostridium difficile Growth In Vitro and Proliferation In Vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 2020, 11, e00019-20. [Google Scholar] [CrossRef]
- Goh, S.; Hussain, H.; Chang, B.J.; Emmett, W.; Riley, T.V.; Mullany, P. Phage ϕC2 Mediates Transduction of Tn6215, Encoding Erythromycin Resistance, between Clostridium difficile Strains. mBio 2013, 4, e00840-13. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Townsend, E.M.; Kelly, L.; Muscatt, G.; Box, J.D.; Hargraves, N.; Lilley, D.; Jameson, E. The Human Gut Phageome: Origins and Roles in the Human Gut Microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 498. [Google Scholar] [CrossRef]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.L.; Chan, P.K.S.; Chan, M.C.W.; Wu, J.C.Y.; et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018, 67, 634–643. [Google Scholar]
- Horgan, M.; O’Sullivan, O.; Coffey, A.; Fitzgerald, G.F.; van Sinderen, D.; McAuliffe, O.; Ross, R.P. Genome analysis of the Clostridium difficile phage PhiCD6356, a temperate phage of the Siphoviridae family. Gene 2010, 462, 34–43. [Google Scholar] [CrossRef]
- He, M.; Sebaihia, M.; Lawley, T.D.; Stabler, R.A.; Dawson, L.F.; Martin, M.J.; Holt, K.E.; Seth-Smith, H.M.; Quail, M.A.; Rance, R.; et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. USA 2010, 107, 7527–7532. [Google Scholar] [CrossRef]
- Ramírez-Vargas, G.; Goh, S.; Rodríguez, C. The Novel Phages phiCD5763 and phiCD2955 Represent Two Groups of Big Plasmidial Siphoviridae Phages of Clostridium difficile. Front. Microbiol. 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Flores, C.O.; Lawley, T.D.; Clokie, M.R.J. Abundant and Diverse Clustered Regularly Interspaced Short Palindromic Repeat Spacers in Clostridium difficile Strains and Prophages Target Multiple Phage Types within This Pathogen. mBio 2014, 5, e01045-01013. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.J.; Barylski, J.; Hargreaves, K.R.; Millard, A.A.; Vinner, G.K.; Clokie, M.R. Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology. Viruses 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Riley, T.V.; Chang, B.J. Isolation and Characterization of Temperate Bacteriophages of Clostridium difficile. Appl. Environ. Microbiol. 2005, 71, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Meessen-Pinard, M.; Sekulovic, O.; Fortier, L.C. Evidence of In Vivo Prophage Induction during Clostridium difficile Infection. Appl. Environ. Microbiol. 2012, 78, 7662–7670. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Colvin, H.V.; Patel, K.V.; Clokie, J.; Clokie, M.R.J. Genetically diverse Clostridium difficile strains harbouring abundant prophages in an estuarine environment. Appl. Environ. Microbiol. 2013, 79, 6236–6243. [Google Scholar] [CrossRef]
- Whittle, M.J.; Bilverstone, T.W.; van Esveld, R.J.; Lücke, A.C.; Lister, M.M.; Kuehne, S.A.; Minton, N.P. A Novel Bacteriophage with Broad Host Range against Clostridioides difficile Ribotype 078 Supports SlpA as the Likely Phage Receptor. Microbiol. Spectr. 2022, 10, e0229521. [Google Scholar] [CrossRef]
- Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef]
- Goh, S.; Chang, B.J.; Riley, T.V. Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 2005, 54, 129–135. [Google Scholar] [CrossRef]
- Govind, R.; Vediyappan, G.; Rolfe, R.D.; Dupuy, B.; Fralick, J.A. Bacteriophage-Mediated Toxin Gene Regulation in Clostridium difficile. J. Virol. 2009, 83, 12037–12045. [Google Scholar] [CrossRef]
- Sekulovic, O.; Meessen-Pinard, M.; Fortier, L.C. Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J. Bacteriol. 2011, 193, 2726–2734. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. What Does the Talking?: Quorum Sensing Signalling Genes Discovered in a Bacteriophage Genome. PLoS ONE 2014, 9, e85131. [Google Scholar] [CrossRef]
- Govind, R.; Fralick, J.A.; Rolfe, R.D. In vivo lysogenization of a Clostridium difficile bacteriophage ΦCD119. Anaerobe 2011, 17, 125–129. [Google Scholar]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Shan, J.; Patel, P.V.; Hickenbotham, P.T.; Nale, J.Y.; Hargreaves, K.R.; Clokie, M.R.J. Prophage carriage and diversity within clinically relevant strains of Clostridium difficile. Appl. Environ. Microbiol. 2012, 78, 6027–6034. [Google Scholar] [CrossRef]
- Nale, J.Y.; Shan, J.; Hickenbotham, P.T.; Fawley, W.N.; Wilcox, M.H.; Clokie, M.R.J. Diverse Temperate Bacteriophage Carriage in Clostridium difficile 027 Strains. PLoS ONE 2012, 7, e37263. [Google Scholar] [CrossRef]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol. 2009, 501, 69–76. [Google Scholar] [CrossRef]
- Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages using the small drop plaque assay system. Methods Mol. Biol. 2009, 501, 81–85. [Google Scholar] [CrossRef]
- Paolo, U.G. Induction by Mitomycin C of recA Protein Synthesis in Bacteria and Spheroplast. J. Biol. Chem. 1982, 257, 14932–14936. [Google Scholar]
- Sangster, W.; Hegarty, J.P.; Stewart, D.B., Sr. Phage tail-like particles kill Clostridium difficile and represent an alternative to conventional antibiotics. Surgery 2015, 157, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Matsushiro, A.; Sato, K.; Miyamoto, H.; Yamamura, T.; Honda, T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J. Bacteriol. 1999, 181, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Clokie, J. A Taxonomic Review of Clostridium difficile Phages and Proposal of a Novel Genus, “Phimmp04likevirus”. Viruses 2015, 7, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Phage Classification and Characterization. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Methods in Molecular Biology™; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 127–140. [Google Scholar]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018-2019update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
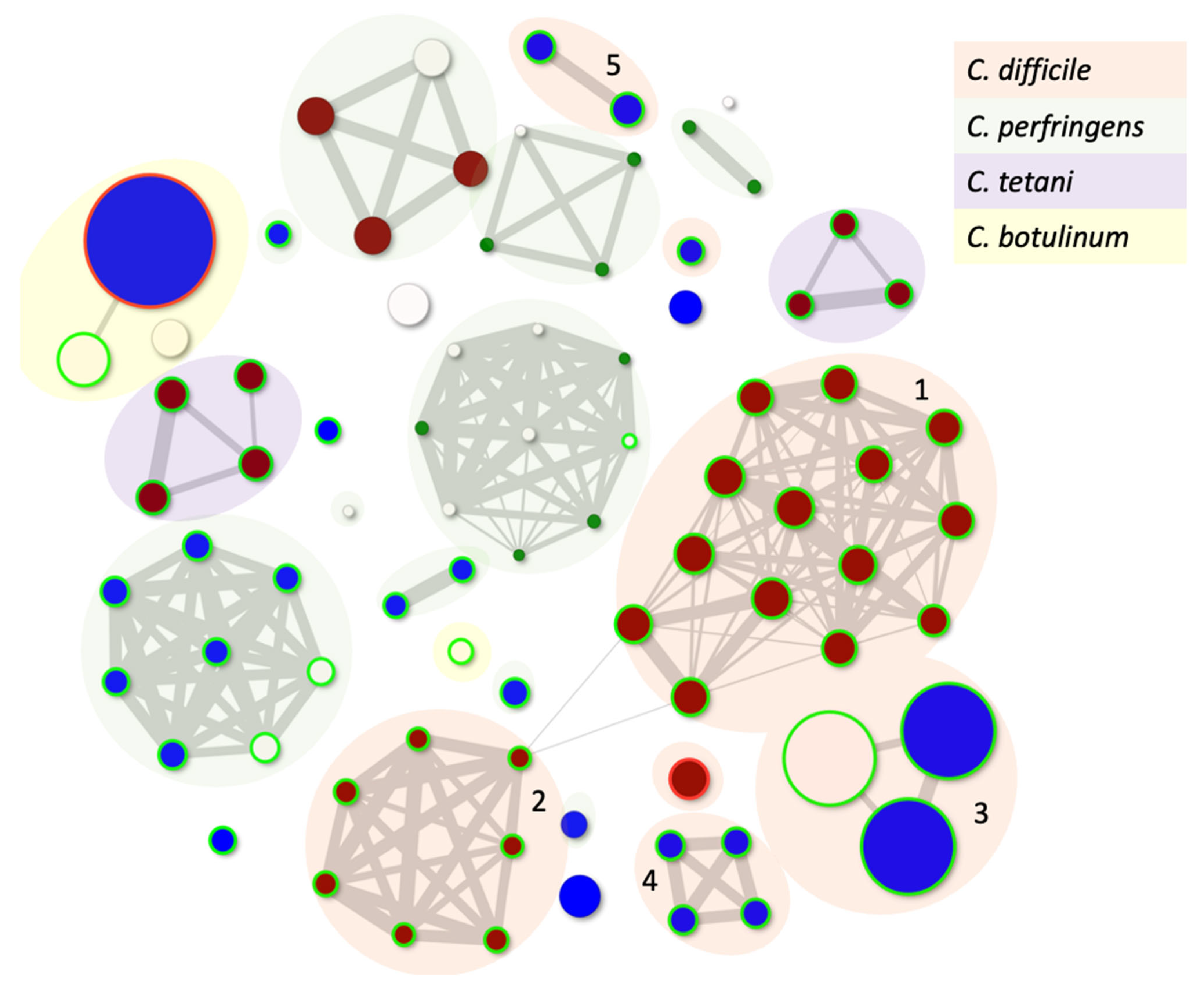
- Rangel-Pineros, G.; Millard, A.; Michniewski, S.; Scanlan, D.; Sirén, K.; Reyes, A.; Petersen, B.; Clokie, M.R.J.; Sicheritz-Pontén, T. From Trees to Clouds: PhageClouds for Fast Comparison of ∼640,000 Phage Genomic Sequences and Host-Centric Visualization Using Genomic Network Graphs. PHAGE 2021, 2, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Sekulovic, O.; Dupuy, B.; Soutourina, O.; Monot, M.; Fortier, L.C. High Prevalence and Genetic Diversity of Large phiCD211 (phiCDIF1296T)-Like Prophages in Clostridioides difficile. Appl. Environ. Microbiol. 2018, 84, e02164-02117. [Google Scholar] [CrossRef]
- Murillo, T.; Ramírez-Vargas, G.; Riedel, T.; Overmann, J.; Andersen, J.M.; Guzmán-Verri, C.; Chaves-Olarte, E.; Rodríguez, C. Two Groups of Cocirculating, Epidemic Clostridiodes difficile Strains Microdiversify through Different Mechanisms. Genome Biol. Evol. 2018, 10, 982–998. [Google Scholar] [CrossRef]
- Dowah, A.S.A.; Clokie, M.R.J. Review of the nature, diversity and structure of bacteriophage receptor binding proteins that target Gram-positive bacteria. Biophys. Rev. 2018, 10, 535–542. [Google Scholar] [CrossRef]
- Mahony, J.; van Sinderen, D. Gram-positive phage-host interactions. Front. Microbiol. 2015, 6, 61. [Google Scholar] [CrossRef]
- Mahony, J.; Cambillau, C.; van Sinderen, D. Host recognition by lactic acid bacterial phages. FEMS Microbiol. Rev. 2017, 41, S16–S26. [Google Scholar] [CrossRef]
- Li, X.; Koç, C.; Kühner, P.; Stierhof, Y.D.; Krismer, B.; Enright, M.C.; Penadés, J.R.; Wolz, C.; Stehle, T.; Cambillau, C.; et al. An essential role for the baseplate protein Gp45 in phage adsorption to Staphylococcus aureus. Sci. Rep. 2016, 6, 26455. [Google Scholar] [CrossRef]
- Habann, M.; Leiman, P.G.; Vandersteegen, K.; Van den Bossche, A.; Lavigne, R.; Shneider, M.M.; Bielmann, R.; Eugster, M.R.; Loessner, M.J.; Klumpp, J. Listeria phage A511, a model for the contractile tail machineries of SPO1-related bacteriophages. Mol. Microbiol. 2014, 92, 84–99. [Google Scholar] [CrossRef]
- São-José, C.; Baptista, C.; Santos, M.A. Bacillus subtilis operon encoding a membrane receptor for bacteriophage SPP1. J. Bacteriol. 2004, 186, 8337–8346. [Google Scholar] [CrossRef]
- Thanki, A.M.; Taylor-Joyce, G.; Dowah, A.; Nale, J.Y.; Malik, D.; Clokie, M.R.J. Unravelling the Links between Phage Adsorption and Successful Infection in Clostridium difficile. Viruses 2018, 10, 411. [Google Scholar] [CrossRef]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef]
- Dingle, K.E.; Elliott, B.; Robinson, E.; Griffiths, D.; Eyre, D.W.; Stoesser, N.; Vaughan, A.; Golubchik, T.; Fawley, W.N.; Wilcox, M.H.; et al. Evolutionary History of the Clostridium difficile Pathogenicity Locus. Genome Biol. Evol. 2014, 6, 36–52. [Google Scholar] [CrossRef]
- Ramesh, V.; Fralick, J.A.; Rolfe, R.D. Prevention of Clostridium difficile -induced ileocecitis with Bacteriophage. Anaerobe 1999, 5, 69–78. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiology and Molecular Biology Reviews 2016, 80, 523–543. [Google Scholar] [CrossRef]
- Fortier, L.-C. Bacteriophages Contribute to Shaping Clostridioides (Clostridium) difficile Species. Front. Microbiol. 2018, 9, 2033. [Google Scholar] [CrossRef]
- Nale, J.Y.; Al-Tayawi, T.S.; Heaphy, S.; Clokie, M.R.J. Impact of Phage CDHS-1 on the Transcription, Physiology and Pathogenicity of a Clostridioides difficile Ribotype 027 Strain, R20291. Viruses 2021, 13, 2262. [Google Scholar] [CrossRef] [PubMed]
- Sekulovic, O.; Fortier, L.C. Global transcriptional response of Clostridium difficile carrying the phiCD38-2 prophage. Appl. Environ. Microbiol. 2014, 81, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Y.; Dong, K.; Kuo, C.J.; Li, C.; Zhu, Y.Q.; Qin, J.; Li, Q.T.; Chang, Y.F.; Guo, X.; et al. Isolation and Characterization of the Novel Phage JD032 and Global Transcriptomic Response during JD032 Infection of Clostridioides difficile Ribotype 078. mSystems 2020, 5, e00017-20. [Google Scholar] [CrossRef] [PubMed]
- Meader, E.; Mayer, M.J.; Gasson, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe 2010, 16, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Normington, C.; Moura, I.B.; Bryant, J.A.; Ewin, D.J.; Clark, E.V.; Kettle, M.J.; Harris, H.C.; Spittal, W.; Davis, G.; Henn, M.R.; et al. Biofilms harbour Clostridioides difficile, serving as a reservoir for recurrent infection. npj Biofilms Microbiomes 2021, 7, 16. [Google Scholar] [CrossRef]
- Frost, L.R.; Cheng, J.K.J.; Unnikrishnan, M. Clostridioides difficile biofilms: A mechanism of persistence in the gut? PLoS Pathogens 2021, 17, e1009348. [Google Scholar] [CrossRef]
- Vuotto, C.; Donelli, G.; Buckley, A.; Chilton, C. Clostridium difficile Biofilm. Adv. Exp. Med. Biol. 2018, 1050, 97–115. [Google Scholar] [CrossRef]
- Dapa, T.; Unnikrishnan, M. Biofilm formation by Clostridium difficile. Gut Microbes 2013, 4, 397–402. [Google Scholar] [CrossRef]
- Shan, J.; Ramachandran, A.; Thanki, A.M.; Vukusic, F.B.I.; Barylski, J.; Clokie, M.R.J. Bacteriophages are more virulent to bacteria with human cells than they are in bacterial culture; insights from HT-29 cells. Sci. Rep. 2018, 8, 5091. [Google Scholar] [CrossRef]
- Drudy, D.; Donoghue, D.P.; Baird, A.; Fenelon, L.; Farrelly, C. Flow cytometric analysis of Clostridium difficile adherence to human intestinal epithelial cells. J. Med. Microbiol. 2001, 50, 526–534. [Google Scholar] [CrossRef]
- Anonye, B.O.; Hassall, J.; Patient, J.; Detamornrat, U.; Aladdad, A.M.; Schüller, S.; Rose, F.R.A.J.; Unnikrishnan, M. Probing Clostridium difficile Infection in Complex Human Gut Cellular Models. Front. Microbiol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Hoffmann, P.; Burmester, M.; Langeheine, M.; Brehm, R.; Empl, M.T.; Seeger, B.; Breves, G. Caco-2/HT29-MTX co-cultured cells as a model for studying physiological properties and toxin-induced effects on intestinal cells. PLoS ONE 2021, 16, e0257824. [Google Scholar] [CrossRef]
- Castro-Córdova, P.; Mora-Uribe, P.; Reyes-Ramírez, R.; Cofré-Araneda, G.; Orozco-Aguilar, J.; Brito-Silva, C.; Mendoza-León, M.J.; Kuehne, S.A.; Minton, N.P.; Pizarro-Guajardo, M.; et al. Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 2021, 12, 1140. [Google Scholar] [CrossRef]
- Cerquetti, M.; Serafino, A.; Sebastianelli, A.; Mastrantonio, P. Binding of Clostridium difficile to Caco-2 epithelial cell line and to extracellular matrix proteins. FEMS Immunol. Med. Microbiol. 2002, 32, 211–218. [Google Scholar] [CrossRef]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Comparison of oritavancin versus vancomycin as treatments for clindamycin-induced Clostridium difficile PCR ribotype 027 infection in a human gut model. J. Antimicrob. Chemother. 2008, 62, 1078–1085. [Google Scholar] [CrossRef]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 2009, 63, 520–525. [Google Scholar] [CrossRef]
- Meader, E.; Mayer, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Evaluation of bacteriophage therapy to control Clostridium difficile and toxin production in an in vitro human colon model system. Anaerobe 2013, 22, 25–30. [Google Scholar] [CrossRef]
- Freeman, J.; Baines, S.D.; Jabes, D.; Wilcox, M.H. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J. Antimicrob. Chemother. 2005, 56, 717–725. [Google Scholar] [CrossRef]
- Tsai, C.J.; Loh, J.M.; Proft, T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 2016, 7, 214–229. [Google Scholar] [CrossRef]
- Cutuli, M.A.; Petronio Petronio, G.; Vergalito, F.; Magnifico, I.; Pietrangelo, L.; Venditti, N.; Di Marco, R. Galleria mellonella as a consolidated in vivo model hosts: New developments in antibacterial strategies and novel drug testing. Virulence 2019, 10, 527–541. [Google Scholar] [CrossRef]
- Piatek, M.; Sheehan, G.; Kavanagh, K. Galleria mellonella: The Versatile Host for Drug Discovery, In Vivo Toxicity Testing and Characterising Host-Pathogen Interactions. Antibiotics 2021, 10, 1545. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, G.; Kavanagh, K. Proteomic Analysis of the Responses of Candida albicans during Infection of Galleria mellonella Larvae. J. Fungi 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Allegra, E.; Titball, R.W.; Carter, J.; Champion, O.L. Galleria mellonella larvae allow the discrimination of toxic and non-toxic chemicals. Chemosphere 2018, 198, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Browne, N.; Heelan, M.; Kavanagh, K. An analysis of the structural and functional similarities of insect hemocytes and mammalian phagocytes. Virulence 2013, 4, 597–603. [Google Scholar] [CrossRef]
- Kavanagh, K.; Reeves, E.P. Exploiting the potential of insects for in vivo pathogenicity testing of microbial pathogens. FEMS Microbiol. Rev. 2004, 28, 101–112. [Google Scholar] [CrossRef]
- Elzinga, J.; van der Oost, J.; de Vos, W.M.; Smidt, H. The Use of Defined Microbial Communities To Model Host-Microbe Interactions in the Human Gut. Microbiol. Mol. Biol. Rev. 2019, 83, e00054-18. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef]
- Best, E.L.; Freeman, J.; Wilcox, M.H. Models for the study of Clostridium difficile infection. Gut Microbes 2012, 3, 145–167. [Google Scholar] [CrossRef]
- Minton, N.P.; Ehsaan, M.; Humphreys, C.M.; Little, G.T.; Baker, J.; Henstra, A.M.; Liew, F.; Kelly, M.L.; Sheng, L.; Schwarz, K.; et al. A roadmap for gene system development in Clostridium. Anaerobe 2016, 41, 104–112. [Google Scholar] [CrossRef]
- Cartman, S.T.; Kelly, M.L.; Heeg, D.; Heap, J.T.; Minton, N.P. Precise Manipulation of the Clostridium difficile Chromosome Reveals a Lack of Association between the <i>tcdC</i> Genotype and Toxin Production. Appl. Environ. Microbiol. 2012, 78, 4683–4690. [Google Scholar] [CrossRef]
- Joseph, R.C.; Kim, N.M.; Sandoval, N.R. Recent Developments of the Synthetic Biology Toolkit for Clostridium. Front. Microbiol. 2018, 9, 154. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The ClosTron: A universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 2007, 70, 452–464. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Minton, N.P. ClosTron-mediated engineering of Clostridium. Bioengineered 2012, 3, 247–254. [Google Scholar] [CrossRef]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The ClosTron: Mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar] [CrossRef]
- Heap, J.T.; Cartman, S.T.; Kuehne, S.A.; Cooksley, C.; Minton, N.P. ClosTron-targeted mutagenesis. Methods Mol. Biol. 2010, 646, 165–182. [Google Scholar] [CrossRef]
- Zhu, D.; Bullock, J.; He, Y.; Sun, X. Cwp22, a novel peptidoglycan cross-linking enzyme, plays pleiotropic roles in Clostridioides difficile. Environ. Microbiol. 2019, 21, 3076–3090. [Google Scholar] [CrossRef]
- Gu, H.; Yang, Y.; Wang, M.; Chen, S.; Wang, H.; Li, S.; Ma, Y.; Wang, J. Novel Cysteine Desulfidase CdsB Involved in Releasing Cysteine Repression of Toxin Synthesis in Clostridium difficile. Front. Cell. Infect. Microbiol. 2017, 7, 531. [Google Scholar] [CrossRef]
- Permpoonpattana, P.; Phetcharaburanin, J.; Mikelsone, A.; Dembek, M.; Tan, S.; Brisson, M.C.; La Ragione, R.; Brisson, A.R.; Fairweather, N.; Hong, H.A.; et al. Functional characterization of Clostridium difficile spore coat proteins. J. Bacteriol. 2013, 195, 1492–1503. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Minton, N.P. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 2009, 78, 79–85. [Google Scholar] [CrossRef]
- Faulds-Pain, A.; Wren, B.W. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl. Environ. Microbiol. 2013, 79, 4768–4771. [Google Scholar] [CrossRef]
- Purdy, D.; O’Keeffe, T.A.; Elmore, M.; Herbert, M.; McLeod, A.; Bokori-Brown, M.; Ostrowski, A.; Minton, N.P. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol. Microbiol. 2002, 46, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.K.; Ehsaan, M.; Philip, S.; Collery, M.M.; Janoir, C.; Collignon, A.; Cartman, S.T.; Minton, N.P. Expanding the Repertoire of Gene Tools for Precise Manipulation of the Clostridium difficile Genome: Allelic Exchange Using pyrE Alleles. PLoS ONE 2013, 8, e56051. [Google Scholar] [CrossRef] [PubMed]
- Thanki, A.M. Development of a Phage-Based Diagnostic Test for the Identification of Clostridium difficile. Ph.D. Thesis, Loughborough University, Loughborough, UK, 2016. [Google Scholar]
- McAllister, K.N.; Bouillaut, L.; Kahn, J.N.; Self, W.T.; Sorg, J.A. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci. Rep. 2017, 7, 14672. [Google Scholar] [CrossRef] [PubMed]
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR–Cas Systems. Viruses 2018, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Dietrich, S.; Hoppert, M.; Hertel, R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses 2018, 10, 241. [Google Scholar] [CrossRef]
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional Analysis of Bacteriophage Immunity through a Type I-E CRISPR-Cas System in Vibrio cholerae and Its Application in Bacteriophage Genome Engineering. J. Bacteriol. 2016, 198, 578–590. [Google Scholar] [CrossRef]
- Hoshiga, F.; Yoshizaki, K.; Takao, N.; Miyanaga, K.; Tanji, Y. Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 2019, 366, fnz041. [Google Scholar] [CrossRef]
- Wang, S.; Hong, W.; Dong, S.; Zhang, Z.T.; Zhang, J.; Wang, L.; Wang, Y. Genome engineering of Clostridium difficile using the CRISPR-Cas9 system. Clin. Microbiol. Infect. 2018, 24, 1095–1099. [Google Scholar] [CrossRef]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Malik, D.J. Approaches for manufacture, formulation, targeted delivery and controlled release of phage-based therapeutics. Curr. Opin. Biotechnol. 2021, 68, 262–271. [Google Scholar] [CrossRef]
- Ma, Y.; Pacan, J.C.; Wang, Q.; Xu, Y.; Huang, X.; Korenevsky, A.; Sabour, P.M. Microencapsulation of bacteriophage felix O1 into chitosan-alginate microspheres for oral delivery. Appl. Environ. Microbiol. 2008, 74, 4799–4805. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Singh, A.; Vandersteegen, K.; Klumpp, J.; Lavigne, R.; Van den Mooter, G. Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections. Eur. J. Pharm. Biopharm. 2013, 84, 578–582. [Google Scholar] [CrossRef]
- Leung, S.S.Y.; Parumasivam, T.; Gao, F.G.; Carter, E.A.; Carrigy, N.B.; Vehring, R.; Finlay, W.H.; Morales, S.; Britton, W.J.; Kutter, E.; et al. Effects of storage conditions on the stability of spray dried, inhalable bacteriophage powders. Int. J. Pharm. 2017, 521, 141–149. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Meeus, J.; Lavigne, R.; Van den Mooter, G. Instability of bacteriophages in spray-dried trehalose powders is caused by crystallization of the matrix. Int. J. Pharm. 2014, 472, 202–205. [Google Scholar] [CrossRef]
- Philip, A.K.; Philip, B. Colon targeted drug delivery systems: A review on primary and novel approaches. Oman Med. J. 2010, 25, 79–87. [Google Scholar] [CrossRef]
- Dini, C.; Islan, G.A.; de Urraza, P.J.; Castro, G.R. Novel biopolymer matrices for microencapsulation of phages: Enhanced protection against acidity and protease activity. Macromol. Biosci. 2012, 12, 1200–1208. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.R.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Amidon, S.; Brown, J.E.; Dave, V.S. Colon-targeted oral drug delivery systems: Design trends and approaches. AAPS PharmSciTech 2015, 16, 731–741. [Google Scholar] [CrossRef]
- Stanford, K.; McAllister, T.A.; Niu, Y.D.; Stephens, T.P.; Mazzocco, A.; Waddell, T.E.; Johnson, R.P. Oral delivery systems for encapsulated bacteriophages targeted at Escherichia coli O157:H7 in feedlot cattle. J. Food Prot. 2010, 73, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Matinkhoo, S.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H.; Vehring, R. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 2011, 100, 5197–5205. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Rezaie-Yazdi, Z.; Leppanen, M.; Stapley, A.G.F.; Leaper, M.C.; Malik, D.J. Microencapsulation of Salmonella-Specific Bacteriophage Felix O1 Using Spray-Drying in a pH-Responsive Formulation and Direct Compression Tableting of Powders into a Solid Oral Dosage Form. Pharmaceuticals 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Vladisavljević, G.T.; Clokie, M.R.J.; Malik, D.J. Microencapsulation of Clostridium difficile specific bacteriophages using microfluidic glass capillary devices for colon delivery using pH triggered release. PLoS ONE 2017, 12, e0186239. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kumari, A.; Kumari Negi, A.; Galav, V.; Thakur, S.; Agrawal, M.; Sharma, V. Nanotechnology Based Approaches in Phage Therapy: Overcoming the Pharmacological Barriers. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phage Name | Morphology | Isolation Method | Source | Accession Number |
|---|---|---|---|---|
| phiCDHS-1 | Siphoviridae | Enrichment | Estuarine | KU057941 |
| CDHM19 | Myoviridae | NC_028996 | ||
| CDHM11 | Myoviridae | NC_029001 | ||
| CDKM15 | Myoviridae | Sediment | KX228400 | |
| CDKM9 | Myoviridae | KX228399 | ||
| phiMMP02 | Myoviridae | Patients | NC_019421.1 | |
| PhiMMP03 | Myoviridae | NC_028959 | ||
| PhiMMP01 | Myoviridae | NC_028883 | ||
| PhiMMP04 | Myoviridae | NC_019422 | ||
| PhiCD418 | Myoviridae | Sewage | MW512573 | |
| PhiCD2301 | Myoviridae | MW512571 | ||
| PhCD08011 | Myoviridae | MW512572 | ||
| PhiCD1801 | Myoviridae | MW512570 | ||
| PhiCD146 | Siphoviridae | Induction (Mitomycin C) | UV 302 nm | NC_028958 |
| PhiCD111 | Siphoviridae | NC_028905 | ||
| PhiCD 24-1 | Siphoviridae | LN681534 | ||
| PhiCD505 | Myoviridae | NC_028764 | ||
| PhiCD481-1 | Myoviridae | NC_028951 | ||
| PhiCD506 | Myoviridae | NC_028838 | ||
| PhiCD38-2 | Siphoviridae | 0.5–5 | NC_015568 | |
| PhiCD6356 | Siphoviridae | NC_015262 | ||
| PhiCD27 | Myoviridae | NC_011398.1 | ||
| phiSemix9P1 | Myoviridae | KX905163.1 | ||
| phiCDHM1 | Myoviridae | NC_024144 | ||
| PhiC2 | Myoviridae | NC_009231.1 | ||
| CDHM13 | Myoviridae | NC_029116 | ||
| CDHM14 | Myoviridae | LK985321 | ||
| phiCDKH01 | Siphoviridae | JACSDL010000003.1 | ||
| JD032 | Myoviridae | MK473382 | ||
| HMC114 | Phage tail-like particles | CM000660.1 | ||
| LIBA6276 | Siphoviridae | Unknown | MF547662.1 | |
| ΦCD1801 | Myoviridae | Enrichment | Sewage | MW512570 |
| ΦCD08011 | Myoviridae | MW512572 | ||
| ΦCD418 | Myoviridae | MW512573 | ||
| ΦCD2301 | Myoviridae | MW512571 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nale, J.Y.; Thanki, A.M.; Rashid, S.J.; Shan, J.; Vinner, G.K.; Dowah, A.S.A.; Cheng, J.K.J.; Sicheritz-Pontén, T.; Clokie, M.R.J. Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages. Viruses 2022, 14, 2772. https://doi.org/10.3390/v14122772
Nale JY, Thanki AM, Rashid SJ, Shan J, Vinner GK, Dowah ASA, Cheng JKJ, Sicheritz-Pontén T, Clokie MRJ. Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages. Viruses. 2022; 14(12):2772. https://doi.org/10.3390/v14122772
Chicago/Turabian StyleNale, Janet Y., Anisha M. Thanki, Srwa J. Rashid, Jinyu Shan, Gurinder K. Vinner, Ahmed S. A. Dowah, Jeffrey K. J. Cheng, Thomas Sicheritz-Pontén, and Martha R. J. Clokie. 2022. "Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages" Viruses 14, no. 12: 2772. https://doi.org/10.3390/v14122772
APA StyleNale, J. Y., Thanki, A. M., Rashid, S. J., Shan, J., Vinner, G. K., Dowah, A. S. A., Cheng, J. K. J., Sicheritz-Pontén, T., & Clokie, M. R. J. (2022). Diversity, Dynamics and Therapeutic Application of Clostridioides difficile Bacteriophages. Viruses, 14(12), 2772. https://doi.org/10.3390/v14122772