
Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Mosquito Infections
2.2. Whole Genome and Small RNA Sequencing
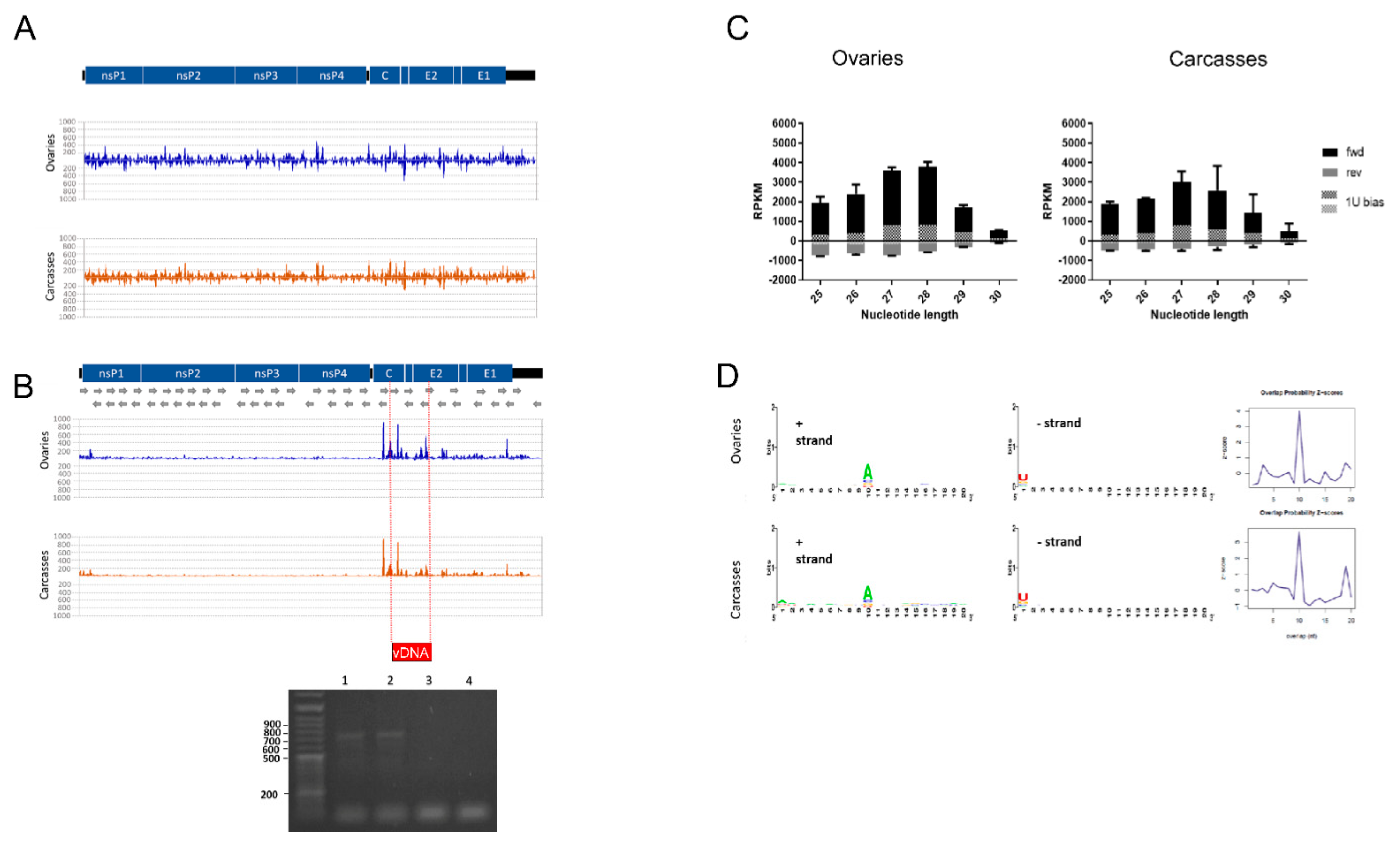
2.3. Detection of vDNA Fragments
2.4. Bioinformatic Analyses of WGS Data
2.5. Bioinformatic Analyses of sRNA Data
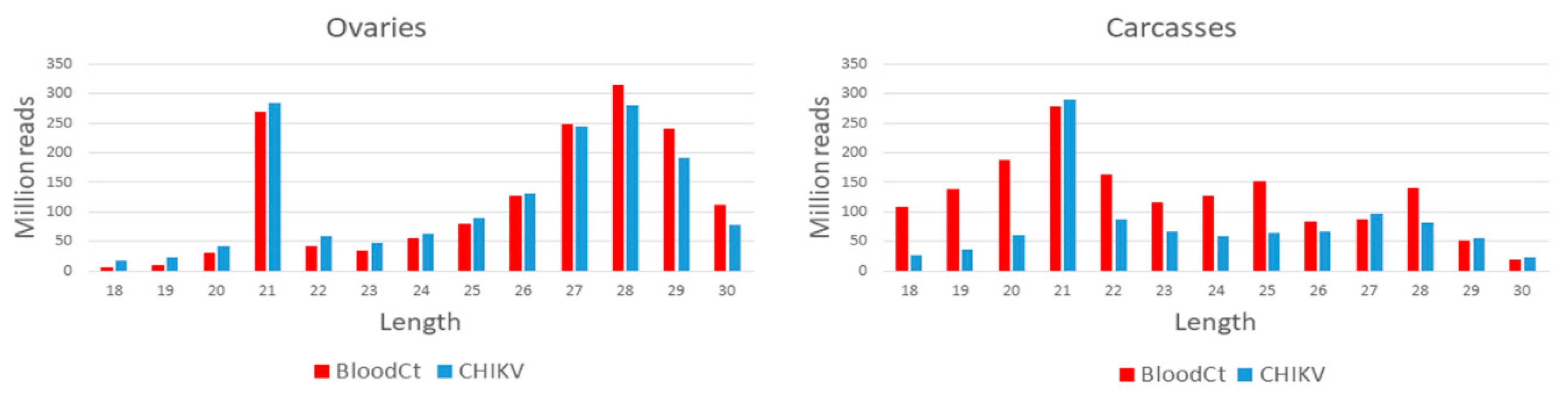
2.5.1. Small RNA Profile of the CHIKV Genome
2.5.2. Small RNA Profile of Mosquito Transcripts, piRNA Clusters and nrEVEs
2.6. Differential Expression of Ae. albopictus Annotated miRNAs
2.7. Quantitative PCR
3. Results and Discussion
3.1. Small RNA Profile of CHIKV, vDNA Fragments, and New nrEVEs
3.2. Viral SmallRNAs on Ae. albopictus Transcripts
3.3. The Profile of SmallRNAs on Ae. albopictus Transcripts
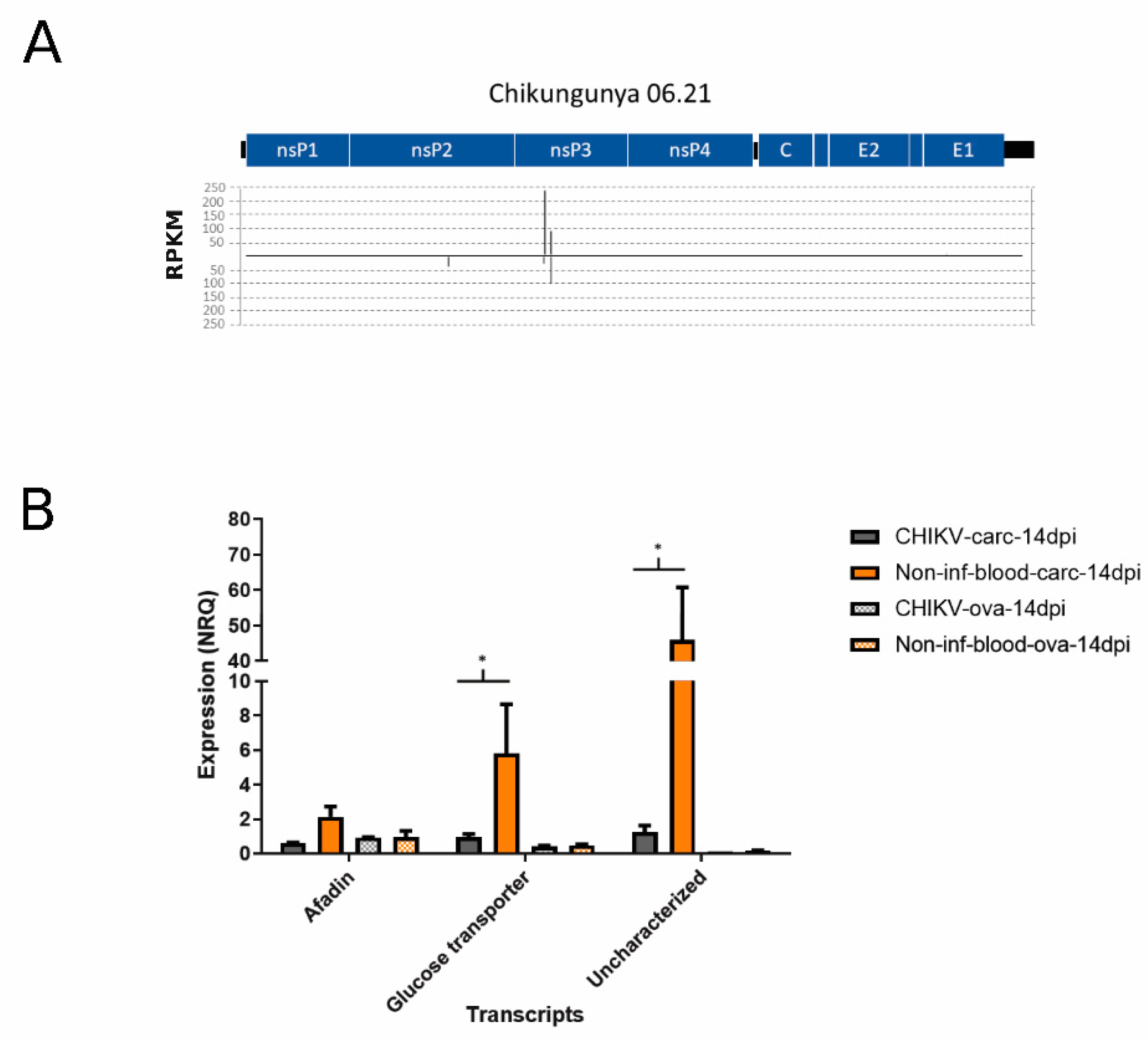
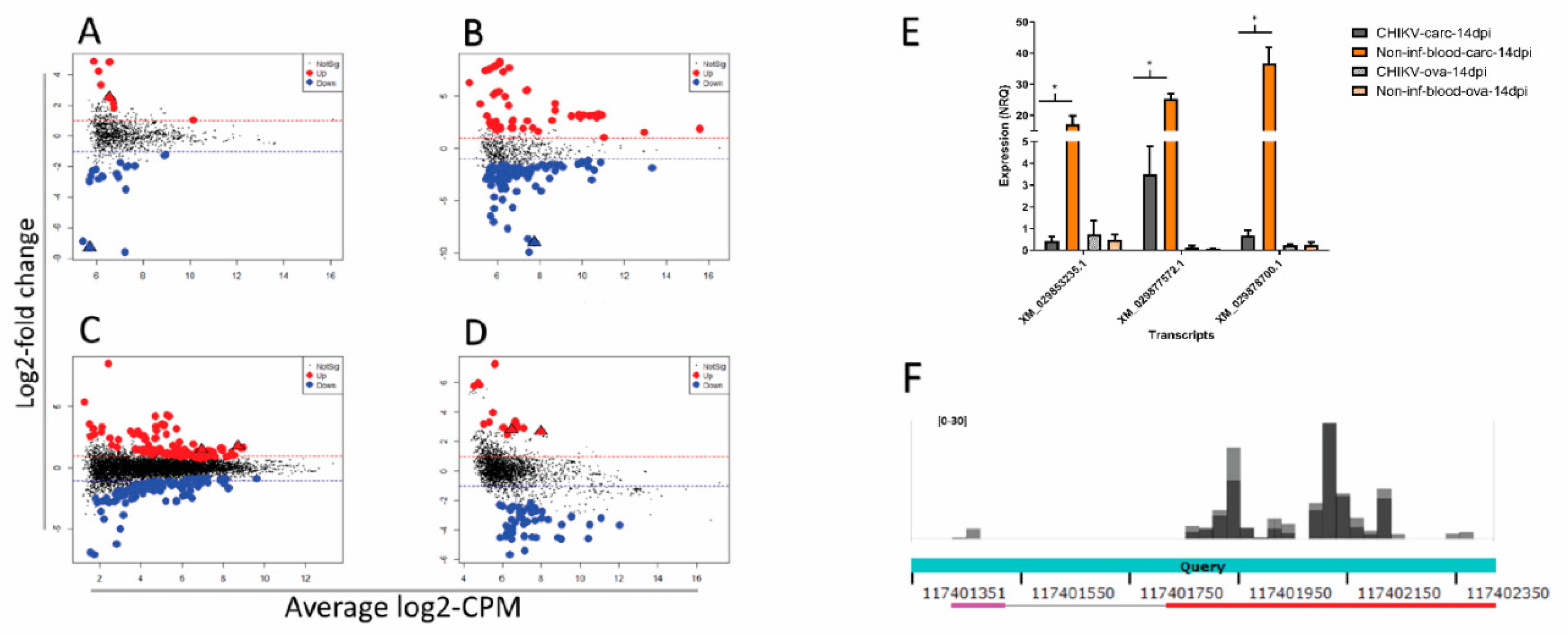
3.4. Differential Abundance of nrEVEs-sRNAs Following Infection and Their Targets on the Ae. albopictus Transcriptome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mason, P.J.; Haddow, A.J. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–1953; An additional note on Chikungunya virus isolations and serum antibodies. Trans. R. Soc. Trop. Med. Hyg. 1957, 51, 238–240. [Google Scholar] [CrossRef]
- Njenga, K.M.; Nderitu, L.; Ledermann, J.P.; Ndirangu, A.; Logue, C.H.; Kelly, C.H.L.; Sang, R.; Sergon, K.; Breiman, R.; Powers, A.M. Tracking epidemic Chikungunya virus into the Indian Ocean from East Africa. J. Gen. Virol. 2008, 89, 2754–2760. [Google Scholar] [CrossRef]
- Zeller, H.; Van Bortel, W.; Sudre, B. Chikungunya: Its history in Africa and Asia and its spread to new regions in 2013–2014. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef] [PubMed]
- Leparc-Goffart, I.; Nougairede, A.; Cassadou, S.; Prat, C.; de Lamballerie, X. Chikungunya in the Americas. Lancet 2014, 383, 514. [Google Scholar] [CrossRef]
- Amraoui, F.; Failloux, A.-B. Chikungunya: An unexpected emergence in Europe. Curr. Opin. Virol. 2016, 21, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Forrester, N.L. Chikungunya: Evolutionary history and recent epidemic spread. Antivir. Res. 2015, 120, 32–39. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Neglected Diseases. Available online: https://www.who.int/neglected_diseases/diseases/en/ (accessed on 7 September 2020).
- Cox, J.; Mota, J.; Sukupolvi-Petty, S.; Diamond, M.S.; Rico-Hesse, R. Mosquito bite delivery of dengue virus enhances immunogenicity and pathogenesis in humanized mice. J. Virol. 2012. [Google Scholar] [CrossRef]
- Liu, J.; Swevers, L.; Kolliopoulou, A.; Smagghe, G. Arboviruses and the challenge to establish systemic and persistent infections in competent mosquito vectors: The interaction with the RNAi mechanism. Front. Physiol. 2019, 10, 890. [Google Scholar] [CrossRef]
- Dubrulle, M.; Mousson, L.; Moutailler, S.; Vazeille, M.; Failloux, A.-B. Chikungunya virus and Aedes mosquitoes: Saliva is infectious as soon as two days after oral infection. PLoS ONE 2009, 4, e5895. [Google Scholar] [CrossRef]
- Merkling, S.H.; van Rij, R.P. Beyond RNAi: Antiviral defense strategies in Drosophila and mosquito. J. Insect Physiol. 2013, 59, 159–170. [Google Scholar] [CrossRef]
- Cheng, G.; Liu, Y.; Wang, P.; Xiao, X. Mosquito defense strategies against viral infection. Trends Parasitol. 2016, 32, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Webster, J.A.; Madzokere, E.T.; Stephenson, E.B.; Herrero, L.J. Mosquito antiviral defense mechanisms: A delicate balance between innate immunity and persistent viral infection. Parasites Vectors 2019, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bosio, C.F.; Fulton, R.E.; Salasek, M.L.; Beaty, B.J.; Black, W.C. 4th Quantitative trait loci that control vector competence for dengue-2 virus in the mosquito Aedes aegypti. Genetics 2000, 156, 687–698. [Google Scholar]
- Bennett, K.E.; Flick, D.; Fleming, K.H.; Jochim, R.; Beaty, B.J.; Black, W.C., 4th. Quantitative trait loci that control dengue-2 virus dissemination in the mosquito Aedes aegypti. Genetics 2005, 170, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.W.E.; Kantor, A.M.; Passarelli, A.L.; Clem, R.J. Tissue barriers to arbovirus infection in mosquitoes. Viruses 2015, 7, 3741–3767. [Google Scholar] [CrossRef]
- Palmer, W.H.; Varghese, F.S.; van Rij, R.P. Natural variation in resistance to virus infection in Dipteran insects. Viruses 2018, 10, 118. [Google Scholar] [CrossRef]
- Garcia-Luna, S.M.; Weger-Lucarelli, J.; Rückert, C.; Murrieta, R.A.; Young, M.C.; Byas, A.D.; Fauver, J.R.; Perera, R.; Flores-Suarez, A.E.; Ponce-Garcia, G.; et al. Variation in competence for ZIKV transmission by Aedes aegypti and Aedes albopictus in Mexico. PLoS Negl. Trop. Dis. 2018, 12, e0006599. [Google Scholar] [CrossRef]
- Dickson, L.B.; Sanchez-Vargas, I.; Sylla, M.; Fleming, K.; Black, W.C., 4th. Vector competence in West African Aedes aegypti is flavivirus species and genotype dependent. PLoS Negl. Trop. Dis. 2014, 8, e3153. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, L.; Scott, T.W. Mode of transmission and the evolution of arbovirus virulence in mosquito vectors. Proc. R. Soc. B Biol. Sci. 2009. [Google Scholar] [CrossRef]
- Salas-Benito, J.S.; De Nova-Ocampo, M. Viral interference and persistence in mosquito-borne flaviviruses. J. Immunol. Res. 2015, 2015, 873404. [Google Scholar] [CrossRef]
- Chotiwan, N.; Andre, B.G.; Sanchez-Vargas, I.; Islam, M.N.; Grabowski, J.M.; Hopf-Jannasch, A.; Gough, E.; Nakayasu, E.; Blair, C.D.; Belisle, J.T.; et al. Dynamic remodeling of lipids coincides with dengue virus replication in the midgut of Aedes aegypti mosquitoes. PLoS Pathog. 2018, 14, e1006853. [Google Scholar] [CrossRef]
- Da Cruz-Nunes, E.; Canuto, G.A.B. Metabolomics applied in the study of emerging arboviruses caused by Aedes aegypti mosquitoes: A review. Electrophoresis 2020, 41, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, L.; Saleh, M.-C. Manipulating mosquito tolerance for arbovirus control. Cell Host Microbe 2019, 26, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.E.; Blair, C.D. Arbovirus-mosquito interactions: RNAi pathway. Curr. Opin. Virol. 2015, 15, 119–126. [Google Scholar] [CrossRef]
- Samuel, G.H.; Adelman, Z.N.; Myles, K.M. Antiviral immunity and virus-mediated antagonism in disease vector mosquitoes. Trends Microbiol. 2018, 26, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Mongelli, V.; Saleh, M.-C. Bugs are not to be silenced: Small RNA pathways and antiviral responses in insects. Annu. Rev. Virol. 2016, 3, 573–589. [Google Scholar] [CrossRef]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Asgari, S. Role of microRNAs in arbovirus/vector interactions. Viruses 2014, 6, 3514–3534. [Google Scholar] [CrossRef]
- Goic, B.; Stapleford, K.A.; Frangeul, L.; Doucet, A.J.; Gausson, V.; Blanc, H.; Schemmel-Jofre, N.; Cristofari, G.; Lambrechts, L.; Vignuzzi, M.; et al. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Joosten, J.; Van Rij, R.P.; Miesen, P. Slicing of viral RNA guided by endogenous piRNAs triggers the production of responder and trailer piRNAs in Aedes mosquitoes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nag, D.K.; Brecher, M.; Kramer, L.D. DNA forms of arboviral RNA genomes are generated following infection in mosquito cell cultures. Virology 2016, 498, 164–171. [Google Scholar] [CrossRef]
- Poirier, E.Z.; Goic, B.; Tomé-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-dependent generation of viral DNA from defective genomes of RNA viruses modulates antiviral immunity in insects. Cell Host Microbe 2018, 23, 353–365.e8. [Google Scholar] [CrossRef]
- Palatini, U.; Masri, R.A.; Cosme, L.V.; Koren, S.; Thibaud-Nissen, F.; Biedler, J.K.; Krsticevic, F.; Johnston, J.S.; Halbach, R.; Crawford, J.E.; et al. Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 2020, 21, 215. [Google Scholar] [CrossRef]
- Palatini, U.; Miesen, P.; Carballar-Lejarazu, R.; Ometto, L.; Rizzo, E.; Tu, Z.; van Rij, R.P.; Bonizzoni, M. Comparative genomics shows that viral integrations are abundant and express piRNAs in the arboviral vectors Aedes aegypti and Aedes albopictus. BMC Genom. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Crava, C.M.; Varghese, F.S.; Pischedda, E.; Halbach, R.; Palatini, U.; Marconcini, M.; Gasmi, L.; Redmond, S.; Afrane, Y.; Ayala, D.; et al. Population genomics in the arboviral vector Aedes aegypti reveals the genomic architecture and evolution of endogenous viral elements. Mol. Ecol. 2021. [Google Scholar] [CrossRef]
- Tassetto, M.; Kunitomi, M.; Whitfield, Z.J.; Dolan, P.T.; Sánchez-Vargas, I.; Garcia-Knight, M.; Ribiero, I.; Chen, T.; Olson, K.E.; Andino, R. Control of RNA viruses in mosquito cells through the acquisition of vDNA and endogenous viral elements. eLife 2019, 8, e41244. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Baidaliuk, A.; Miesen, P.; Van Rij, R.P.; Lambrechts, L.; Saleh, M.-C. Non-retroviral endogenous viral element limits cognate virus replication in Aedes aegypti ovaries CFAV-eve efficient viral replication CFAV. Curr. Biol. 2020, 30, 3495–3506.e6. [Google Scholar] [CrossRef]
- Houé, V.; Gabiane, G.; Dauga, C.; Suez, M.; Madec, Y.; Mousson, L.; Marconcini, M.; Yen, P.-S.; de Lamballerie, X.; Bonizzoni, M.; et al. Evolution and biological significance of flaviviral elements in the genome of the arboviral vector Aedes albopictus. Emerg. Microbes Infect. 2019, 8, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, E.; Scolari, F.; Valerio, F.; Carballar-Lejarazu, R.; Catapano, P.L.; Waterhouse, R.M.; Bonizzoni, M. Insights into an unexplored component of the mosquito repeatome: Distribution and variability of viral sequences integrated into the genome of the arboviral vector Aedes albopictus. Front. Genet. 2019, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.P.; Miesen, P.; Overheul, G.J.; Van Rij, R.P.; Van Oers, M.M.; Pijlman, G.P. Mosquito small RNA responses to West Nile and insect-specific virus infections in Aedes and Culex mosquito cells. Viruses 2019, 11, 271. [Google Scholar] [CrossRef]
- Miesen, P.; Joosten, J.; van Rij, R.P. PIWIs go viral: Arbovirus-derived piRNAs in vector mosquitoes. PLoS Pathog. 2016, 12, e1006017. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.-E.; Borchert, G.M. Viral microRNAs, host microRNAs regulating viruses, and bacterial microRNA-like RNAs. Methods Mol. Biol. 2017, 1617, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Frangeul, L.; Blanc, H.; Saleh, M. Differential Small RNA Responses against co-infecting insect-specific viruses in Aedes albopictus mosquitoes. Viruses 2020, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.D. Deducing the role of virus genome-derived PIWI-associated RNAs in the mosquito-arbovirus arms race. Front. Genet. 2019, 10, 1114. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Olson, K.E.; Blair, C.D.; Garcia-Blanco, M.A.; Cullen, B.R. A “microRNA-like” small RNA expressed by Dengue virus? Proc. Natl. Acad. Sci. USA 2014, 111, E2359. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Asgari, S. MicroRNA-like viral small RNA from Dengue virus 2 autoregulates its replication in mosquito cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2746–2751. [Google Scholar] [CrossRef]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef]
- Sigle, L.T.; McGraw, E.A. Expanding the canon: Non-classical mosquito genes at the interface of arboviral infection. Insect Biochem. Mol. Biol. 2019, 109, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Mousson, L.; Martin, E.; Zouache, K.; Madec, Y.; Mavingui, P.; Failloux, A.B. Wolbachia modulates Chikungunya replication in Aedes albopictus. Mol. Ecol. 2010, 19, 1953–1964. [Google Scholar] [CrossRef]
- Marconcini, M.; Hernandez, L.; Iovino, G.; Houé, V.; Valerio, F.; Palatini, U.; Pischedda, E.; Crawford, J.E.; White, B.J.; Lin, T.; et al. Polymorphism analyses and protein modelling inform on functional specialization of Piwi clade genes in the arboviral vector Aedes albopictus. PLoS Negl. Trop. Dis. 2019, 13, e0007919. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Broad Inst. Harv. MIT 2013, 00, 1–3. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Szymczak, S.; Ellinghaus, D.; Hemmrich, G.; Rühlemann, M.; Kraemer, L.; Mucha, S.; Wienbrandt, L.; Stanulla, M.; Franke, A.; et al. Vy-PER: Eliminating false positive detection of virus integration events in next generation sequencing data. Sci. Rep. 2015, 5, 11534. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, E.; Crava, C.; Carlassara, M.; Zucca, S.; Gasmi, L.; Bonizzoni, M. ViR: A tool to solve intrasample variability in the prediction of viral integration sites using whole genome sequencing data. BMC Bioinform. 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Rosenkranz, D.; Han, C.T.; Roovers, E.F.; Zischler, H.; Ketting, R.F. Piwi proteins and piRNAs in mammalian oocytes and early embryos: From sample to sequence. Genom. Data 2015, 5, 309–313. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Uhrig, S.; Klein, H. PingPongPro: A tool for the detection of piRNA-mediated transposon-silencing in small RNA-Seq data. Bioinformatics 2018, 35, 335–336. [Google Scholar] [CrossRef]
- Antoniewski, C. Computing siRNA and piRNA overlap signatures. Methods Mol. Biol. 2014, 6, 135–146. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- BBMAP. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 15 September 2020).
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Huang, K.-Y.; Lee, T.-Y.; Teng, Y.-C.; Chang, T.-H. ViralmiR: A support-vector-machine-based method for predicting viral microRNA precursors. BMC Bioinform. 2015, 16 (Suppl. S1), S1–S9. [Google Scholar] [CrossRef]
- Tav, C.; Tempel, S.; Poligny, L.; Tahi, F. miRNAFold: A web server for fast miRNA precursor prediction in genomes. Nucleic Acids Res. 2016, 44, W181–W184. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.P.; Willmann, M.R.; Meyers, B.C. Experimental design, preprocessing, normalization and differential expression analysis of small RNA sequencing experiments. Silence 2011, 2, 2. [Google Scholar] [CrossRef]
- Locati, M.D.; Terpstra, I.; de Leeuw, W.C.; Kuzak, M.; Rauwerda, H.; Ensink, W.A.; van Leeuwen, S.; Nehrdich, U.; Spaink, H.P.; Jonker, M.J.; et al. Improving small RNA-seq by using a synthetic spike-in set for size-range quality control together with a set for data normalization. Nucleic Acids Res. 2015, 43, e89. [Google Scholar] [CrossRef]
- Akkouche, A.; Mugat, B.; Barckmann, B.; Varela-Chavez, C.; Li, B.; Raffel, R.; Pélisson, A.; Chambeyron, S. Piwi is required during drosophila embryogenesis to license dual-strand piRNA clusters for transposon repression in adult ovaries. Mol. Cell 2017, 66, 411–419.e4. [Google Scholar] [CrossRef]
- Venny. Available online: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 3 August 2020).
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- FASTX. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 3 August 2020).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Batz, Z.A.; Goff, A.C.; Armbruster, P.A. MicroRNAs are differentially abundant during Aedes albopictus diapause maintenance but not diapause induction. Insect Mol. Biol. 2017, 26, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tu, S.; Stubna, M.; Wu, W.-S.; Huang, W.-C.; Weng, Z.; Lee, H.-C. The piRNA targeting rules and the resistance to piRNA silencing in endogenous genes. Science 2018, 359, 587–592. [Google Scholar] [CrossRef]
- Lavezzo, E.; Falda, M.; Fontana, P.; Bianco, L.; Toppo, S. Enhancing protein function prediction with taxonomic constraints—The Argot2.5 web server. Methods 2016, 93, 15–23. [Google Scholar] [CrossRef]
- Reynolds, J.A.; Poelchau, M.F.; Rahman, Z.; Armbruster, P.A.; Denlinger, D.L. Transcript profiling reveals mechanisms for lipid conservation during diapause in the mosquito, Aedes albopictus. J. Insect Physiol. 2012, 58, 966–973. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Hellström, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef]
- Morazzani, E.M.; Wiley, M.R.; Murreddu, M.G.; Adelman, Z.N.; Myles, K.M. Production of virus-derived ping-pong-dependent piRNA-like small RNAs in the mosquito soma. PLoS Pathog. 2012, 8, e1002470. [Google Scholar] [CrossRef]
- ter Horst, A.M.; Nigg, J.C.; Dekker, F.M.; Falk, B.W. Endogenous viral elements are widespread in arthropod genomes and commonly give rise to PIWI-interacting RNAs. J. Virol. 2019, 93, e02124-18. [Google Scholar] [CrossRef]
- Lee, R.C.H.; Chu, J.J.H. Proteomics profiling of chikungunya-infected Aedes albopictus C6/36 cells reveal important mosquito cell factors in virus replication. PLoS Negl. Trop. Dis. 2015, 9, e0003544. [Google Scholar] [CrossRef]
- Patramool, S.; Surasombatpattana, P.; Luplertlop, N.; Sévéno, M.; Choumet, V.; Thomas, F.; Missé, D. Proteomic analysis of an Aedes albopictus cell line infected with Dengue serotypes 1 and 3 viruses. Parasit. Vectors 2011, 4, 138. [Google Scholar] [CrossRef]
- Tchankouo-Nguetcheu, S.; Khun, H.; Pincet, L.; Roux, P.; Bahut, M.; Huerre, M.; Guette, C.; Choumet, V. Differential protein modulation in midguts of Aedes aegypti infected with chikungunya and dengue 2 viruses. PLoS ONE 2010, 5, e13149. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.B.; Wahl, A.S.; Freund, S.; Genzel, Y.; Reichl, U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst. Biol. 2010, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Mondotte, J.A.; Gausson, V.; Frangeul, L.; Suzuki, Y.; Vazeille, M.; Mongelli, V.; Blanc, H.; Failloux, A.-B.; Saleh, M.-C. Evidence for long-lasting transgenerational antiviral immunity in insects. Cell Rep. 2020, 33, 108506. [Google Scholar] [CrossRef] [PubMed]
- Halbach, R.; Miesen, P.; Joosten, J.; Taşköprü, E.; Rondeel, I.; Pennings, B.; Vogels, C.B.F.; Merkling, S.H.; Koenraadt, C.J.; Lambrechts, L.; et al. A satellite repeat-derived piRNA controls embryonic development of Aedes. Nature 2020, 580, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.O.; Adamec, J.; Kuhn, R.J. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 2012, 8, e1002584. [Google Scholar] [CrossRef]
- Vial, T.; Tan, W.-L.; Deharo, E.; Missé, D.; Marti, G.; Pompon, J. Mosquito metabolomics reveal that dengue virus replication requires phospholipid reconfiguration via the remodeling cycle. Proc. Natl. Acad. Sci. USA 2020, 117, 27627–27636. [Google Scholar] [CrossRef]
- Gottipati, K.; Woodson, M.; Choi, K.H. Membrane binding and rearrangement by chikungunya virus capping enzyme nsP1. Virology 2020, 544, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shrinet, J.; Jain, S.; Jain, J.; Bhatnagar, R.K.; Sunil, S. Next generation sequencing reveals regulation of distinct Aedes microRNAs during chikungunya virus development. PLoS Negl. Trop. Dis. 2014, 8, e2616. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.L.; Harrison, T.; Hess, A.M.; Ebel, G.D. MicroRNA levels are modulated in Aedes aegypti after exposure to Dengue-2. Insect Mol. Biol. 2014, 23, 132–139. [Google Scholar] [CrossRef]
- Saldaña, M.A.; Etebari, K.; Hart, C.E.; Widen, S.G.; Wood, T.G.; Thangamani, S.; Asgari, S.; Hughes, G.L. Zika virus alters the microRNA expression profile and elicits an RNAi response in Aedes aegypti mosquitoes. PLoS Negl. Trop. Dis. 2017, 11, e0005760. [Google Scholar] [CrossRef]
- Gainetdinov, I.; Colpan, C.; Arif, A.; Cecchini, K.; Zamore, P.D. A single mechanism of biogenesis, initiated and directed by PIWI proteins, explains piRNA production in most animals unified piRNA biogenesis model. Mol. Cell 2018, 71, 775–790.e5. [Google Scholar] [CrossRef] [PubMed]
- Miesen, P.; Ivens, A.; Buck, A.H.; van Rij, R.P. Small RNA profiling in dengue virus 2-infected Aedes mosquito cells reveals viral piRNAs and novel host miRNAs. PLoS Negl. Trop. Dis. 2016, 10, 1–22. [Google Scholar] [CrossRef]
- Sánchez-Vargas, I.; Scott, J.C.; Poole-Smith, B.K.; Franz, A.W.E.; Barbosa-Solomieu, V.; Wilusz, J.; Olson, K.E.; Blair, C.D. Dengue virus type 2 infections of Aedes aegypti are modulated by the mosquito’s RNA interference pathway. PLoS Pathog. 2009, 5, e1000299. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.C.; Brackney, D.E.; Campbell, C.L.; Bondu-Hawkins, V.; Hjelle, B.; Ebel, G.D.; Olson, K.E.; Blair, C.D. Comparison of dengue virus type 2-specific small RNAs from RNA interference-competent and -incompetent mosquito cells. PLoS Negl. Trop. Dis. 2010, 4, e848. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.M.; Prasad, A.N.; Ptitsyn, A.; Ebel, G.D.; Olson, K.E.; Barbacioru, C.; Monighetti, C.; Campbell, C.L. Small RNA profiling of Dengue virus-mosquito interactions implicates the PIWI RNA pathway in anti-viral defense. BMC Microbiol. 2011, 11, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Nag, D.K.; Kramer, L.D. Patchy DNA forms of the Zika virus RNA genome are generated following infection in mosquito cell cultures and in mosquitoes. J. Gen. Virol. 2017, 98, 2731–2737. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
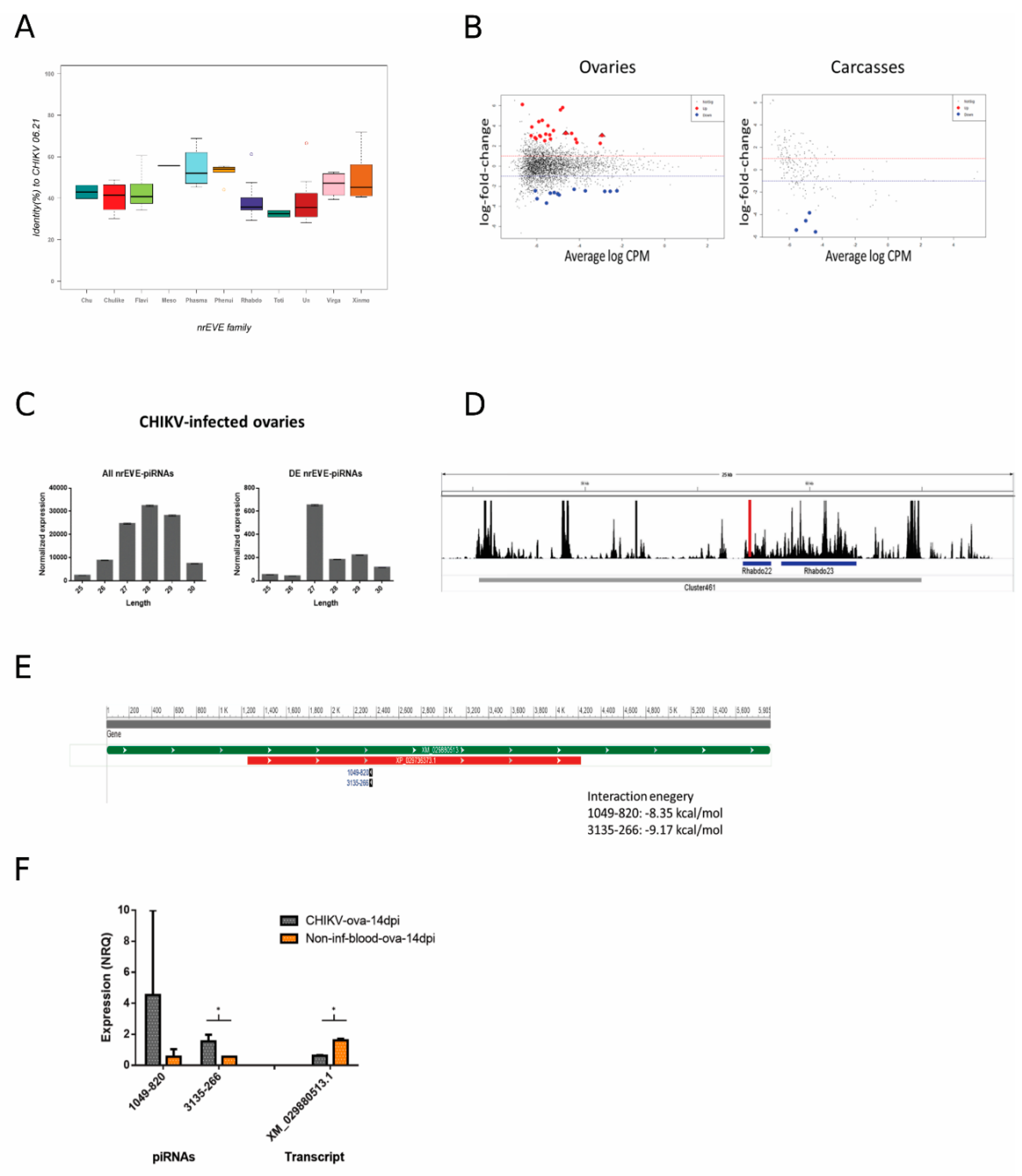{kind=link}
| piRNAs | siRNA | |||
|---|---|---|---|---|
| Sample | +RPKM | −RPKM | +RPKM | −RPKM |
| Ovaries | 14,025 ± 1007 | 3036 ± 61 | 28,453 ± 1308 | 33,094 ± 766 |
| Carcasses | 11,610 ± 2993 | 1852 ± 613 | 38,731 ± 2230 | 26,837 ± 127 |
| miRNA | Total Count | Log2FC | FDR |
|---|---|---|---|
| Ovaries | |||
| aal-miR-210 | 791 | 4.396 | 0.003 |
| aal-miR-124 | 2592 | 4.209 | 0.001 |
| aal-miR-1000 | 1147 | 3.177 | 0.014 |
| aal-miR-219 | 115 | 2.980 | 0.014 |
| aal-miR-932 | 2858 | 2.890 | 0.001 |
| aal-miR-981b | 260 | 2.864 | 0.026 |
| aal-miR-193 | 93 | 2.725 | 0.028 |
| aal-miR-285 | 2257 | 2.179 | 0.012 |
| aal-miR-2941 | 94,365 | −1.026 | 0.026 |
| aal-miR-7 | 6849 | −1.054 | 0.012 |
| aal-miR-316 | 2558 | −1.200 | 0.012 |
| aal-miR-9b | 4903 | −1.247 | 0.006 |
| Carcasses | |||
| aal-miR-new6 | 235 | −2.399 | 0.016 |
| aal-miR-1891 | 15,269 | −1.929 | 0.016 |
| Term | Total Count | KEGG ID | p-Value |
|---|---|---|---|
| Over-Expressed miRNAs Targets | |||
| Lysine degradation | 9 | aag00310 | 0.000 |
| mTOR signaling pathway | 9 | aag04150 | 0.000 |
| Toll and Imd signaling pathway | 3 | aag04624 | 0.003 |
| Phosphatidylinositol signaling system | 3 | aag04070 | 0.006 |
| Apoptosis | 3 | aag04214 | 0.007 |
| Terpenoid backbone biosynthesis | 2 | aag00900 | 0.014 |
| AGE-RAGE signaling pathway | 2 | aag04933 | 0.019 |
| MAPK signaling pathway | 3 | aag04013 | 0.022 |
| Endocytosis | 3 | aag04144 | 0.045 |
| Under-Expressed miRNAs Targets | |||
| SNARE interactions in vesicular transport | 3 | aag04130 | 0.000 |
| Proteasome | 3 | aag03050 | 0.001 |
| Phagosome | 3 | aag04145 | 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marconcini, M.; Pischedda, E.; Houé, V.; Palatini, U.; Lozada-Chávez, N.; Sogliani, D.; Failloux, A.-B.; Bonizzoni, M. Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes. Viruses 2021, 13, 553. https://doi.org/10.3390/v13040553
Marconcini M, Pischedda E, Houé V, Palatini U, Lozada-Chávez N, Sogliani D, Failloux A-B, Bonizzoni M. Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes. Viruses. 2021; 13(4):553. https://doi.org/10.3390/v13040553
Chicago/Turabian StyleMarconcini, Michele, Elisa Pischedda, Vincent Houé, Umberto Palatini, Nabor Lozada-Chávez, Davide Sogliani, Anna-Bella Failloux, and Mariangela Bonizzoni. 2021. "Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes" Viruses 13, no. 4: 553. https://doi.org/10.3390/v13040553
APA StyleMarconcini, M., Pischedda, E., Houé, V., Palatini, U., Lozada-Chávez, N., Sogliani, D., Failloux, A.-B., & Bonizzoni, M. (2021). Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes. Viruses, 13(4), 553. https://doi.org/10.3390/v13040553