Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related mortality. Almost half of HCC cases are associated with hepatitis B virus (HBV) infections, which often lead to HBV sequence integrations in the human genome. Accurate identification of HBV integration sites at a single nucleotide resolution is critical for developing a better understanding of the cancer genome landscape and of the disease itself. Here, we performed further analyses and characterization of HBV integrations identified by our recently reported VIcaller platform in recurrent or known HCC genes (such as TERT, MLL4, and CCNE1) as well as non-recurrent cancer-related genes (such as CSMD2, NKD2, and RHOU). Our pathway enrichment analysis revealed multiple pathways involving the alcohol dehydrogenase 4 gene, such as the metabolism pathways of retinol, tyrosine, and fatty acid. Further analysis of the HBV integration sites revealed distinct patterns involving the integration upper breakpoints, integrated genome lengths, and integration allele fractions between tumor and normal tissues. Our analysis also implies that the VIcaller method has diagnostic potential through discovering novel clonal integrations in cancer-related genes. In conclusion, although VIcaller is a hypothesis free virome-wide approach, it can still be applied to accurately identify genome-wide integration events of a specific candidate virus and their integration allele fractions.
1. Introduction
Hepatocellular carcinoma (HCC), a primary liver malignancy and leading cause of cancer-related deaths, is a major global health concern. The onset of HCC is often preceded by chronic liver conditions (such as hepatitis, cirrhosis or fibrosis [1,2]), which can further complicate anti-cancer treatment regimens [3]. Extensive research has established that chronic Hepatitis B virus (HBV) infection is a leading risk factor for the initiation and progression of HCC [4] and may account for approximately half of all HCC cases [5]. With about 400 million global cases of HBV infection [5], HBV-associated HCC remains highly prevalent, particularly in parts of Asia and Sub-Saharan Africa [6]. Despite the strong correlation between HBV infections and HCC onset, the precise genetic and genomic mechanisms underlying this relationship are still poorly understood [5].
Following infection, HBVs commonly integrate their DNA into the human genome. Such integration events have been identified in 75–90% of HCC tissues [7]. These integrations may further lead to development of host-virus fusion transcripts, particularly if the integrations occur within genic regions. The HBV genome is circular and partially double stranded and is approximately 3200 base pairs (bp) in length. It contains four overlapping open reading frames, namely surface, core, polymerase, and X [8]. The HBV X region, located near the nucleotide 1800 (nt 1374–1838) region on the HBV reference genome [9], is often retained following integration events and is selectively over-expressed in HCC [10,11]; thus, it may contribute to the high rate of metastasis in HBV-associated HCC patients [12]. Systematic research on the identification and characterization of the patterns of integration breakpoints on both the human and HBV genomes is critical for understanding the mechanistic processes of HBV integrations. We recently found that the cellular proportion of each integration event, which can be estimated based on its integration allele fraction, serves as important evidence to identify integration events involved in early-stage tumorigenesis [13]. Such analyses may provide new opportunities to understand the genomic landscape of HBV integrations and their involvement in the early stages of HCC development, which may help identify new diagnostic biomarkers and potential targets for therapeutic intervention.
An increasing number of studies have focused on the detection of infectious pathogens using cancer high-throughput sequencing (HTS) data [14]. HTS reads uniquely mapped to a viral reference genome are strong evidence of the presence of the virus in a sample; however, this analysis is compounded by two major practical challenges. First, the detected virus-mappable reads may have been derived from environmental microbes, synthetic DNA [15], or commonly-used cell lines such as HeLa cells [16]. Second, the presence of virus-mappable reads alone may not be conclusive for its role in tumorigenesis since viral infection may have occurred after oncogenesis [16]. To overcome these challenges, we recently developed a novel strategy and bioinformatics platform, viral integration caller (VIcaller), for identifying clonal viral integrations in the human genome and providing estimated integration allele frequencies [13]. We recently reported our preliminary findings on the analysis of whole genome sequencing (WGS) data from 88 HBV-associated HCC patients [9] using the VIcaller approach, and demonstrated high sensitivity and precision for the identification of integration events [13]. In this study, we report our in-depth analyses of these HBV integration events detected by VIcaller, as well as the characterization and pathway enrichment analyses of these integration events.
2. Methods
2.1. Detection of HBV Integrations Using VIcaller
A total of 88 HCC samples with both tumor and paired normal tissues were previously analyzed using the VIcaller platform [13]. In brief, we first submitted the paired-end reads in FASTQ format to VIcaller’s “detect” function to screen for integrations of the candidate virus, i.e., HBV (NC_003977.2), with the parameters: “-d WGS -m standard -r -a -q 20”. We then validated all identified candidate HBV integrations using the VIcaller “validate” function with the default parameters. Only the successfully validated HBV integration candidates were included for further analyses. Lastly, we calculated the integration allele fraction of each detected HBV integration event using the VIcaller “calculate” function with default parameters. To identify novel HBV integration events in the 88 HCC samples, we subsequently compared our detected HBV integrations with those presented by Sung et al. [9]. The resulting HBV integrations were then classified as “consistent”, if detected both by VIcaller and Sung et al. “novel”, if detected by VIcaller but not by Sung et al., and “missing” if detected by Sung et al. but not VIcaller [13].
2.2. Repeat Sequence Comparative Analysis
The repeat sequence annotation file generated by RepeatMasker [17] was downloaded from the UCSC Genome Browser (build hg19). The repeat density was examined using the BEDtools2 window function [18], and we determined the proportion of repeat sequences covering a 2000 bp window centered at each HBV integration site. We compared the repeat densities of novel, missing, and consistent HBV integrations with random positions across the human genome using a student’s t-test.
2.3. Upper and Lower Breakpoint Analyses
The genomic coordinates of the upper and lower breakpoints of each integration on the HBV reference genome (NC_003977.2) were analyzed separately. The HBV upper breakpoint is the genomic coordinate of the sequence junction connected upstream (5′) orientation on the human sequence, while the lower breakpoint is the other HBV sequence junction connected downstream (3′) on the human sequence. When the breakpoint locations were detected from chimeric reads only (without split reads; chimeric and split reads were depicted in Supplementary Figure S1 of our previous paper [19]), the nearest location to the breakpoint was adopted. HBV integrations with one breakpoint and those with two breakpoints were analyzed separately. The lengths of inserted HBV genomes or sequences were estimated based on the HBV integration events with both upper and lower breakpoints detected.
2.4. Gene Ontology and Pathway Enrichment Analyses of Novel HBV Integrations
We first converted the identified novel HBV integrations into variant call format, treating HBV integrations as insertions relative to the human reference genome, and then performed functional annotation using CADD [20] (https://cadd.gs.washington.edu). For the human genes in which the integrations occurred, we manually obtained and curated protein functions and associated diseases or biological functions through literature searches. To identify novel pathways involving genes other than the known HCC-associated genes (i.e., TERT, MLL4, and CCNE1), our pathway enrichment analysis was carried out based only on non-recurrent human genes with HBV integrations (i.e., genes observed only once in our examined samples) using the KEGG pathway database [21,22], as described in our previous paper [22]. Briefly, the overrepresentation of pathway-associated genes from our gene set was modelled according to a hypergeometric distribution function to generate enrichment p values; and the enrichment ratio was defined as expected divided by observed number of genes per pathway.
2.5. Integration Allele Fraction Analysis
The allele fraction of each HBV integration detected in the tumor and paired normal tissues of each subject (i.e., HCC sample) was calculated by VIcaller. The HCC samples were ranked first by the highest allele fraction in the tumor tissues; then by the highest in the normal tissues having no HBV integrations detected in the tumors. The HBV integrations with high allele fractions (i.e., ≥90%), but which were supported by only a very small number of reads (i.e., fewer than four supporting reads), were excluded from subsequent analyses.
2.6. Tumor Grade Analysis
The tumor pathology grades (i.e., poor, moderate, and high levels) were obtained for each subject from the Sung et al., study [9]. A student’s t-test was used to compare the average numbers of HBV integrations between each pair of the distinct pathology grade groups. All 88 analyzed HCC patients were from chronically HBV infected patients. Additional demographic and clinicopathologic characteristics of the samples were described in Supplementary Table S1 of the Sung et al., study [9].
3. Results
3.1. Analyses of Novel HBV Integrations Identified by VIcaller
In general, HBV integrations identified in genomic repetitive regions are considered less accurate than those identified in other genomic regions due to higher alignment errors of HTS reads in these regions (Supplementary Figure S1). We combined the HBV integrations identified from HBsAg-positive and negative tumor and normal tissues, and then compared the density values of repeat sequences among four groups, including our identified novel and consistent HBV integrations, missing integrations, and randomly sampled genomic positions. We found that the novel and consistent HBV integrations showed decreased repeat sequence density compared to the randomly sampled positions, although the p values were statistically insignificant (p values = 0.08 and 0.1, respectively). By comparison, the repeat sequence density of the missing integrations trended higher than our randomly sampled genomic positions and consistent HBV integrations (p values = 0.09 and 0.08, respectively) (Figure 1A). We further observed that most of the samples with novel HBV integrations (e.g., samples 90T and 73T) showed significantly upregulated TERT gene expression levels, implying that the novel HBV integrations detected by VIcaller are accurate and TERT upregulation is likely involved in the disease mechanism in these samples (Figure 1B).
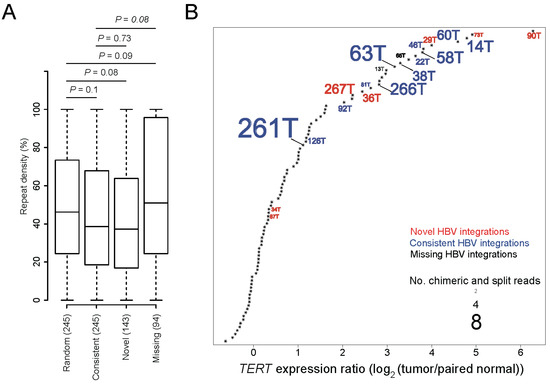
Figure 1.
Accuracy of the novel hepatitis B virus (HBV) integrations detected by VIcaller. (A) Comparison of the repeat sequence density values among the novel, consistent, and missing HBV integration events and randomly selected genomic positions. “Novel” HBV integrations are those detected only by VIcaller but not Sung et al. “Consistent” HBV integrations are those detected by both VIcaller and Sung et al. “Missing” HBV integrations are those detected only by Sung et al. but not VIcaller. The p values were computed using a student’s t-test. (B) Most of the TERT gene expression levels of the novel HBV integrations are significantly up-regulated and thus ranked among the top upregulations. Only samples with HBV integrations in TERT are labeled in the plot. Red, blue, and black correspond to novel, consistent, and missing integrations, respectively. The font size has been scaled to reflect the number of supporting reads per integration.
3.2. Charaterization of HBV Integration Breakpoints
We first characterized the upper and lower breakpoints on the HBV reference genome of the 388 HBV integrations [13] identified in the tumor and paired normal tissues of the 88 subjects. We found that both upper and lower breakpoints were highly enriched at ~1.8 kb, with the upper breakpoints also enriched between 1.8 kb and 3.2 kb compared to the lower breakpoints (Supplementary Figure S3A). These patterns were consistent between the HBV integrations with one breakpoint, and those with two identified breakpoints (Supplementary Figure S3B,C).
We then characterized breakpoint hotspots of HBV integrations between the tumor and paired normal tissues. The upper and lower breakpoints on the HBV genome were analyzed separately. We observed distinct patterns for the upper breakpoints on the HBV genome between the tumor (at ~1.8 kb) and normal (at ~2.2 kb, core gene) tissues (Figure 2A). In contrast, the lower breakpoint hotspots were located at ~1.8 kb (core and X genes) in both tumor and normal tissues (Figure 2B), which was consistent with previous results [9]. Moreover, we found that the length of inserted HBV sequences detected in tumor tissues were significantly shorter than those detected in normal tissues (p value = 6.4 × 10−5; Figure 2C,D); and the HBV integrations in the normal tissues were prone to have more complete HBV genome sequences, with an enrichment of the upper breakpoints at ~2.2 kb.
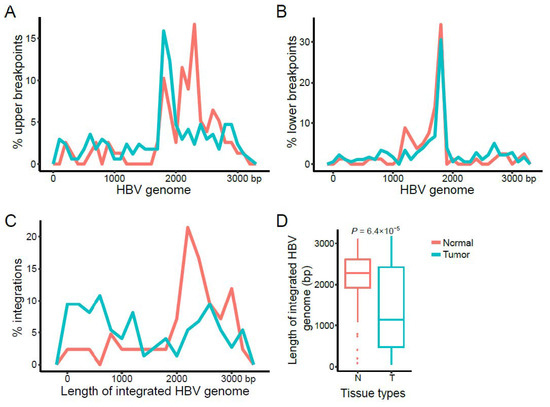
Figure 2.
Characteristics of the HBV breakpoints and inserted HBV genome length. Distribution of the (A) upper and (B) lower breakpoints on the HBV genome. (C,D) Distribution and comparison of the lengths of the inserted HBV sequences between tumor and normal tissues. The p values were computed using a student’s t-test.
3.3. Annotation of Novel Non-Recurrent Genic HBV Integrations and Gene Pathway Analysis
We performed gene annotation analysis of all novel non-recurrent HBV integrations, i.e., integrations in genes other than TERT, MLL4, and CCNE1. We identified 48 genic HBV integrations in 18 tumor samples and 29 normal samples (Table 1). For example, we detected an integration event in the intronic region of the CSMD2 gene, supported by five chimeric and split reads. CSMD2 may act as a tumor suppressor gene for colorectal cancer [23]. We detected another integration in the RHOU gene, which has been associated with basal cell carcinoma [24]. We also detected an integration in the intronic region of NKD2 supported by four chimeric and split reads. NKD2 is a wnt antagonist shown to escort TGF-α-containing exocytic vesicles [25]. Dysregulation of NKD2 has been suggested to be involved in tumorigenesis of multiple cancers, including gastric cancer [26], esophageal cancer [27], and HCC [28], among others.

Table 1.
Characterization of all the novel non-recurrent genic HBV integrations identified by VIcaller.
We then carried out gene pathway enrichment analysis for all the non-recurrent genes with any HBV integrations using the KEGG pathway database (Table 2). We found enrichment of multiple pathways involving the alcohol dehydrogenase 4 (ADH4) gene. Among these pathways, the retinol metabolism pathway was the most significant, followed by the tyrosine metabolism and fatty acid metabolism pathways (p = 0.0005, 0.004, and 0.004; and enrichment ratios = 43.95, 45.73, and 43.61, respectively). We also observed enrichment of pathways involving ITGA11, such as focal adhesion and extra cellular matrix-receptor interaction (p = 0.004 and 0.006; and enrichment ratios = 14.06 and 22.06, respectively). Additionally, other important pathways related to metabolism were also significantly enriched, such as arachidonic acid metabolism, drug metabolism, and purine metabolism (p = 0.004, 0.005, and 0.01; and enrichment ratios = 31.78, 25.69, and 11.57, respectively).
Table 2.
Pathway enrichment analysis of all genes with non-recurrent HBV integrations.
3.4. Characteristics of Integrations Involved in Early-Stage Tumorigenesis
We analyzed the integration allele fraction of each HBV integration event in both the tumor and its paired normal tissues for each sample. We found that HBV integrations had higher allele fractions in the tumor tissue compared to its paired adjacent normal tissue in nearly all samples (Figure 3A,B). We excluded six HBV integration events because each had fewer than four supporting reads, even they showed extremely high integration allele fractions (i.e., 100% for five integrations and ~90% for one integration). The HBV integrations in the three well-known recurrent genes, TERT, MLL4, and CCNE1, always had the highest integration allele fractions [13] in each sample. Besides the integrations in these three genes, we identified seven HBV integrations with high integration allele fractions in other genes, most of which have been reported to be associated with cancer, including GAS7, SPECC1 (NSP), RSPO2, NRG1, PRDM16, ARID1B, and AFF1, as initially described in our recent method paper [13]. The HBV integrations in GAS7, NRG1, PRDM16, and ARID1B are among the highest integration allele fractions in each sample and no HBV integrations were found in any of the three recurrent genes (i.e., TERT, MLL4, and CCNE1) in these samples. In comparison to the three recurrent genes, the non-recurrent cancer-related genes had comparably high HBV integration allele fractions, e.g., also significantly higher than intergenic integrations and integrations in non-cancer-related genes (Figure 3C,D). The results imply that the HBV integrations in these cancer-related genes may also be involved in the early-stage tumorigenesis and potentially play vital roles in HCC.
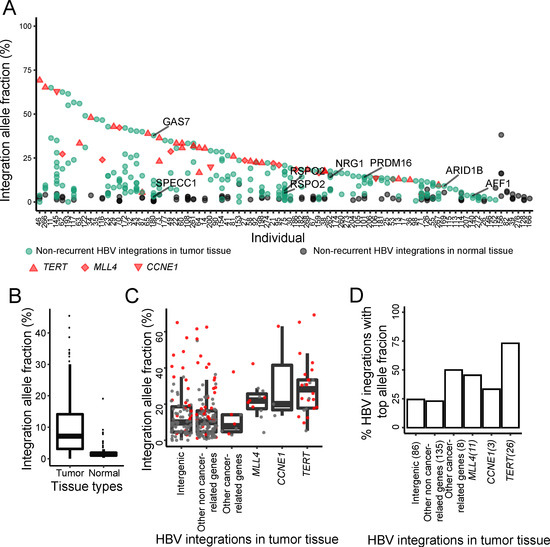
Figure 3.
Integration allele fractions of HBV integrations in tumor and paired normal tissues. (A) HBV integrations in tumor and its paired normal tissue of each subject. The HBV integrations in the TERT, MLL4, and CCNE1 (recurrent) genes are shown in red and the HBV integrations in other (non-recurrent) cancer-related genes are in green. (B) Comparison of the HBV integration allele fractions between tumor and normal tissues. (C) Comparison of the HBV integration allele fractions among different groups (the integration event with the top allele fraction was used for each subject). (D) Comparison between the number of HBV integrations with top cellular proportion between different groups.
3.5. Comparison of HBV Integration Abundance among Tumor Grades
Well-differentiated cancer cells tend to grow and spread more slowly than poorly differentiated or undifferentiated cancer cells [69]. We compared the number of HBV integrations per sample detected by VIcaller among three tumor groups, including poor, moderately, and highly differentiated tumors. The numbers of HBV integrations distinguished highly differentiated tumors (mean ± standard deviation = 2.1 ± 2.1) from moderately differentiated tumors (3.8 ± 3.5) with p value of 0.04 (Figure 4). This preliminary finding, if proven, may be crucial as patients with highly differentiated tumors have better prognosis than those with moderately differentiated tumors. By comparison, no statistically significant difference was reported based on the HBV integrations detected by Sung et al. [9] (p > 0.05; Supplementary Figure S4).
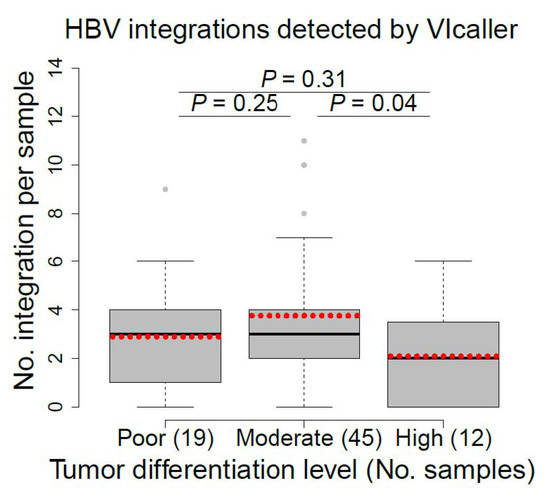
Figure 4.
Comparison of the number of HBV integrations detected by VIcaller among poor, moderate, and highly differentiated tumors. Student’s t test was used to compare each group pair. The dotted red line indicates the average number of HBV integrations for each patient group.
4. Discussion
Chronic HBV infections play a considerable role in the pathophysiology of HCC. The large-scale efforts of vaccination have reduced the incidence of HBV infections; however, chronic HBV infections are still persistent in 3.5% of the global population [70]. HBV infections often subsequently lead to HBV sequence integrations in the human genomes. It is imperative to accurately identify and characterize precise HBV integration events. Understanding the HBV integration sites in HCC genomes may provide new targets for therapeutic development. In this study, we carried out a comprehensive analysis of the HBV integrations that we previously detected in 88 HCC patients using our virome-wide approach, VIcaller. We compared our findings with those reported in a previous study that used an HBV-specific approach. We focused on the novel integration events identified by our VIcaller approach. Since the contributions of TERT and MLL4 have been reported previously [7,71,72,73], we primarily examined and characterized the novel non-recurrent genic HBV integrations. Furthermore, it is notable that the VIcaller method has diagnostic potential through discovering novel integrations in known cancer-related genes. For example, we found intronic TERT integrations that have not been reported previously in samples 73T and 90T, and these findings provide evidence supporting a possible genetic diagnosis or explanation (which still needs further investigation) for the corresponding HCC patients for the first time.
We identified HBV integrations with high integration allele fractions in tumors in seven non-recurrent cancer-related genes, including GAS7 [74,75], NSP [76], RSPO2 [77,78], NRG1 [79], PRDM16 [80], ARID1B [81], and AFF1 [82]. GAS7 has been shown to be directly regulated by P53 and is part of a critical mechanism that mediates breast cancer metastasis [74]; NRG1 fusions were identified as drivers for lung adenocarcinoma [79]; And the involvement of PRDM16 in leukemia [80], ARID1B in ovarian cancer [81], and AFF1 in leukemia [82] has also been reported; It has been demonstrated that a GAS7-mediated pathway suppressed proliferation of HCC cells following treatment with oxaliplatin, an alkylating anti-neoplastic agent, and the inhibition of GAS7 negated the beneficial effects of the drug [83]. Similarly, NRG1 is associated with promoting metastasis of HCC cells by increasing epithelial-mesenchymal transition, thus increasing migratory behavior of the cancer cells [84] (genetic variants in NRG1 have also been associated with schizophrenia [85]).
We observed some unexpected HBV integrations only in normal tissues but not in paired tumor tissues. Most of these integrations had very low integration allele fractions (<5%), including those located in cancer-related genes. For example, we found an unexpected integration in the MAGI1 gene only in a normal tissue; however, only two chimeric and split reads were observed for this integration and its integration allele fraction was only 1.04%. MAGI1 has been associated with HCC [34]. We observed another unexpected integration in the intronic region of RTN4 only in a normal tissue, though its integration allele fraction was only 2.35%. RTN4 has been demonstrated to induce apoptosis in cancer cells but play an opposite role in normal cells [32]. Similarly, we found an unexpected integration in the intronic regions of EWSR1 only in normal tissue, with an integration allele fraction of only 3.13%. EWSR1 has been implicated in pancreatic cancer [67]. We also found an integration (integration allele fraction was 1.61%) only in normal tissue in DACH2, a biomarker for muscle-invasive urothelial carcinoma [68]. Further analyses are needed to verify these potential integration events, including, but not limited to, the use of single cell sequencing to study tissue microenvironment [86,87]. The observations of integrations in normal tissues in MAGI1, RTN4, EWSR1, and DACH1 highlight the need to screen larger cohorts of healthy tissues in future studies, similar to the current biobank efforts, which may distinguish viral integrations involved in early stages of tumorigenesis from random viral integration events.
We also observed that the lengths of integrated HBV sequences in tumor tissues were generally shorter than those integrated in normal tissues (Figure 2D). Further studies are needed to better understand how HBV integrations play a role in HCC, including lengths of HBV integrations, functional roles of inserted HBV DNA regions, cellular proportions of HBV integrations, and other covariates or factors. For example, it is known that the HBV X region is highly correlated with HCC onset [88]. We found that many HBV integrations in tumor tissues contained HBV enhancer or promoter regions, including parts of the HBV X region [13]. Similar research has been shown in Merkle cell carcinoma. Before Merkel cell polyomavirus can transform cells, truncated large T antigen gene sequences integrate into the host genome, leading to cellular transformation and tumor proliferation [88]. In addition, the HBV integrations in tumors had higher integration allele fractions (Figure 3A,B). By comparison, although the lengths of HBV integrations in normal tissues were longer, the total number of integration events was smaller, and their integration allele fractions were also lower. These results together further support the importance of including our proposed integration allele fraction analysis in studies aimed at identifying oncoviruses and cancer-related genes.
Furthermore, our pathway analysis of the novel non-recurrent genic HBV integrations revealed many critical metabolic pathways potentially involved in HCC. Notably, many pathways involving ADH4 were among the most enriched hits. Proper ADH4 functions are critical for alcohol metabolism [89], and functional changes in ADH4 [90,91] and other alcohol dehydrogenase-related genes [92,93] are associated with alcohol dependence or alcohol-induced medical diseases. Alcohol is classified as group 1 carcinogen [94]. Excessive intake of alcohol leads to fatty liver, cirrhosis, and eventual development of HCC [94,95,96]. Moreover, many recent publications have shown that aberrations in FAK expression contribute to the onset and invasiveness of HCC [97,98,99], which is supported by our findings of an enrichment of the focal adhesion pathway involving genes such as ITGA11, VCL, and LAMA.
The applications of the VIcaller method and platform are not limited to analysis of HBV integrations or HCC but can be directly applied to detect other oncoviruses in other cancer types. Approximately 15% of the global cancer incidences have viral etiology [100]. For example, EBV infections, detected in more than 95% of the world population, are associated with multiple cancers, such as non-Hodgkin’s lymphoma, nasopharyngeal carcinoma, and gastric cancer [101]; also, approximately 99% of cervical cancer cases are associated with human papillomaviruses, as are 50% of penile, vulvar, and vaginal cancers [88]. Discovering new oncogenic effects of human viruses is critical since such findings may eventually lead to development of vaccination strategies to reduce virus-mediated cancer mortality. VIcaller allows using cancer genome data for identifying virome-wide viral infections, particularly integration sites and their fusion transcripts, and for determining early-stage clonal integrations involved in early-stage tumorigenesis. For instance, we found that HBV integrations in the TERT promoter regions were prone to have the same orientation as TERT (e.g., 22 out of the 29 integration events were in the same 3′ to 5′ orientation as TERT in the human genome). This supports possible formation of fusion transcripts, although other mechanisms [102,103,104] may also be possible. This also emphasizes the importance of examining sequence orientations of viral integrations and identifying both upper and lower breakpoints when possible.
For future research, it will be worthwhile to further investigate the precise HBV genotypes (i.e., 10 HBV strains from A-J [105]), since the epidemiological and pathological HBV strain variations may impact the progression and clinical outcomes of HCC [106]. The HBV subtypes [107] can be identified by employing the available online genotyping tools such as HBVdb, HBV STAR or HepSEQ. Furthermore, a multidisciplinary analysis, such as integrated genomic, transcriptomic (e.g., long-read DNA and RNA sequencing), epigenomic, and proteomic investigations, may provide further functional validation of the biological roles of viral integrations in human diseases and health.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/13/2/245/s1, Figure S1: HBV integrations detected in repeat regions, Figure S2: comparison of numbers of supporting reads, Figure S3: comparison of upper and lower breakpoints, Figure S4: comparison of numbers of HBV integrations.
Author Contributions
Conceptualization, D.L.; methodology, D.L., and X.C.; software, X.C. and A.S.; validation, X.C., P.P.M., A.S., and D.L.; formal analysis, X.C. and A.S.; investigation, D.L., X.C., and P.P.M.; resources, D.L. and X.C.; data curation, X.C. and A.S.; writing—original draft preparation, P.P.M. and X.C.; writing—review and editing, P.P.M., X.C., D.L., and A.S.; visualization, X.C. and A.S.; supervision, D.L.; project administration, D.L.; funding acquisition, D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Start-up Fund of The University of Vermont, and partially by research grant from the Department of Defense Lung Cancer Research Program (LC190467).
Acknowledgments
The whole-genome sequencing data of the patients was obtained through the European Nucleotide Archive (accession number ERP001196). The normalized gene expression data was obtained through the Gene Expression Omnibus database (accession number GSE25097). The authors acknowledge the Vermont Advanced Computing Core and the Massachusetts Green High-Performance Computer C3DDB for computing resources. The authors thank Jason Kost, Jian Cao, Chao Cheng, Wing-Kin Sung, Guangchen Liu, and Michael Mariani for their discussions pertaining to the analysis.
Conflicts of Interest
Arvis Sulovari is currently an employee of Cajal Neuroscience, Inc. and has received compensation from it. Sulovari conducted the research while he was at the University of Vermont, and thus Cajal Neuroscience played no roles in the research of this manuscript. Other authors declare no potential conflict of interest.
References
- Jiang, D.; Deng, J.; Dong, C.; Ma, X.; Xiao, Q.; Zhou, B.; Yang, C.; Wei, L.; Conran, C.; Zheng, S.L.; et al. Knowledge-based analyses reveal new candidate genes associated with risk of hepatitis B virus related hepatocellular carcinoma. BMC Cancer 2020, 20, 403. [Google Scholar] [CrossRef]
- Liu, Z.; Chalasani, N.; Lin, J.; Gawrieh, S.; He, Y.; Tseng, Y.J.; Liu, W. Integrative omics analysis identifies macrophage migration inhibitory factor signaling pathways underlying human hepatic fibrogenesis and fibrosis. J. Bio-X Res. 2019, 2, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Malek, N.P. Early Detection of Hepatocellular Carcinoma: How to Screen and Follow up Patients with Liver Cirrhosis According to the GERMAN S3 Guideline? Diagnostics (Basel) 2015, 5, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar] [PubMed]
- Zhang, X.; Wang, L.; Yan, Y. Identification of potential key genes and pathways in hepatitis B virus-associated hepatocellular carcinoma by bioinformatics analyses. Oncol. Lett. 2020, 19, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Li, C.; Liang, L.; Xing, H.; Sun, L.Y.; Quan, B.; Wu, H.; Xu, X.F.; Wu, M.C.; Pawlik, T.M.; et al. Early and Late Recurrence of Hepatitis B Virus-Associated Hepatocellular Carcinoma. Oncologist 2020, 25, e1541. [Google Scholar] [CrossRef]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Chisari, F.V.; Ferrari, C.; Mondelli, M.U. Hepatitis B virus structure and biology. Microb. Pathog. 1989, 6, 311–325. [Google Scholar] [CrossRef]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Li, C.; Lin, C.; Cong, X.; Jiang, Y. PDK1-WNK1 signaling is affected by HBx and involved in the viability and metastasis of hepatic cells. Oncol. Lett. 2018, 15, 5940–5946. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Yao, Q.; Liu, J.S.; Wang, Y.Y.; Sun, W.M. Hepatitis B virus X protein promotes epithelial-mesenchymal transition and metastasis in hepatocellular carcinoma cell line HCCLM3 by targeting HMGA2. Oncol. Lett. 2018, 16, 5709–5714. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kost, J.; Sulovari, A.; Wong, N.; Liang, W.S.; Cao, J.; Li, D. A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome. Res. 2019, 29, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sultmann, H.; Moch, H.; Pathogens, P.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef]
- Chen, X.; Li, D. Sequencing facility and DNA source associated patterns of virus-mappable reads in whole-genome sequencing data. Genomics 2020, 113, 1189–1198. [Google Scholar] [CrossRef]
- Cao, J.; Li, D. Searching for human oncoviruses: Histories, challenges, and opportunities. J. Cell Biochem. 2018, 119, 4897–4906. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Version open-4.0.5. 2013–2015. Institute for Systems Biology. 2015. Available online: http://repeatmasker.org (accessed on 6 November 2013).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Chen, X.; Kost, J.; Li, D. Comprehensive comparative analysis of methods and software for identifying viral integrations. Brief Bioinform. 2019, 20, 2088–2097. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Kanehisa, M. Post-Genome Informatics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Sulovari, A.; Chen, Y.H.; Hudziak, J.J.; Li, D. Atlas of human diseases influenced by genetic variants with extreme allele frequency differences. Hum. Genet. 2017, 136, 39–54. [Google Scholar] [CrossRef]
- Zhang, R.; Song, C. Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumour Biol. 2014, 35, 4419–4423. [Google Scholar] [CrossRef] [PubMed]
- Chahal, H.S.; Wu, W.; Ransohoff, K.J.; Yang, L.; Hedlin, H.; Desai, M.; Lin, Y.; Dai, H.J.; Qureshi, A.A.; Li, W.Q.; et al. Genome-wide association study identifies 14 novel risk alleles associated with basal cell carcinoma. Nat. Commun. 2016, 7, 12510. [Google Scholar] [CrossRef]
- Hu, T.; Li, C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef]
- Jia, Y.; Cao, B.; Yang, Y.; Linghu, E.; Zhan, Q.; Lu, Y.; Yu, Y.; Herman, J.G.; Guo, M. Silencing NKD2 by promoter region hypermethylation promotes gastric cancer invasion and metastasis by up-regulating SOX18 in human gastric cancer. Oncotarget 2015, 6, 33470–33485. [Google Scholar] [CrossRef][Green Version]
- Cao, B.; Yang, W.; Jin, Y.; Zhang, M.; He, T.; Zhan, Q.; Herman, J.G.; Zhong, G.; Guo, M. Silencing NKD2 by Promoter Region Hypermethylation Promotes Esophageal Cancer Progression by Activating Wnt Signaling. J. Thorac. Oncol. 2016, 11, 1912–1926. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, S.; Chen, Y.; Hu, B.; Lu, C. Low expression of NKD2 is associated with enhanced cell proliferation and poor prognosis in human hepatocellular carcinoma. Hum. Pathol. 2018, 72, 80–90. [Google Scholar] [CrossRef]
- Havik, B.; Le Hellard, S.; Rietschel, M.; Lybaek, H.; Djurovic, S.; Mattheisen, M.; Muhleisen, T.W.; Degenhardt, F.; Priebe, L.; Maier, W.; et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol. Psychiatry 2011, 70, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Stacey, S.N.; Sulem, P.; Gudbjartsson, D.F.; Jonasdottir, A.; Thorleifsson, G.; Gudjonsson, S.A.; Masson, G.; Gudmundsson, J.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. Germline sequence variants in TGM3 and RGS22 confer risk of basal cell carcinoma. Hum. Mol. Genet. 2014, 23, 3045–3053. [Google Scholar] [CrossRef] [PubMed]
- Watari, A.; Yutsudo, M. Multi-functional gene ASY/Nogo/RTN-X/RTN4: Apoptosis, tumor suppression, and inhibition of neuronal regeneration. Apoptosis Int. J. Program. Cell Death 2003, 8, 5–9. [Google Scholar] [CrossRef]
- Castelletti, F.; Donadelli, R.; Banterla, F.; Hildebrandt, F.; Zipfel, P.F.; Bresin, E.; Otto, E.; Skerka, C.; Renieri, A.; Todeschini, M.; et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc. Natl. Acad. Sci. USA 2008, 105, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, T.; Wang, Z. Downregulation of MAGI1 associates with poor prognosis of hepatocellular carcinoma. J. Investig. Surg. 2012, 25, 93–99. [Google Scholar] [CrossRef]
- Genetics of Personality Consortium; de Moor, M.H.; van den Berg, S.M.; Verweij, K.J.; Krueger, R.F.; Luciano, M.; Arias Vasquez, A.; Matteson, L.K.; Derringer, J.; Esko, T.; et al. Meta-analysis of Genome-wide Association Studies for Neuroticism, and the Polygenic Association with Major Depressive Disorder. JAMA Psychiatry 2015, 72, 642–650. [Google Scholar] [CrossRef]
- Konno, K.; Matsuda, K.; Nakamoto, C.; Uchigashima, M.; Miyazaki, T.; Yamasaki, M.; Sakimura, K.; Yuzaki, M.; Watanabe, M. Enriched expression of GluD1 in higher brain regions and its involvement in parallel fiber-interneuron synapse formation in the cerebellum. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 7412–7424. [Google Scholar] [CrossRef]
- Luo, X.; Kranzler, H.R.; Zuo, L.; Lappalainen, J.; Yang, B.Z.; Gelernter, J. ADH4 gene variation is associated with alcohol dependence and drug dependence in European Americans: Results from HWD tests and case-control association studies. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2006, 31, 1085–1095. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhang, W.; Sehmi, J.; Li, X.; Wass, M.N.; Van der Harst, P.; Holm, H.; Sanna, S.; Kavousi, M.; Baumeister, S.E.; et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011, 43, 1131–1138. [Google Scholar] [CrossRef]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef]
- Pyun, J.A.; Kim, S.; Cha, D.H.; Kwack, K. Epistasis between IGF2R and ADAMTS19 polymorphisms associates with premature ovarian failure. Hum. Reprod. 2013, 28, 3146–3154. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, T.; Polvi, A.; Rydman, P.; Vendelin, J.; Pulkkinen, V.; Salmikangas, P.; Makela, S.; Rehn, M.; Pirskanen, A.; Rautanen, A.; et al. Characterization of a common susceptibility locus for asthma-related traits. Science 2004, 304, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.; Cule, E.; Moncrieffe, H.; Hinks, A.; Ursu, S.; Patrick, F.; Kassoumeri, L.; Flynn, E.; Bulatovic, M.; Wulffraat, N.; et al. Genome-wide data reveal novel genes for methotrexate response in a large cohort of juvenile idiopathic arthritis cases. Pharm. J. 2014, 14, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Ji, M.Y.; Shen, L.; Qiu, S.; Guo, X.F.; Dong, W.G. Decreased EGR3 expression is related to poor prognosis in patients with gastric cancer. J. Mol. Histol. 2013, 44, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, Y.T.; Xie, X.; Yang, Y.N.; Ma, X.; Zheng, Y.Y.; Pan, S.; Liu, F.; Chen, B.D. Association of Egr3 genetic polymorphisms and coronary artery disease in the Uygur and Han of China. Lipids Health Dis. 2014, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.D.; Goodarzi, M.O.; Langefeld, C.D.; Wang, N.; Guo, X.; Taylor, K.D.; Fingerlin, T.E.; Norris, J.M.; Buchanan, T.A.; Xiang, A.H.; et al. Genetic Variants Associated with Quantitative Glucose Homeostasis Traits Translate to Type 2 Diabetes in Mexican Americans: The GUARDIAN (Genetics Underlying Diabetes in Hispanics) Consortium. Diabetes 2015, 64, 1853–1866. [Google Scholar] [CrossRef]
- Majumder, S.; Fisk, H.A. VDAC3 and Mps1 negatively regulate ciliogenesis. Cell Cycle 2013, 12, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Comuzzie, A.G.; Cole, S.A.; Laston, S.L.; Voruganti, V.S.; Haack, K.; Gibbs, R.A.; Butte, N.F. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS ONE 2012, 7, e51954. [Google Scholar] [CrossRef]
- Park, B.H.; Lee, Y.H. Phosphorylation of SAV1 by mammalian ste20-like kinase promotes cell death. BMB Rep. 2011, 44, 584–589. [Google Scholar] [CrossRef]
- Zi, M.; Maqsood, A.; Prehar, S.; Mohamed, T.M.; Abou-Leisa, R.; Robertson, A.; Cartwright, E.J.; Ray, S.G.; Oh, S.; Lim, D.S.; et al. The mammalian Ste20-like kinase 2 (Mst2) modulates stress-induced cardiac hypertrophy. J. Biol. Chem. 2014, 289, 24275–24288. [Google Scholar] [CrossRef]
- Ohshima, K.; Kanto, K.; Hatakeyama, K.; Ide, T.; Wakabayashi-Nakao, K.; Watanabe, Y.; Sakura, N.; Terashima, M.; Yamaguchi, K.; Mochizuki, T. Exosome-mediated extracellular release of polyadenylate-binding protein 1 in human metastatic duodenal cancer cells. Proteomics 2014, 14, 2297–2306. [Google Scholar] [CrossRef]
- Ilmer, M.; Boiles, A.R.; Regel, I.; Yokoi, K.; Michalski, C.W.; Wistuba, I.I.; Rodriguez, J.; Alt, E.; Vykoukal, J. RSPO2 Enhances Canonical Wnt Signaling to Confer Stemness-Associated Traits to Susceptible Pancreatic Cancer Cells. Cancer Res. 2015, 75, 1883–1896. [Google Scholar] [CrossRef]
- Wu, Y.W.; Prakash, K.M.; Rong, T.Y.; Li, H.H.; Xiao, Q.; Tan, L.C.; Au, W.L.; Ding, J.Q.; Chen, S.D.; Tan, E.K. Lingo2 variants associated with essential tremor and Parkinson’s disease. Hum. Genet. 2011, 129, 611–615. [Google Scholar] [CrossRef]
- Perroud, N.; Uher, R.; Ng, M.Y.; Guipponi, M.; Hauser, J.; Henigsberg, N.; Maier, W.; Mors, O.; Gennarelli, M.; Rietschel, M.; et al. Genome-wide association study of increasing suicidal ideation during antidepressant treatment in the GENDEP project. Pharm. J. 2012, 12, 68–77. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Hertz, D.L.; Roy, S.; Motsinger-Reif, A.A.; Drobish, A.; Clark, L.S.; McLeod, H.L.; Carey, L.A.; Dees, E.C. CYP2C8*3 increases risk of neuropathy in breast cancer patients treated with paclitaxel. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. ESMO 2013, 24, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, Z.; Feng, J.; Hsu, F.C.; Jin, G.; Kim, S.T.; Zhang, Z.; Gronberg, H.; Zheng, L.S.; Isaacs, W.B.; et al. A genome-wide search for loci interacting with known prostate cancer risk-associated genetic variants. Carcinogenesis 2012, 33, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.P.; Younkin, S.G.; et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Chakarova, C.; Murphy, C.; Becker, M.; Lenassi, E.; Arno, G.; Lek, M.; MacArthur, D.G.; Consortium, U.C.-E.; Bhattacharya, S.S.; et al. Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am. J. Hum. Genet. 2014, 94, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Q.; Popova, S.N.; Brown, E.R.; Barsyte-Lovejoy, D.; Navab, R.; Shih, W.; Li, M.; Lu, M.; Jurisica, I.; Penn, L.Z.; et al. Integrin alpha 11 regulates IGF2 expression in fibroblasts to enhance tumorigenicity of human non-small-cell lung cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11754–11759. [Google Scholar] [CrossRef]
- Abitbol, M.; Thibaud, J.L.; Olby, N.J.; Hitte, C.; Puech, J.P.; Maurer, M.; Pilot-Storck, F.; Hedan, B.; Dreano, S.; Brahimi, S.; et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14775–14780. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.W.; Go, M.J.; Jin, H.S.; Lim, J.E.; Lee, J.Y.; Han, B.G.; Hwang, S.Y.; Lee, S.H.; Park, H.K.; Cho, Y.S.; et al. Genetic variations in ATP2B1, CSK, ARSG and CSMD1 loci are related to blood pressure and/or hypertension in two Korean cohorts. J. Hum. Hypertens. 2010, 24, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Mosca, S.J.; Tetreault, M.; Dempsey, J.C.; Ishak, G.E.; Hartley, T.; Phelps, I.G.; Lamont, R.E.; O’Day, D.R.; Basel, D.; et al. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am. J. Hum. Genet. 2014, 95, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, J.A.; Ng, M.C.; Palmer, N.D.; Keaton, J.M.; Larsen, C.P.; Hicks, P.J.; Consortium, T.D.G.; Langefeld, C.D.; Freedman, B.I.; Bowden, D.W. Coding variants in nephrin (NPHS1) and susceptibility to nephropathy in African Americans. Clin. J. Am. Soc. Nephrol. CJASN 2014, 9, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Liu, Y.; White, C.C.; Feitosa, M.; Smith, A.V.; Heard-Costa, N.; Lohman, K.; Consortium, G.; Consortium, M.; Consortium, G.; et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012, 8, e1002695. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rizzato, C.; Kraft, P.; Kooperberg, C.; Petersen, G.M.; Wang, Z.; Arslan, A.A.; Beane-Freeman, L.; Bracci, P.M.; Buring, J.; et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat. Genet. 2014, 46, 994–1000. [Google Scholar] [CrossRef]
- He, Y.D.; Luo, Z.H.; Yang, M.; Ruan, X.X.; Liu, S.Y.; Wu, Z.Q.; Deng, W.M.; Shao, C.K.; Su, Z.L.; He, D.; et al. Prospective validation of DACH2 as a novel biomarker for prediction of metastasis and prognosis in muscle-invasive urothelial carcinoma of the bladder. Biochem. Biophys. Res. Commun. 2015, 459, 416–423. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Zhang, X.; Li, J.; Wei, X.; Cheng, J.; Ling, Q.; Xie, H.; Zhou, L.; Xu, X.; et al. Diagnostic Value of Preoperative Needle Biopsy for Tumor Grading Assessment in Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0144216. [Google Scholar] [CrossRef]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Chaturvedi, V.K.; Singh, A.; Dubey, S.K.; Hetta, H.F.; John, J.; Singh, M.P. Molecular mechanistic insight of hepatitis B virus mediated hepatocellular carcinoma. Microb. Pathog. 2019, 128, 184–194. [Google Scholar] [CrossRef]
- Furuta, M.; Tanaka, H.; Shiraishi, Y.; Unida, T.; Imamura, M.; Fujimoto, A.; Fujita, M.; Sasaki-Oku, A.; Maejima, K.; Nakano, K.; et al. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 2018, 9, 25075–25088. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef]
- Chang, J.W.; Kuo, W.H.; Lin, C.M.; Chen, W.L.; Chan, S.H.; Chiu, M.F.; Chang, I.S.; Jiang, S.S.; Tsai, F.Y.; Chen, C.H.; et al. Wild-type p53 upregulates an early onset breast cancer-associated gene GAS7 to suppress metastasis via GAS7-CYFIP1-mediated signaling pathway. Oncogene 2018, 37, 4137–4150. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yin, C.; Qi, J.; Duan, D.; Jiang, X.; Yu, J.; Luo, Z. miR-362-5p promotes cell proliferation and cell cycle progression by targeting GAS7 in acute myeloid leukemia. Hum. Cell 2020, 33, 405–415. [Google Scholar] [CrossRef]
- D’Agostino, L.; Giordano, A. NSP 5a3a: A potential novel cancer target in head and neck carcinoma. Oncotarget 2010, 1, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cao, J.; Zhang, N.; Tu, M.; Xu, F.; Wei, S.; Chen, X.; Xu, Y. Identification of RSPO2 Fusion Mutations and Target Therapy Using a Porcupine Inhibitor. Sci. Rep. 2018, 8, 14244. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Sakamoto, T.; Hashimoto, T.; Tsukamoto, S.; Iwasa, S.; Saito, Y.; Sekine, S. Identification of a novel PRR15L-RSPO2 fusion transcript in a sigmoid colon cancer derived from superficially serrated adenoma. Virchows. Arch. 2019, 475, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lee, D.; Hong, D.W.; Hong, S.H.; Hwang, J.A.; Lee, B.I.; You, H.J.; Lee, G.K.; Kim, I.H.; Lee, Y.S.; et al. Oncogenic function and clinical implications of SLC3A2-NRG1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget 2016, 7, 69450–69465. [Google Scholar] [CrossRef]
- Corrigan, D.J.; Luchsinger, L.L.; Justino de Almeida, M.; Williams, L.J.; Strikoudis, A.; Snoeck, H.W. PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J. Clin. Investig. 2018, 128, 3250–3264. [Google Scholar] [CrossRef]
- Han, X.; Chen, W.; Chen, P.; Zhou, W.; Rong, Y.; Lv, Y.; Li, J.A.; Ji, Y.; Chen, W.; Lou, W.; et al. Aberration of ARID1A Is Associated with the Tumorigenesis and Prognosis of Sporadic Nonfunctional Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, 514–523. [Google Scholar] [CrossRef]
- Domer, P.H.; Fakharzadeh, S.S.; Chen, C.S.; Jockel, J.; Johansen, L.; Silverman, G.A.; Kersey, J.H.; Korsmeyer, S.J. Acute mixed-lineage leukemia t(4;11)(q21;q23) generates an MLL-AF4 fusion product. Proc. Natl. Acad. Sci. USA 1993, 90, 7884–7888. [Google Scholar] [CrossRef]
- Li, D.; Zhang, B.; Hu, C. Oxaliplatin inhibits proliferation and migration of human hepatocellular carcinoma cells via GAS7C and the N-WASP/FAK/F-actin pathway. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.M.; Li, L.X.; Bian, X.Y.; Shi, X.J.; Lu, L.L.; Zhou, H.X.; Pan, T.J.; Zhou, J.; Fan, J.; Wu, W.Z. miR-296-5p suppresses EMT of hepatocellular carcinoma via attenuating NRG1/ERBB2/ERBB3 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 294. [Google Scholar] [CrossRef]
- Li, D.; Collier, D.A.; He, L. Meta-analysis shows strong positive association of the neuregulin 1 (NRG1) gene with schizophrenia. Hum. Mol. Genet. 2006, 15, 1995–2002. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, N.; Cai, Y.; Lei, F.; Weitz, D.A. Single-cell sequencing leads a new era of profiling transcriptomic landscape. J. Bio-X Res. 2018, 1, 2–6. [Google Scholar] [CrossRef]
- Li, G.; Yang, Y.; Van Buren, E.; Li, Y. Dropout imputation and batch effect correction for single-cell RNA sequencing data. J. Bio-X Res. 2019, 2, 169–177. [Google Scholar] [CrossRef]
- Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Yin, S.J.; Chou, C.F.; Lai, C.L.; Lee, S.L.; Han, C.L. Human class IV alcohol dehydrogenase: Kinetic mechanism, functional roles and medical relevance. Chem. Biol. Interact 2003, 143–144, 219–227. [Google Scholar] [CrossRef]
- Edenberg, H.J.; Xuei, X.; Chen, H.J.; Tian, H.; Wetherill, L.F.; Dick, D.M.; Almasy, L.; Bierut, L.; Bucholz, K.K.; Goate, A.; et al. Association of alcohol dehydrogenase genes with alcohol dependence: A comprehensive analysis. Hum. Mol. Genet. 2006, 15, 1539–1549. [Google Scholar] [CrossRef]
- Guindalini, C.; Scivoletto, S.; Ferreira, R.G.; Breen, G.; Zilberman, M.; Peluso, M.A.; Zatz, M. Association of genetic variants in alcohol dehydrogenase 4 with alcohol dependence in Brazilian patients. Am. J. Psychiatry 2005, 162, 1005–1007. [Google Scholar] [CrossRef]
- Li, D.; Zhao, H.; Gelernter, J. Strong association of the alcohol dehydrogenase 1B gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol. Psychiatry 2011, 70, 504–512. [Google Scholar] [CrossRef]
- Li, D.; Zhao, H.; Gelernter, J. Strong protective effect of the aldehyde dehydrogenase gene (ALDH2) 504lys (*2) allele against alcoholism and alcohol-induced medical diseases in Asians. Hum. Genet. 2012, 131, 725–737. [Google Scholar] [CrossRef]
- Matsushita, H.; Takaki, A. Alcohol and hepatocellular carcinoma. BMJ Open Gastroenterol. 2019, 6, e000260. [Google Scholar] [CrossRef]
- Guan, X.; Xing, F.; Li, Y. Alcohol consumption increases the incidence of hepatocellular carcinoma in patients with hepatitis B cirrhosis but not in patients with hepatitis C cirrhosis. Eur. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Ramadori, P.; Cubero, F.J.; Liedtke, C.; Trautwein, C.; Nevzorova, Y.A. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers (Basel) 2017, 9, 130. [Google Scholar] [CrossRef]
- Gnani, D.; Romito, I.; Artuso, S.; Chierici, M.; De Stefanis, C.; Panera, N.; Crudele, A.; Ceccarelli, S.; Carcarino, E.; D’Oria, V.; et al. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death Differ. 2017, 24, 889–902. [Google Scholar] [CrossRef]
- Panera, N.; Crudele, A.; Romito, I.; Gnani, D.; Alisi, A. Focal Adhesion Kinase: Insight into Molecular Roles and Functions in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2017, 18, 99. [Google Scholar] [CrossRef]
- Shang, N.; Wang, H.; Bank, T.; Perera, A.; Joyce, C.; Kuffel, G.; Zilliox, M.J.; Cotler, S.J.; Ding, X.; Dhanarajan, A.; et al. Focal Adhesion Kinase and beta-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 2019, 70, 1631–1645. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Viruses in human cancers. Eur. J. Cancer 1999, 35, 1878–1885. [Google Scholar] [CrossRef]
- Butt, A.Q.; Miggin, S.M. Cancer and viruses: A double-edged sword. Proteomics 2012, 12, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Freeberg, M.A.; Winans, S.J.; Taylor, J.; Beemon, K.L. A Novel Long Non-Coding RNA in the hTERT Promoter Region Regulates hTERT Expression. Noncoding RNA 2017, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef]
- Zhang, H.; Weng, X.; Ye, J.; He, L.; Zhou, D.; Liu, Y. Promoter hypermethylation of TERT is associated with hepatocellular carcinoma in the Han Chinese population. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Kao, J.H. Hepatitis B virus genotypes and variants. Cold Spring Harb. Perspect. Med. 2015, 5, a021436. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).