
A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest

Abstract
:1. Introduction
2. Materials and Methods
2.1. Using Amino Acid Mutations in ACE2 RBD and NTD for Variant Determination
2.2. Patient Samples Used for Method Development
2.3. RNA Extraction from Nasopharyngeal Swab Specimens
2.4. PCR Primers
2.5. PCR Conditions
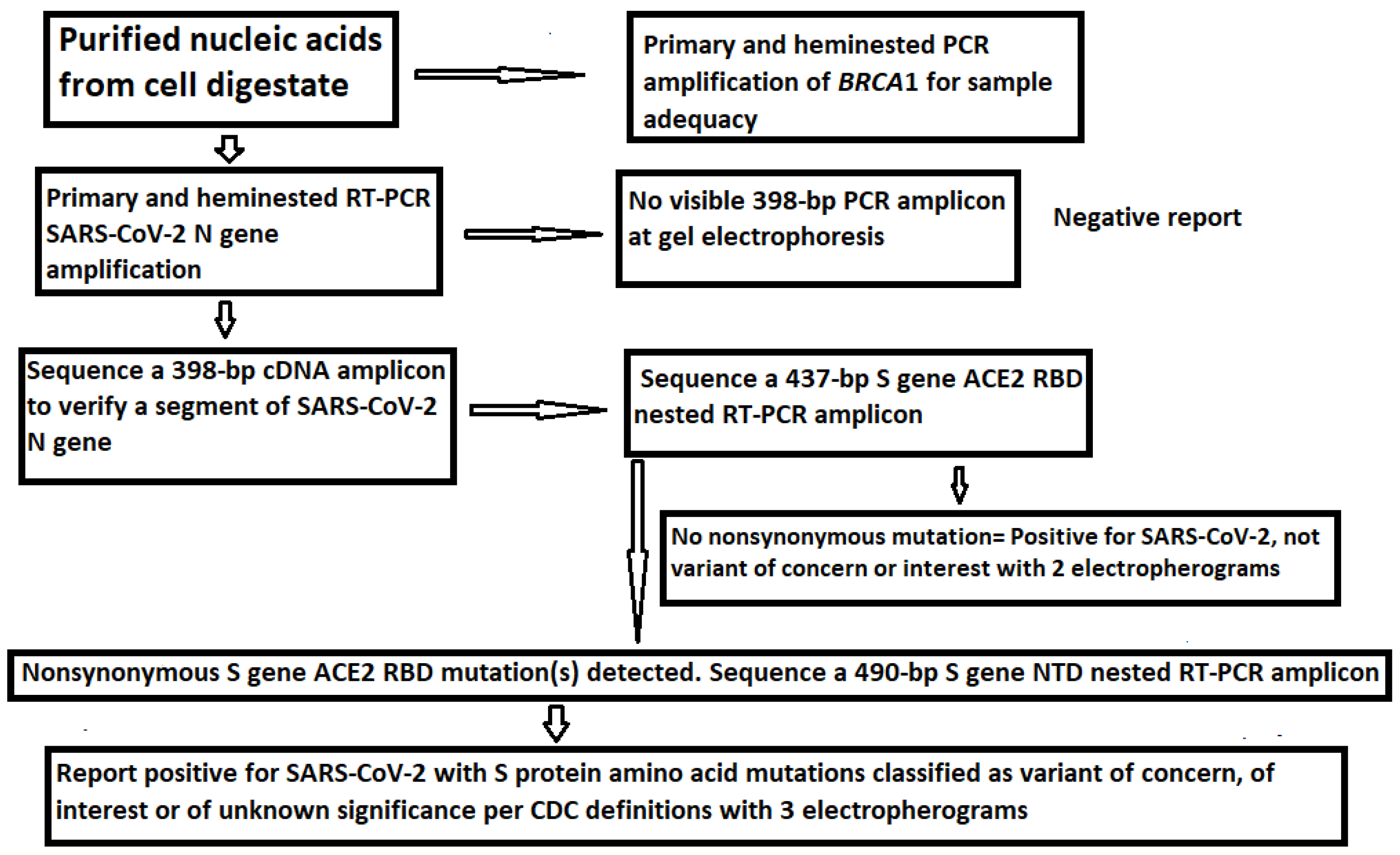
2.6. DNA Sequencing
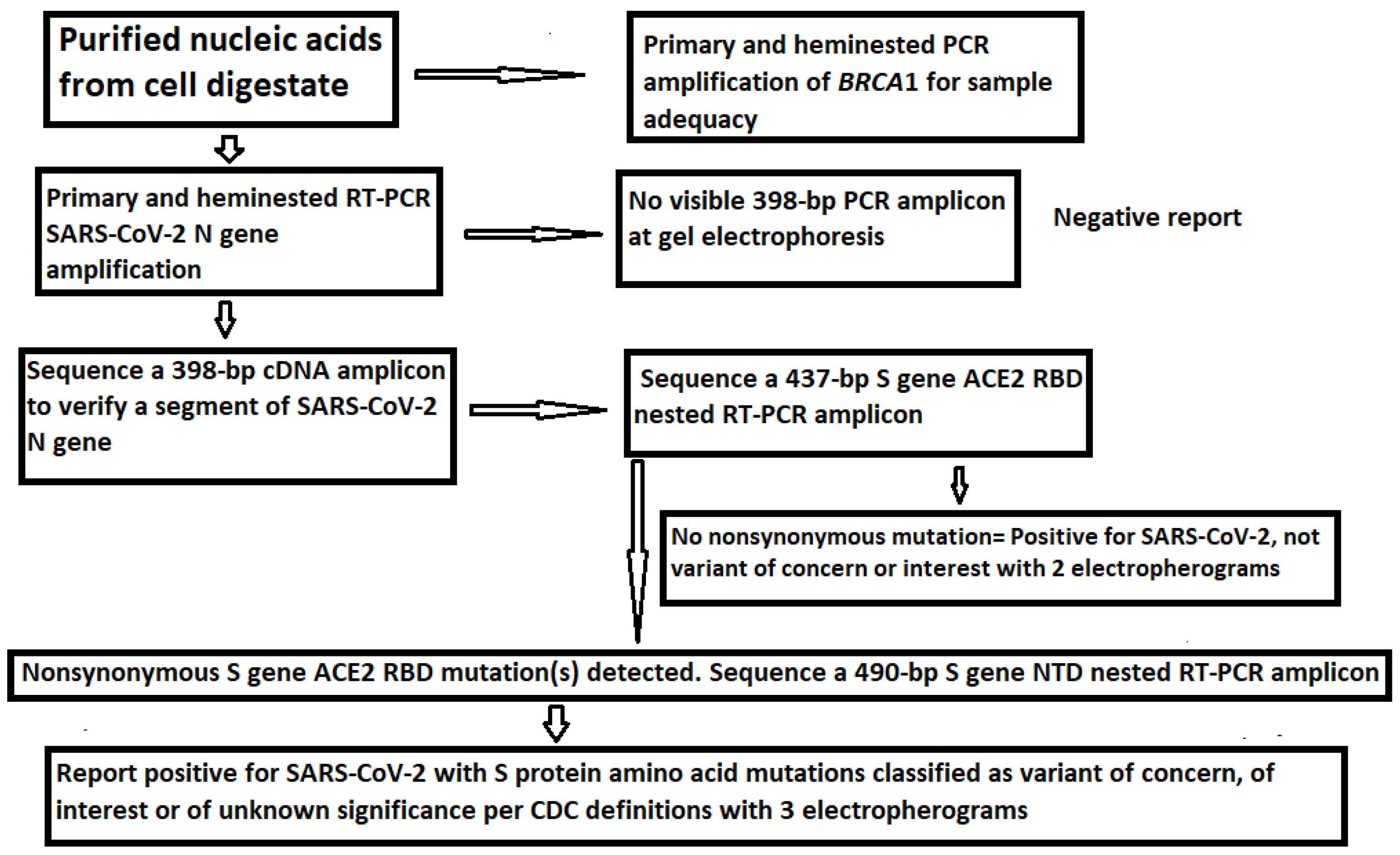
3. Results
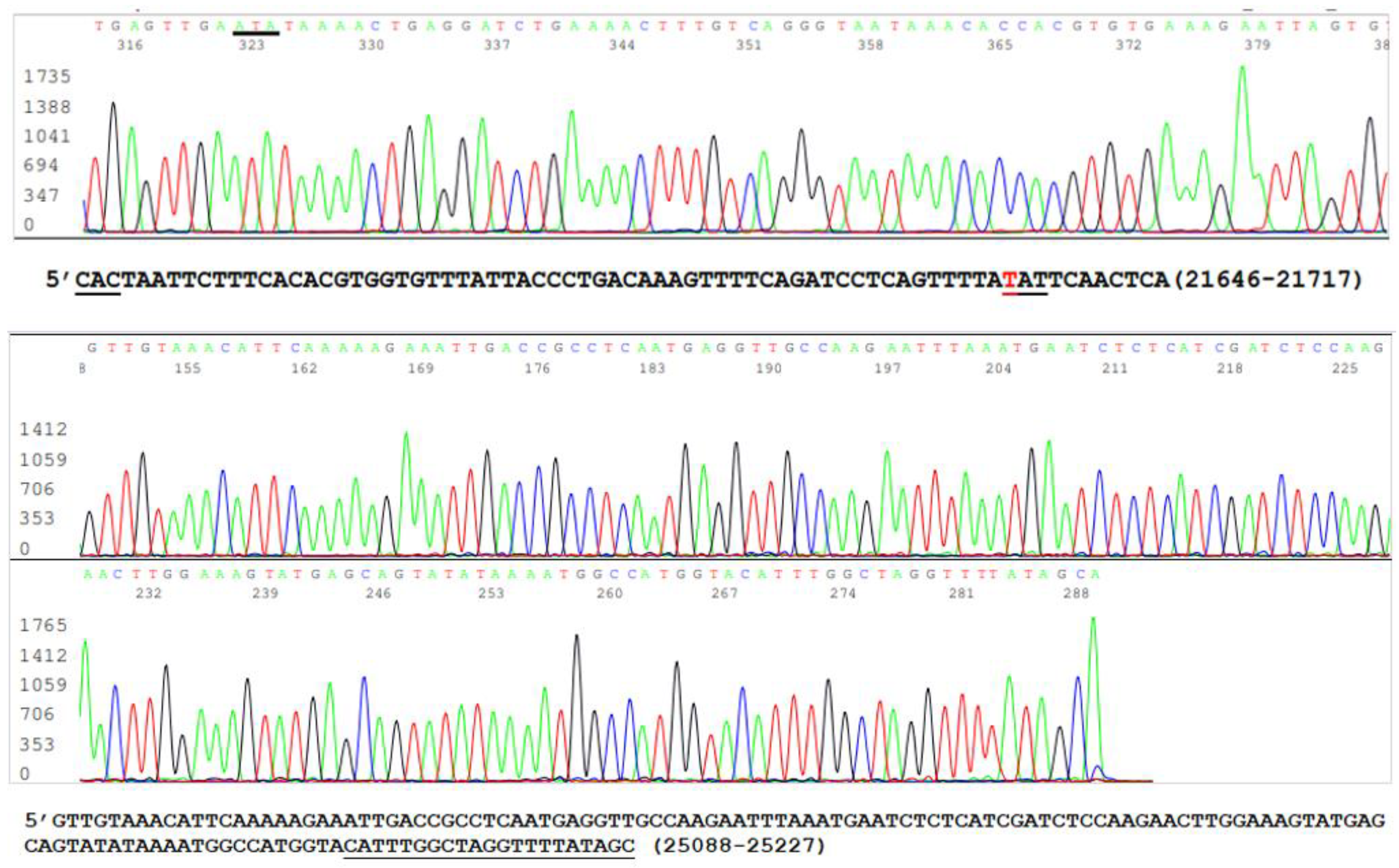
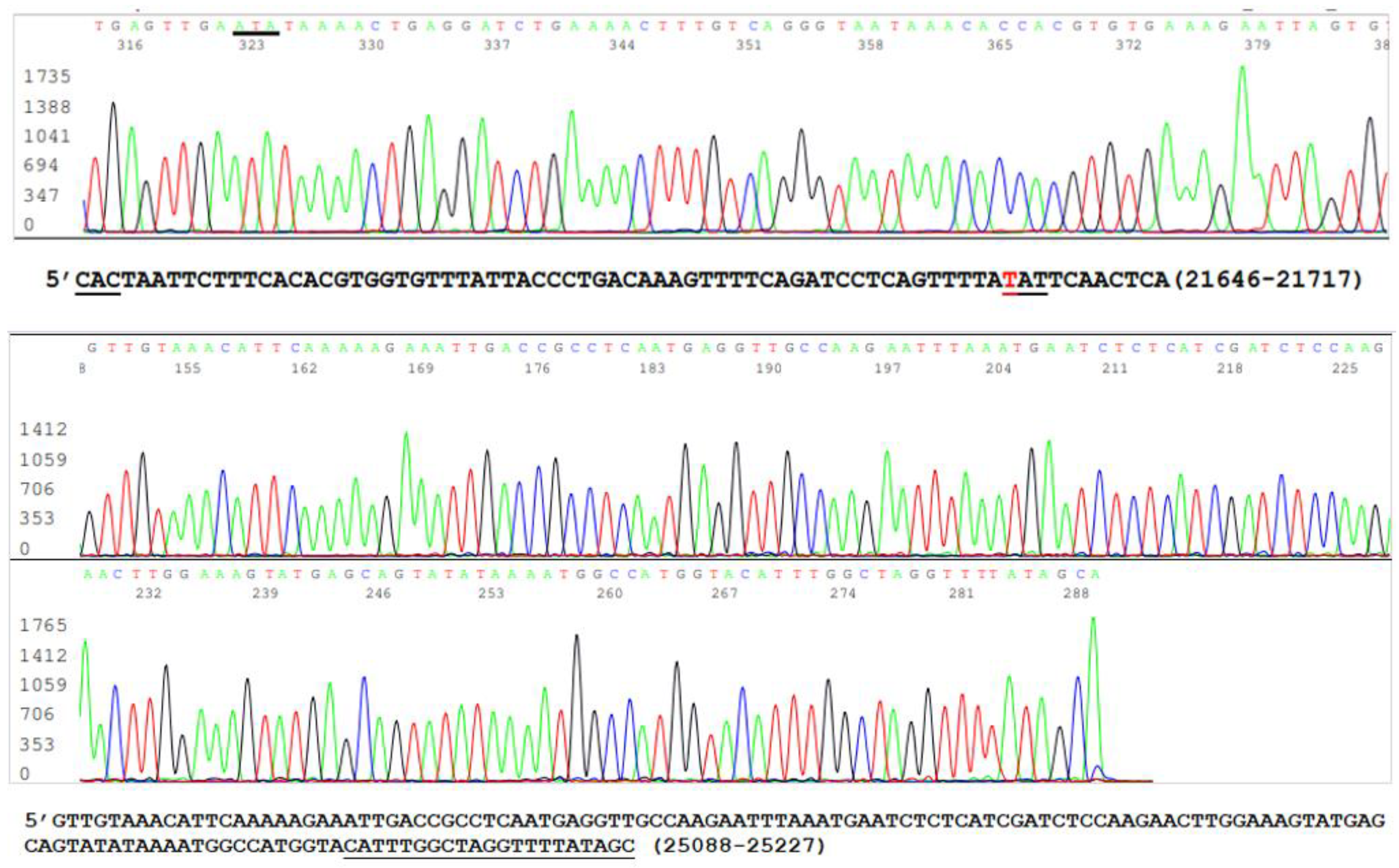
3.1. The Samples Positive for N Gene Also Contained an Intact S Gene
3.2. Limitations of the Size of Diagnostic RT-PCR Amplicon
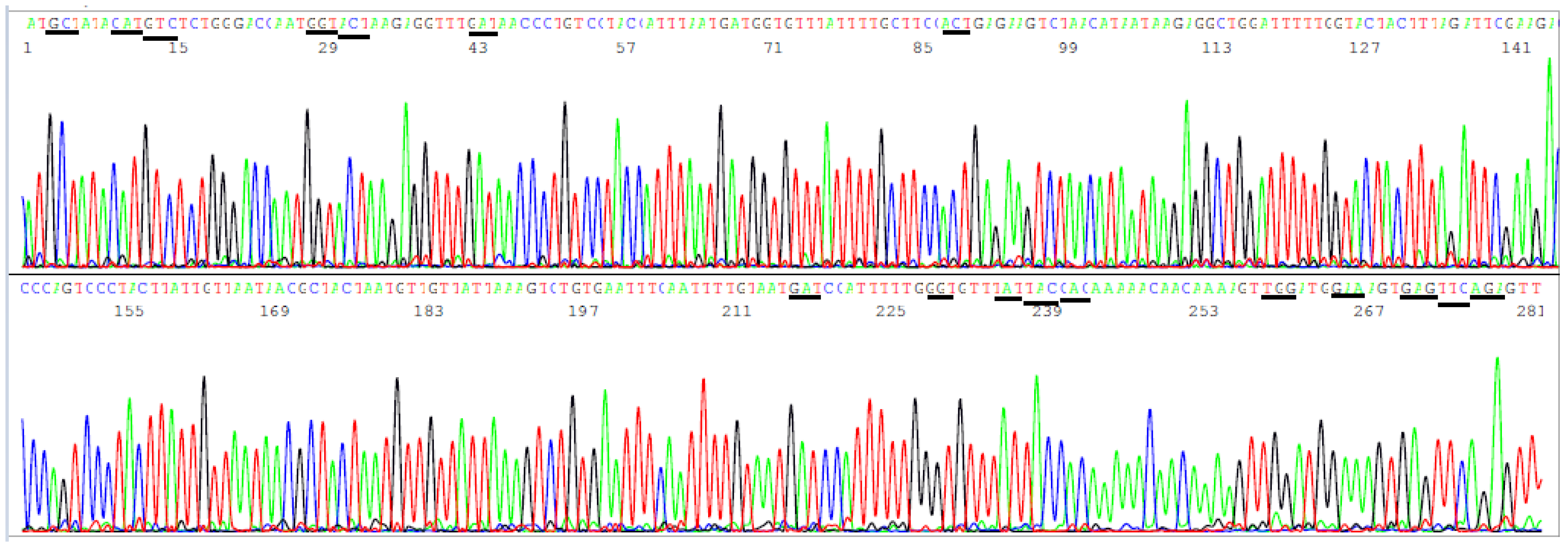
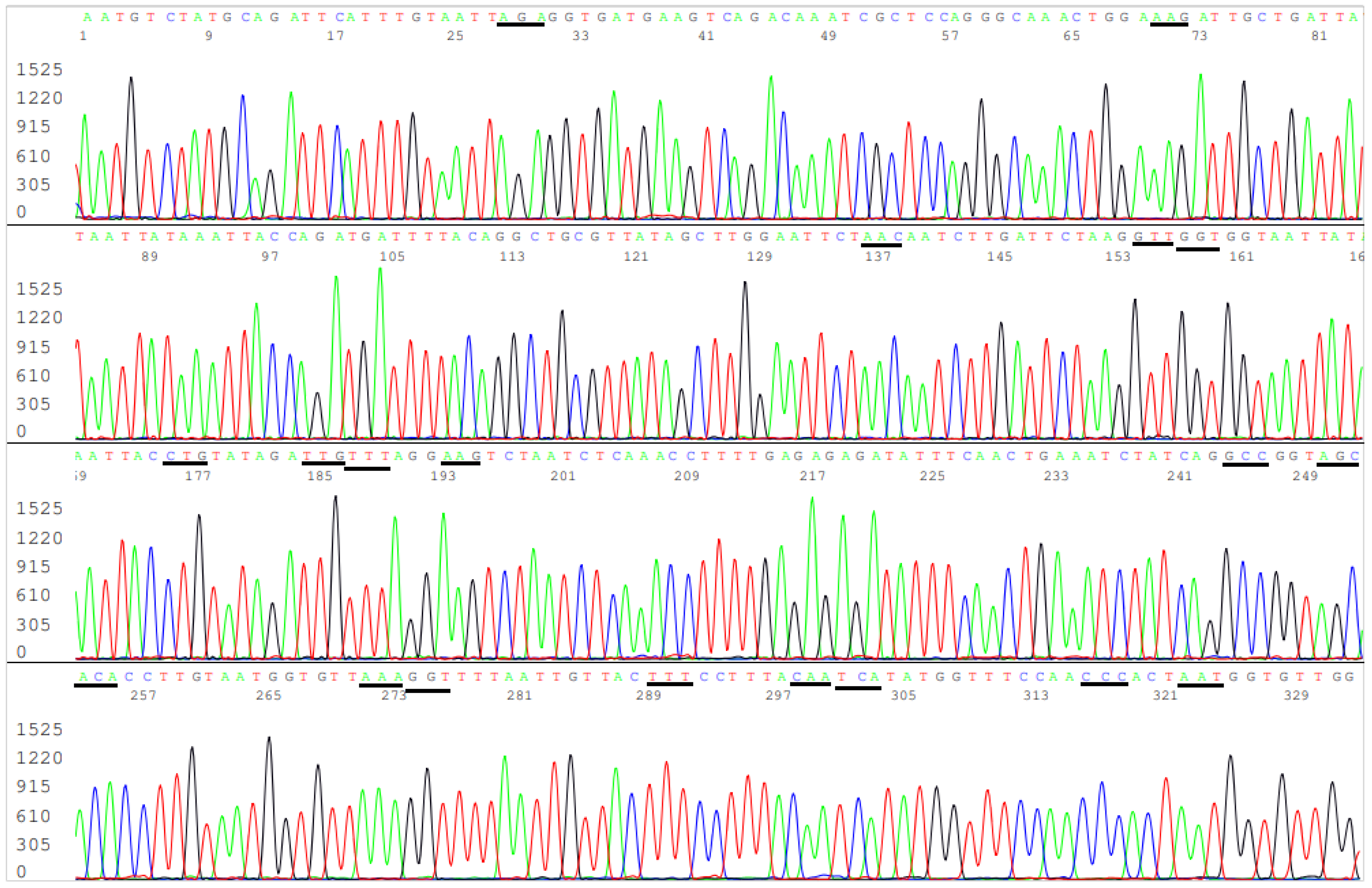
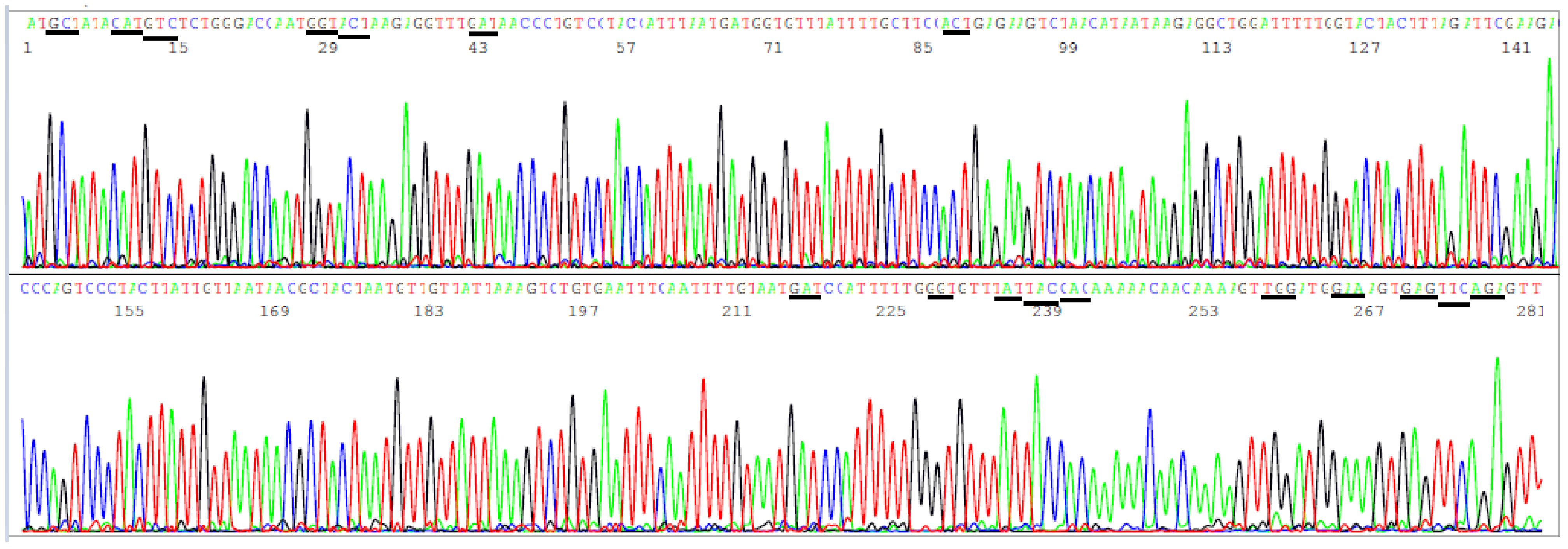
3.3. Target Amplicon Sequencing of the ACE2 RBD Region
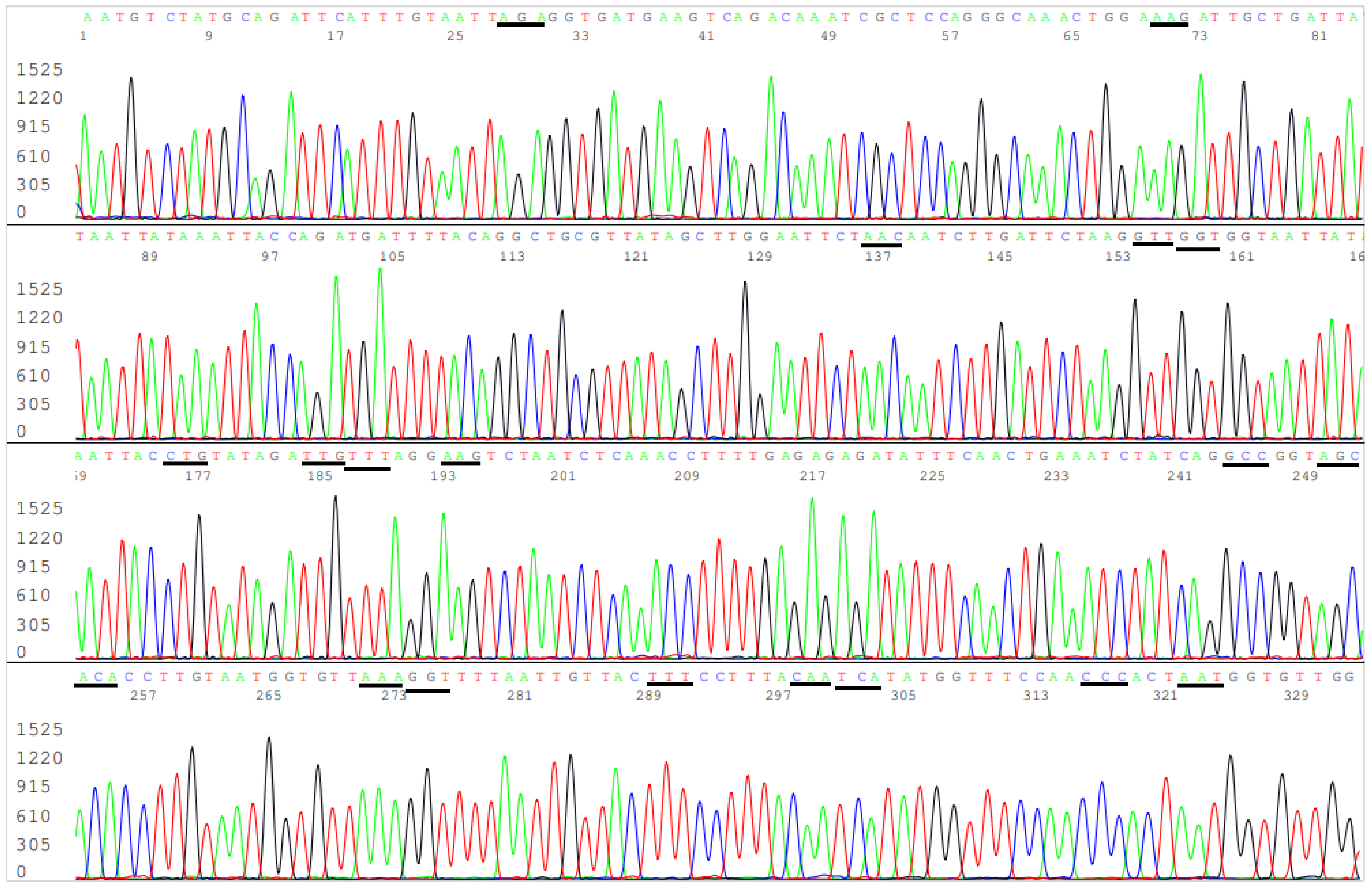
3.4. Target Amplicon Sequencing of the NTD Region
3.5. Determination of Variants by Sequencing of the ACE2 RBD and NTD
4. Discussion
4.1. Accurate Categorization of VOCs and VOIs on All Samples Positive for SARS-CoV-2
4.2. The Current Confusion of Delta Variant Testing
4.3. Accumulation of Mutations in Viruses Is a Function of Passages
- L452R and T478K without concomitant mutations in the NTD (Sequence ID: MZ637393).
- E156del, F157del, R158G without concomitant mutations in the ACE2 RBD (Sequence ID: MZ340544).
- G142D, E156del, F157del, R158G without concomitant mutations in the ACE2 RBD (Sequence ID: MZ341068).
- T95I, G142D, E156del, F157del, R158G, E484K (Sequence ID: MZ531409).
- T95I, L452R (Sequence ID: MZ086521).
4.4. The Delta Variant Scare Is Not Supported by Facts
4.5. Routine Sequencing on All Positive Samples for Variant Determination
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronavirus Updates. Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 7 September 2021).
- John, G.; Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Williams, C.; Chaubey, A.; Rojiani, A.M.; Kolhe, R. Next-Generation Sequencing (NGS) in COVID-19: A Tool for SARS-CoV-2 Diagnosis, Monitoring New Strains and Phylodynamic Modeling in Molecular Epidemiology. Curr. Issues Mol. Biol. 2021, 43, 845–867. [Google Scholar] [CrossRef] [PubMed]
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 15 August 2021).
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 25 August 2021).
- Lee, Y.N.; CNBC. 3 Charts Show How Far Covid Delta Variant Has Spread around the World. PUBLISHED THU, 5 August. 2021. Available online: https://www.cnbc.com/2021/08/06/covid-charts-show-how-far-delta-variant-has-spread-around-the-world.html (accessed on 7 September 2021).
- WHO. Episode #45-Delta Variant. 5 July 2021. Science Conversation. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/media-resources/science-in-5/episode-45—delta-variant (accessed on 7 September 2021).
- World Health Organization. Regional Office for Europe. Methods for the Detection and Identification of SARS-CoV-2 Variants. March 2021. Available online: https://apps.who.int/iris/handle/10665/340067 (accessed on 7 September 2021).
- Australian Government Department of Health. Coronavirus Disease 2019 (COVID-19) CDNA National Guidelines for Public Health Units. 24 June 2021. Available online: https://www1.health.gov.au/internet/main/publishing.nsf/Content/7A8654A8CB144F5FCA2584F8001F91E2/$File/COVID-19-SoNG-v4.7.pdf (accessed on 20 August 2021).
- Buchan, B.W.; Wolk, D.M.; Yao, J.D. Rapid Communication. SARS-CoV-2 Variant Testing. Ver.1, Released 4.28.2021. The Association for Molecular Pathology (AMP), Infectious Diseases Society of America (IDSA), and Pan American Society for Clinical Virology (PASCV). Available online: https://www.amp.org/AMP/assets/File/clinical-practice/COVID/AMP_RC_VariantTestingforSARSCOV2_4_28_21.pdf?pass=91 (accessed on 7 September 2021).
- Tartof, S.Y.; Slezak, J.M.; Fischer, H.; Hong, V.; Ackerson, B.K.; Ranasinghe, O.N.; Frankland, T.B.; Ogun, O.A.; Zamparo, J.M.; Gray, S.; et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: A retrospective cohort study. Lancet 2021, 398, 1407–1416. [Google Scholar] [CrossRef]
- Abnizova, I.; Te Boekhorst, R.; Orlov, Y. Computational Errors and Biases in Short Read Next Generation Sequencing. J. Proteomics Bioinform. 2017, 10, 1–17. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19): Weekly Epidemiological Update (11 May 2021). Available online: https://reliefweb.int/report/world/coronavirus-disease-covid-19-weekly-epidemiological-update-11-may-2021 (accessed on 25 August 2021).
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Technical Briefing 15. 11 June 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993879/Variants_of_Concern_VOC_Technical_Briefing_15.pdf (accessed on 25 August 2021).
- FDA. Clinical Laboratory Improvement Amendments (CLIA). Available online: https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clinical-laboratory-improvement-amendments-clia (accessed on 7 September 2021).
- Liu, Y.; Arase, N.; Kishikawa, J.; Hirose, M.; Li, S.; Tada, A.; Matsuoka, S.; Arakawa, A.; Akamatsu, K.; Ono, C.; et al. The SARS-CoV-2 Delta Variant Is Poised to Acquire Complete Resistance to Wild-Type Spike Vaccines. BioRxiv 2021. [Google Scholar] [CrossRef]
- Li, D.; Edwards, R.J.; Manne, K.; Martinez, D.R.; Schafer, A.; Alam, S.M.; Wiehe, K.; Lu, X.; Parks, R.; Sutherland, L.L.; et al. In vitro and in vivo functions of SARS-CoV-2 infection enhancing and neutralizing antibodies. Cell 2021, 184, 4203–4219.e32. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e4213. [Google Scholar] [CrossRef]
- FDA to CDC. Letter Dated 1 December 2020. Available online: https://www.fda.gov/media/134919/download (accessed on 7 September 2021).
- Lee, S.H. qPCR is not PCR Just as a Straightjacket is not a Jacket-the Truth Revealed by SARS-CoV-2 False- Positive Test Results. COVID-19 Pandemic Case Stud. Opin. 2021, 2, 230–278. [Google Scholar]
- FDA. In Vitro Diagnostics EUAs. Molecular Diagnostic Template for Laboratories. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizationsmedical-devices/vitro-diagnostics-euas (accessed on 5 August 2021).
- Lee, S.H. Testing for SARS-CoV-2 in cellular components by routine nested RT-PCR followed by DNA sequencing. Int. J. Geriatr. Rehabil. 2020, 2, 69–96. [Google Scholar]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266–1273. [Google Scholar] [CrossRef]
- Lou, D.I.; McBee, R.M.; Le, U.Q.; Stone, A.C.; Wilkerson, G.K.; Demogines, A.M.; Sawyer, S.L. Rapid evolution of BRCA1 and BRCA2 in humans and other primates. BMC Evol. Biol. 2014, 14, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarridge, J.E., 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Koblížek, M.; Li, Y.; Liu, Y.; Feng, F.; Ji, J.; Jian, J.; Wu, Z. Long PCR-RFLP of 16S-ITS-23S rRNA genes: A high-resolution molecular tool for bacterial genotyping. J. Appl. Microbiol. 2013, 114, 433–447. [Google Scholar] [CrossRef] [PubMed]
- China National Center for Bioinformation. 2019 Novel Coronavirus Resource (2019nCoVR). Available online: https://ngdc.cncb.ac.cn/ncov// (accessed on 23 August 2021).
- Scribner, H. You’re Not Allowed to Know If You Have the Delta Variant. Deseret News. 24 August 2021. Available online: https://www.deseret.com/coronavirus/2021/8/24/22637867/delta-variant-covid-test-results-how-to-know (accessed on 7 September 2021).
- Fichera, A. Factcheck. Sequencing Used to Identify Delta, Other Coronavirus Variants. 4 August 2021. Available online: https://www.factcheck.org/2021/08/scicheck-sequencing-used-to-identify-delta-other-coronavirus-variants/ (accessed on 7 September 2021).
- Martichoux, A.; Wire, N.M. How Do You Know If You Have the Delta Variant of COVID-19? 8 August 2021. Available online: https://www.news10.com/news/how-do-you-know-if-you-have-the-delta-variant-of-covid-19/ (accessed on 7 September 2021).
- Chow, D.; NBC News. Where’s the Data on Delta? Lack of Testing, Info Makes It Hard to See Virus’s Full Scope. 19 August 2021. Available online: https://www.nbcnews.com/science/science-news/delta-variant-response-hindered-covid-test-limitations-lack-data-rcna1692 (accessed on 7 September 2021).
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018, 8, 10950. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.-L.; Wang, Y.-M.; Wu, Z.-Q.; Xiang, Z.-C.; Guo, L.; Xu, T.; Jiang, Y.-Z.; Xiong, Y.; Li, Y.-J.; Li, X.-W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- McCarty, S.C.; Atlas, R.M. Effect of amplicon size on PCR detection of bacteria exposed to chlorine. PCR Methods Appl. 1993, 3, 181–185. [Google Scholar] [CrossRef]
- Drosten, C.; Preiser, W.; Günther, S.; Schmitz, H.; Doerr, H.W. Severe acute respiratory syndrome: Identification of the etiological agent. Trends Mol. Med. 2003, 9, 325–327. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- CDC. SARS-CoV Specific RT-PCR Primers. Available online: https://www.who.int/csr/sars/CDCprimers.pdf?ua=1 (accessed on 7 September 2021).
- CDC. Severe Acute Respiratory Syndrome (SARS). Available online: https://www.cdc.gov/sars/about/faq.html (accessed on 7 September 2021).
- La Rosa, G.; Brandtner, D.; Mancini, P.; Veneri, C.; Bonanno Ferraro, G.; Bonadonna, L.; Lucentini, L.; Suffredini, E. Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology. Water 2021, 13, 2503. [Google Scholar] [CrossRef]
- Kramvis, A.; Bukofzer, S.; Kew, M.C. Comparison of hepatitis B virus DNA extractions from serum by the QIAamp blood kit, GeneReleaser, and the phenol-chloroform method. J. Clin. Microbiol. 1996, 34, 2731–2733. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, A.; Mallah, S.I.; Alqahtani, M. COVID-19 viral load not associated with disease severity: Findings from a retrospective cohort study. BMC Infect. Dis. 2021, 21, 688. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- CDC. COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker/#variant-proportions (accessed on 7 September 2021).
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [Green Version]
- BEI Resources. NR-54000 SARS-Related Coronavirus 2, Isolate hCoV19/England/204820464/2020 (Viruses). Available online: https://www.beiresources.org/Catalog/animalviruses/NR-54000.aspx (accessed on 7 September 2021).
- Rotondo, J.C.; Martini, F.; Maritati, M.; Mazziotta, C.; Di Mauro, G.; Lanzillotti, C.; Barp, N.; Gallerani, A.; Tognon, M.; Contini, C. SARS-CoV-2 Infection: New Molecular, Phylogenetic, and Pathogenetic Insights. Efficacy of Current Vaccines and the Potential Risk of Variants. Viruses 2021, 13, 1687. [Google Scholar] [CrossRef] [PubMed]
- Reuters. SA Reaches Grim Milestone of 1 Million Covid-19 Cases. 27 December 2020. Available online: https://www.iol.co.za/news/south-africa/kwazulu-natal/sa-reaches-grim-milestone-of-1-million-covid-19-cases-a01906de-5442-451c-932f-84d6fc381b84 (accessed on 7 September 2021).
- O’Connell, O. Independent News. Will the Delta Variant Scare American Diners and Shoppers into Staying Home? Monday 26 July 2021. Available online: https://www.independent.co.uk/news/world/americas/covid-delat-variant-us-economy-b1890897.html (accessed on 7 September 2021).
- Fact Check-the Delta Variant Death Toll Is Not Zero in the United States, as Posts Claim. Available online: https://www.reuters.com/article/factcheck-delta-zero/fact-check-the-delta-variant-death-toll-is-not-zero-in-the-united-states-as-posts-claim-idUSL1N2OO2I3 (accessed on 7 September 2021).
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Technical Briefing 18. 9 July 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1001358/Variants_of_Concern_VOC_Technical_Briefing_18.pdf (accessed on 7 September 2021).
- Zhang, Z.; Yao, W.; Wang, Y.; Long, C.; Fu, X. Wuhan and Hubei COVID-19 Mortality Analysis Reveals the Critical Role of Timely Supply of Medical Resources. J. Infect. 2020, 81, 147–178. [Google Scholar] [CrossRef]
- Centers for Medicare and Medicaid Services. US Government. CLIA SARS-CoV-2 Variant Testing Frequently Asked Question. Date: 3/19/2021. Does a Facility that Performs Surveillance Testing to Identify SARS-CoV-2 Genetic Variants Need a CLIA Certificate? Available online: https://www.cms.gov/files/document/clia-sars-cov-2-variant.pdf?ACSTrackingID=USCDC_2146-DM52811&ACSTrackingLa-bel=Laboratory%20Update%3A%20CMS%20Posts%20FAQ%20for%20Reporting%20Sequencing%20Results%20for%20SARS-CoV-2%20Variants&deliveryName=USCDC_2146-DM52811 (accessed on 7 September 2021).
- CDC. Common Human Coronaviruses. Available online: https://www.cdc.gov/coronavirus/general-information.html (accessed on 7 September 2021).
- Leung, N.H.L. Transmissibility and transmission of respiratory viruses. Nat. Rev. Microbiol. 2021, 19, 528–545. [Google Scholar] [CrossRef]
- CDC. Covid Data Tracker Weekly Review. Interpretive Summary for 3 September 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/covid-data/covidview/index.html (accessed on 7 September 2021).
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. BioRxiv 2021. [Google Scholar] [CrossRef]
- Urhan, A.; Abeel, T. Emergence of novel SARS-CoV-2 variants in the Netherlands. Sci. Rep. 2021, 11, 6625. [Google Scholar] [CrossRef]
- eCDC. SARS-CoV-2 Variants of Concern as of 6 September 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 7 September 2021).
- Scheepers, C.; Everatt, J.; Amoako, D.G.; Mnguni, A.; Ismail, A.; Mahlangu, B.; Wibmer, C.K.; Wilkinson, E.; Tegally, H.; San, J.E.; et al. The Continuous Evolution of SARS-CoV-2 in South Africa: A New Lineage with Rapid Accumulation of Mutations of Concern and Global Detection. Available online: https://www.medrxiv.org/content/10.1101/2021.08.20.21262342v2 (accessed on 7 September 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Variant Labels | Constant AA Mutations | Potential Additional AA Mutations | Pango Lineage | Location of First Identification |
|---|---|---|---|---|
| Alpha | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H | (E484K *), (S494P *) | B.1.1.7 | UK |
| Beta | D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V | B.1.351 | South Africa | |
| Gamma | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I | P.1 | Japan/Brazil | |
| Delta | T19R, T95I, G142D, E156-, F157-, R158G, L452R, T478K, D614G, P681R, D950N | (V70F *), (A222V *), (W258L *) | B.1.617.2 | India |
| Delta plus | T19R, T95I, G142D, E156-, F157-, R158G, K417N, L452R, T478K, D614G, P681R, D950N | (V70F *), (A222V *), (W258L *) | B.1.617.2.1 | India |
| Epsilon | L452R, D614G | B.1.427 | California | |
| Epsilon | S13I, W152C, L452R, D614G | B.1.429 | California | |
| Eta | A67V, 69del, 70del, 144del, E484K, D614G, Q677H, F888L | B.1.525 | UK/Nigeria | |
| Iota | L5F, T95I, D253G, E484K, D614G, A701V | (D80G *), (Y144- *), (F157S *), (L452R *), (S477N *), (T859N *), (D950H *), (Q957R *) | B.1.526 | New York |
| Kappa | G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H | (T95I*) | B.1.617.1 | India |
| Kappa | T19R, G142D, L452R, E484Q, D614G, P681R, D950N | B.1.617.3 | India | |
| Lambda | G75V, T76I, Δ246-252, L452Q, F490S, D614G, T859N | C.37 | Peru |
| S Gene Segment | Oligo-Nucleotide | Sequence | Amplicon Size BP | Location of Primer |
|---|---|---|---|---|
| N-terminal domain | SB5 Primary F | 5′-AACCAGAACTCAATTACCCCC | 21619–21639 | |
| SB6 Primary R | 5′-TTTGAAATTACCCTGTTTTCC | 505 | 22103–22123 | |
| SB7 Nested F | 5′-TCAATTACCCCCTGCATACAC | 21628–21648 | ||
| SB8 Nested R | 5′-ATTACCCTGTTTTCCTTCAAG | 490 | 22097–22117 | |
| ACE2 receptor | SS1 Primary F | 5′-TGTGTTGCTGATTATTCTGTC | 22643–22663 | |
| binding domain | SS2 Primary R | 5′-AAAGTACTACTACTCTGTATG | 460 | 23082–23102 |
| SS3 Nested F | 5′-ATTCTGTCCTATATAATTCCG | 22656–22676 | ||
| SS4 Nested R | 5′-TACTCTGTATGGTTGGTAACC | 437 | 23072–23092 | |
| C-terminal domain | VF1 Primary F | 5′-AATCATTACTACAGACAACAC | 24901–24921 | |
| VF2 Primary R | 5′-CAATCAAGCCAGCTATAAAAC | 338 | 25218–25238 | |
| VF3 Nested F | 5′-AGACAACACATTTGTGTCTGG | 24913–24933 | ||
| VF4 Nested R | 5′-GCTATAAAACCTAGCCAAATG | 315 | 25207–25227 |
| WHO Name Variant | Pango Lineage | ACE2 RBD Mutations | NTD Mutations | Location of First Identification |
|---|---|---|---|---|
| Alpha | B.1.1.7 | N501Y | 69del, 70del, 144del | UK |
| Beta | B.1.351 | K417N, E484K, N501Y | D80A | South Africa |
| Gamma | P.1 | K417T, E484K, N501Y | D138Y | Japan/Brazil |
| Delta | B.1.617.2 | L452R, T478K | T95I, G142D, E156del, F157del, R158G | India |
| Delta plus | B.1.617.2.1 | K417N, L452R, T478K | T95I, G142D, E156del, F157del, R158G | India |
| Epsilon | B.1.427 | L452R | California, USA | |
| Epsilon | B.1.429 | L452R | W152C | California, USA |
| Eta | B.1.525 | E484K | A67V, 69del, 70del, 144del | UK/Nigeria |
| Iota | B.1.526 | E484K | T95I | New York, USA |
| Kappa | B.1.617.1 | L452R, E484Q | G142D, E154K | India |
| Kappa | B.1.617.3 | L452R, E484Q | G142D | India |
| Lambda | C.37 | L452Q, F490S | G75V, T76I | Peru |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H. A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest. Viruses 2021, 13, 2386. https://doi.org/10.3390/v13122386
Lee SH. A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest. Viruses. 2021; 13(12):2386. https://doi.org/10.3390/v13122386
Chicago/Turabian StyleLee, Sin Hang. 2021. "A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest" Viruses 13, no. 12: 2386. https://doi.org/10.3390/v13122386
APA StyleLee, S. H. (2021). A Routine Sanger Sequencing Target Specific Mutation Assay for SARS-CoV-2 Variants of Concern and Interest. Viruses, 13(12), 2386. https://doi.org/10.3390/v13122386