Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Compete Genome Dataset and Recombination Analysis
- Coding and non-coding regions;
- Each ORF and the remainder of the genome;
- Between the ends of the coding regions (external 25%) and the middle of these regions.
2.2. Gene Evolution Rate
2.3. Spike Dataset
2.4. Homology Modelling
3. Results
3.1. Complete Genomes
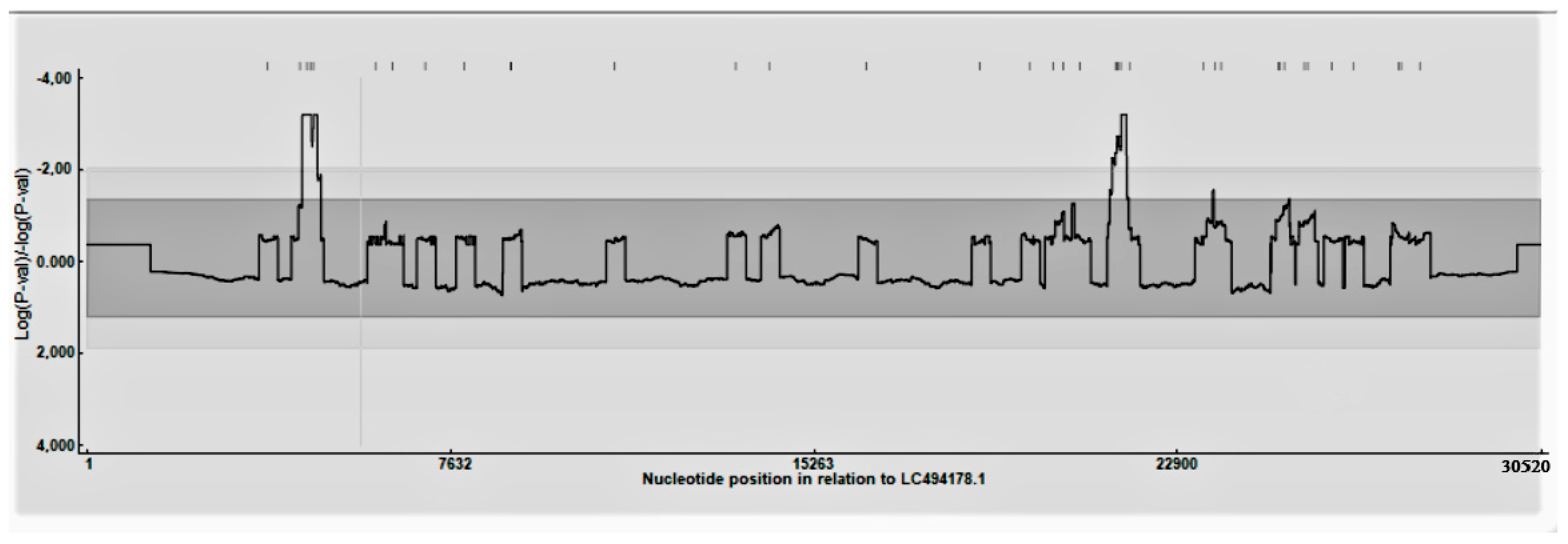
3.1.1. Recombination Rate Analysis
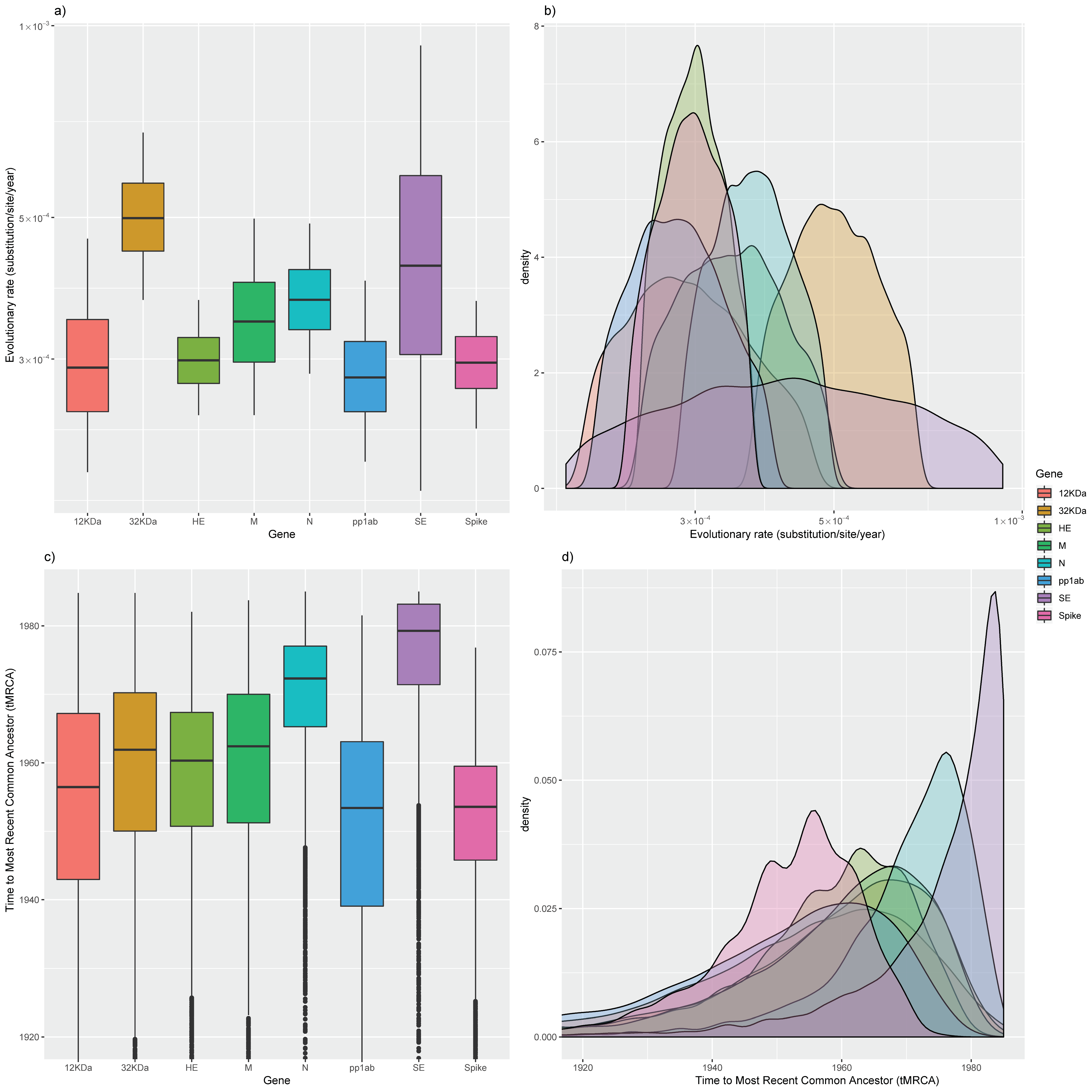
3.1.2. Gene Evolutionary Rate
3.2. Spike Protein
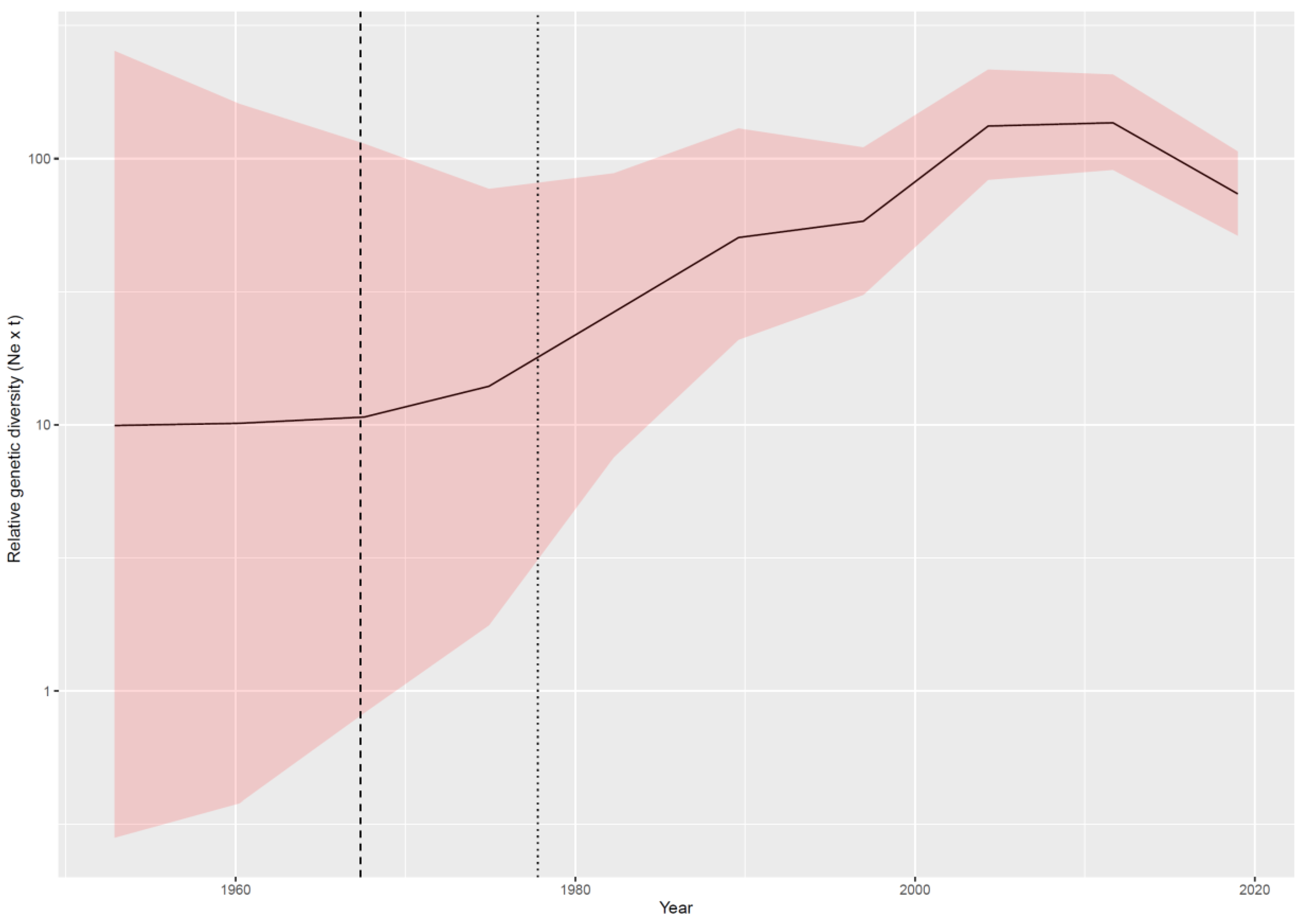
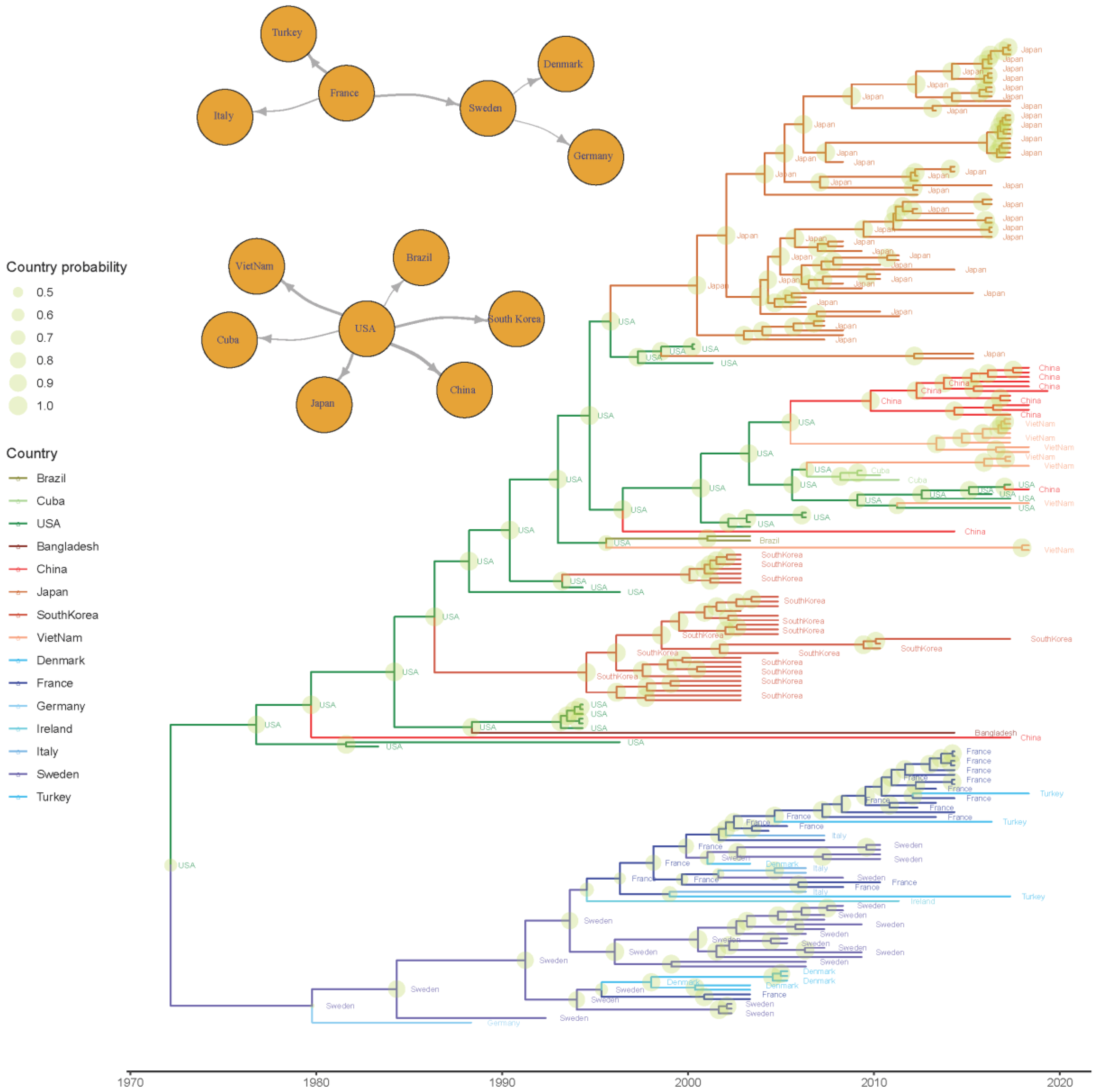
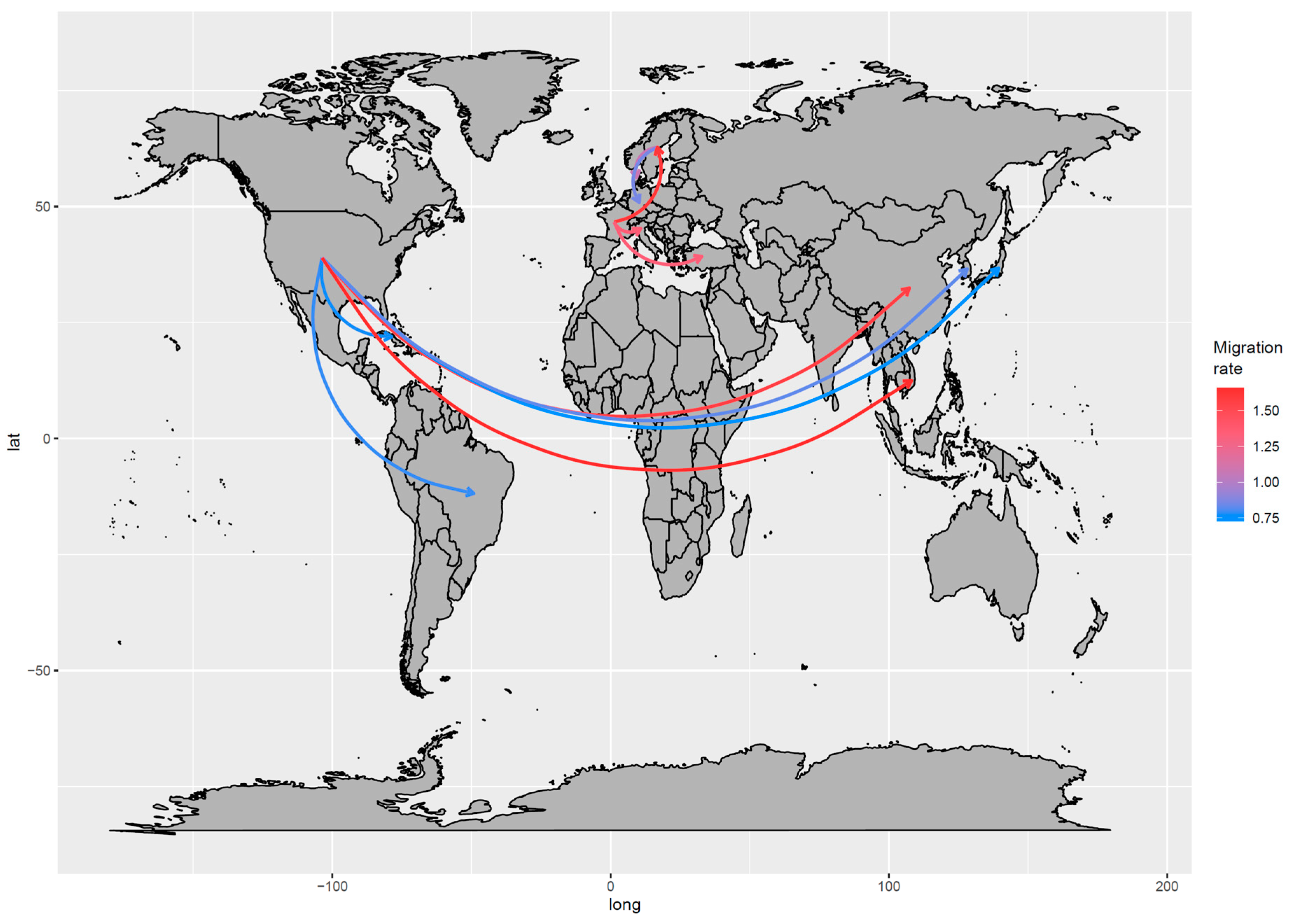
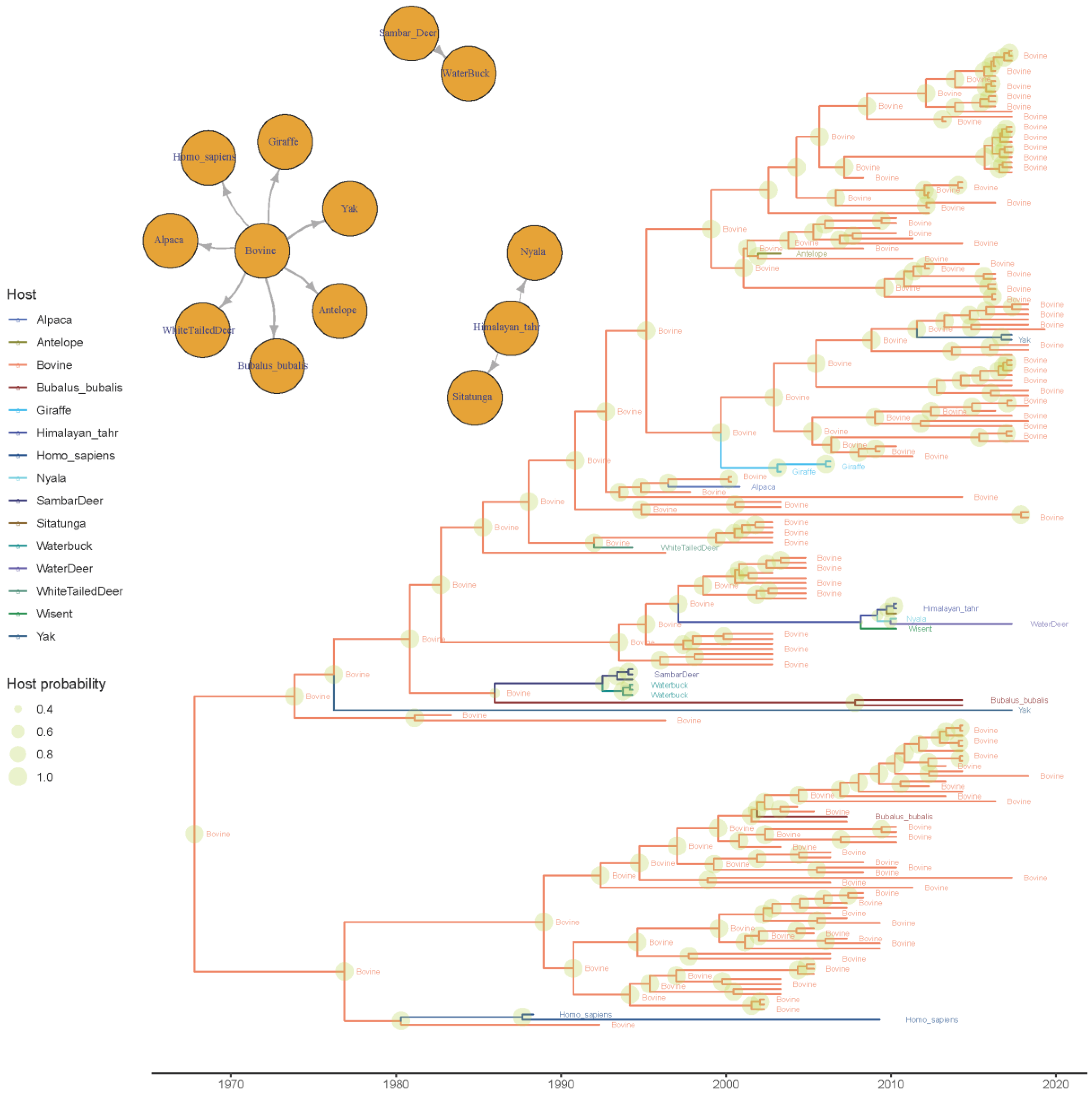
3.2.1. Phylodynamic Analysis
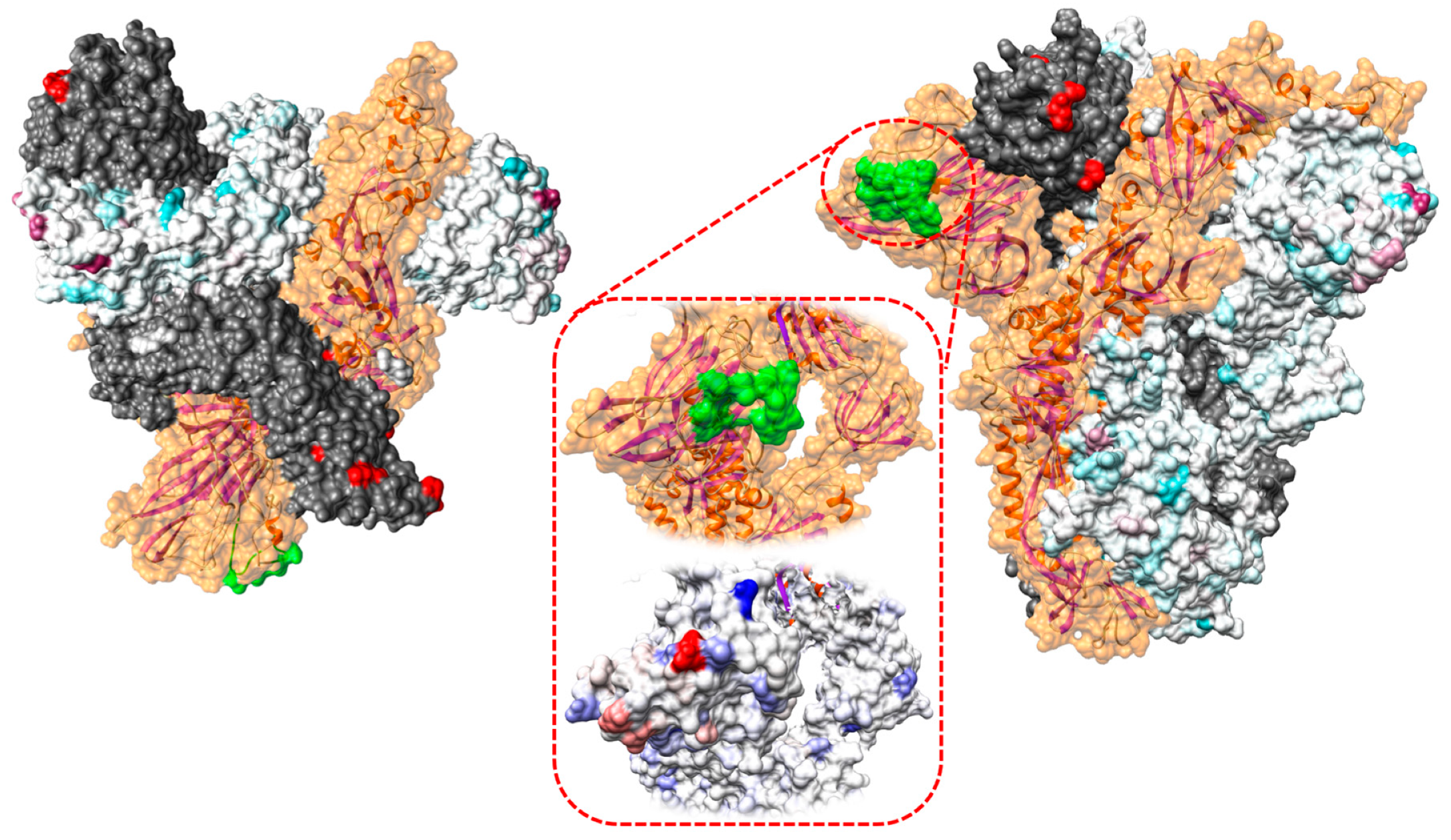
3.2.2. Selective Pressures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suzuki, T.; Otake, Y.; Uchimoto, S.; Hasebe, A.; Goto, Y. Genomic characterization and phylogenetic classification of bovine coronaviruses through whole genome sequence analysis. Viruses 2020, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. The Molecular biology of coronaviruses. Adv. Virus Res. 2006, 65, 193–292. [Google Scholar] [CrossRef]
- Li, F. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J. Virol. 2012, 86, 2856–2858. [Google Scholar] [CrossRef]
- Saif, L.J. Bovine respiratory coronavirus. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. Evolution and Emergence of RNA Viruses; Oxford University Press: Oxford, UK, 2009; ISBN 0199211124. [Google Scholar]
- Ellis, J. What is the evidence that bovine coronavirus is a biologically significant respiratory pathogen in cattle? Can. Vet. J. 2019, 60, 147–152. [Google Scholar]
- Fulton, R.W. Bovine respiratory disease research (1983–2009). Anim. Health Res. Rev. 2009, 10, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hasoksuz, M.; Lathrop, S.; Al-Dubaib, M.A.; Lewis, P.; Saif, L.J. Antigenic variation among bovine enteric coronaviruses (BECV) and bovine respiratory coronaviruses (BRCV) detected using monoclonal antibodies Brief Report. Arch. Virol. 1999, 144, 2441–2447. [Google Scholar] [CrossRef]
- Salem, E.; Dhanasekaran, V.; Cassard, H.; Hause, B.; Maman, S.; Meyer, G.; Ducatez, M.F. Global transmission, spatial segregation, and recombination determine the long-term evolution and epidemiology of bovine coronaviruses. Viruses 2020, 12, 534. [Google Scholar] [CrossRef]
- Amer, H.M. Bovine-like coronaviruses in domestic and wild ruminants. Anim. Health Res. Rev. 2019, 19, 113–124. [Google Scholar] [CrossRef]
- Van der Fels-Klerx, H.J.; Sorensen, J.T.; Jalvingh, A.W.; Huirne, R.B.M. An economic model to calculate farm-specific losses due to bovine respiratory disease in dairy heifers. Prev. Vet. Med. 2001, 51, 75–94. [Google Scholar] [CrossRef]
- Patel, S.J.; Wellington, M.; Shah, R.M.; Ferreira, M.J. Antibiotic stewardship in food-producing animals: Challenges, progress, and opportunities. Clin. Ther. 2020, 42, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- More, S.J. European perspectives on efforts to reduce antimicrobial usage in food animal production. Ir. Vet. J. 2020, 73, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Standley, K. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. (outlines version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Lett, J.-M.; Varsani, A.; Martin, D.P. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 2009, 83, 2697–2707. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Estimating Phylogenetic Trees and Networks Using SplitsTree4. Available online: Www.Splitstree.Org (accessed on 4 June 2020).
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W.; Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 2013, 30, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the Skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef]
- Poon, A.F.Y.; Lewis, F.I.; Frost, S.D.W.; Kosakovsky Pond, S.L. Spidermonkey: Rapid detection of co-evolving sites using Bayesian graphical models. Bioinformatics 2008, 24, 1949–1950. [Google Scholar] [CrossRef]
- Avino, M.; Poon, A.F.Y. Detecting amino acid coevolution with Bayesian graphical models. In Computational Methods in Protein Evolution; Humana Press: New York, NY, USA, 2019; Volume 1851. [Google Scholar] [CrossRef]
- Murrell, B.; de Oliveira, T.; Seebregts, C.; Kosakovsky Pond, S.L.; Scheffler, K. Modeling HIV-1 drug resistance as episodic directional selection. PLoS Comput. Biol. 2012, 8, e1002507. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Lu, M.; Dittmer, U.; Yang, D. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J. Med. Virol. 2020, 92, 491–494. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780128152010. [Google Scholar]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Franzo, G.; Legnardi, M.; Tucciarone, C.M.; Drigo, M.; Martini, M.; Cecchinato, M. Evolution of Infectious Bronchitis Virus in the Field after Homologous Vaccination Introduction. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6842459/ (accessed on 7 April 2020).
- Franzo, G.; Listorti, V.; Naylor, C.J.; Lupini, C.; Laconi, A.; Felice, V.; Drigo, M.; Catelli, E.; Cecchinato, M. Molecular investigation of a full-length genome of a Q1-like IBV strain isolated in Italy in 2013. Virus Res. 2015, 210, 77–80. [Google Scholar] [CrossRef]
- Franzo, G.; Cecchinato, M.; Tosi, G.; Fiorentini, L.; Faccin, F.; Tucciarone, C.M.; Trogu, T.; Barbieri, I.; Massi, P.; Moreno, A. GI-16 lineage (624/I or Q1), there and back again: The history of one of the major threats for poultry farming of our era. PLoS ONE 2018, 13, e0203513. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Posada, D. Analysing recombination in nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 943–955. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Kin, N.; Miszczak, F.; Diancourt, L.; Caro, V.; Moutou, F.; Vabret, A.; Ar Gouilh, M. Comparative molecular epidemiology of two closely related coronaviruses, bovine coronavirus (BCoV) and human coronavirus OC43 (HCoV-OC43), reveals a different evolutionary pattern. Infect. Genet. Evol. 2016, 40, 186–191. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef]
- Moreno, A.; Franzo, G.; Massi, P.; Tosi, G.; Blanco, A.; Antilles, N.; Biarnes, M.; Majó, N.; Nofrarías, M.; Dolz, R.; et al. A novel variant of the infectious bronchitis virus resulting from recombination events in Italy and Spain. Avian Pathol. 2017, 46. [Google Scholar] [CrossRef]
- Bidokhti, M.R.M.M.; Tråvén, M.; Krishna, N.K.; Munir, M.; Belák, S.; Alenius, S.; Cortey, M.; Trå, M.; Krishna, N.K.; Munir, M.; et al. Evolutionary dynamics of bovine coronaviruses: Natural selection pattern of the spike gene implies adaptive evolution of the strains. J. Gen. Virol. 2013, 94, 2036–2049. [Google Scholar] [CrossRef]
- Alejandra Tortorici, M.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef]
- Camps, M.; Herman, A.; Loh, E.; Loeb, L.A. Genetic constraints on protein evolution. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 313–326. [Google Scholar] [CrossRef]
- Zhang, X.M.; Herbst, W.; Kousoulas, K.G.; Storz, J. Biological and genetic characterization of a hemagglutinating coronavirus isolated from a diarrhoeic child. J. Med. Virol. 1994, 44, 152–161. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Maes, P.; Van Reeth, K.; Nauwynck, H.; Pensaert, M.; Van Ranst, M. Evolutionary history of the closely related group 2 coronaviruses: Porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J. Virol. 2006, 80, 7270–7274. [Google Scholar] [CrossRef] [PubMed]
- Kaneshima, T.; Hohdatsu, T.; Hagino, R.; Hosoya, S.; Nojiri, Y.; Murata, M.; Takano, T.; Tanabe, M.; Tsunemitsu, H.; Koyama, H. The infectivity and pathogenicity of a group 2 bovine coronavirus in pups. J. Vet. Med. Sci. 2007, 69, 301–303. [Google Scholar] [CrossRef][Green Version]
- Decaro, N.; Buonavoglia, C. An update on canine coronaviruses: Viral evolution and pathobiology. Vet. Microbiol. 2008, 132, 221–234. [Google Scholar] [CrossRef]
- Vilček, S.; Jacková, A.; Kolesárová, M.; Vlasáková, M. Genetic variability of the S1 subunit of enteric and respiratory bovine coronavirus isolates. Acta Virol. 2017, 61, 212–216. [Google Scholar] [CrossRef]
- Gélinas, A.M.; Boutin, M.; Sasseville, A.M.J.; Dea, S. Bovine coronaviruses associated with enteric and respiratory diseases in Canadian dairy cattle display different reactivities to anti-HE monoclonal antibodies and distinct amino acid changes in their HE, S and ns4.9 protein. Virus Res. 2001, 76, 43–57. [Google Scholar] [CrossRef]
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef]
- Kauder, S.E.; Racaniello, V.R. Poliovirus tropism and attenuation are determined after internal ribosome entry. J. Clin. Investig. 2004, 113, 1743–1753. [Google Scholar] [CrossRef]
- Gunn, L.; Collins, P.J.; O’connell, M.J.; O’shea, H. Phylogenetic investigation of enteric bovine coronavirus in Ireland reveals partitioning between European and global strains. Ir. Vet. J. 2015, 68. [Google Scholar] [CrossRef]
- Hardstaff, J.L.; Häsler, B.; Rushton, J.R. Livestock trade networks for guiding animal health surveillance. BMC Vet. Res. 2015, 11, 82. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think globally, act locally: Phylodynamic reconstruction of infectious bronchitis virus (IBV) QX genotype (GI-19 lineage) reveals different population dynamics and spreading patterns when evaluated on different epidemiological scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Dotto, G.; Gigli, A.; Ceglie, L.; Drigo, M. International trades, local spread and viral evolution: The case of porcine circovirus type 2 (PCV2) strains heterogeneity in Italy. Infect. Genet. Evol. 2015, 32, 409–415. [Google Scholar] [CrossRef] [PubMed]
- U.S.-Vietnam Economic and Trade Relations: Issues for the 111th Congress—EveryCRSReport.com. Available online: https://www.everycrsreport.com/reports/R40755.html (accessed on 5 September 2020).
- González-Corzo, M.A.; González, A.N. U.S. agricultural exports to Cuba: Composition, trends, and prospects for the future. Mag. Food Farm Resour. Issues 2013, 28, 1–6. [Google Scholar]
- Hall, M.D.; Woolhouse, M.E.J.; Rambaut, A. The effects of sampling strategy on the quality of reconstruction of viral population dynamics using Bayesian skyline family coalescent methods: A simulation study. Virus Evol. 2016, 2, vew003. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Protein | Mean | Median | Lower 95HDP | Higher 95HDP |
|---|---|---|---|---|---|
| Evolutionary rate | pp1ab | 2.86 × 10−4 | 2.80 × 10−4 | 2.12 × 10−4 | 3.86 × 10−4 |
| 32 KDa | 5.06 × 10−4 | 4.98 × 10−4 | 3.81 × 10−4 | 6.61 × 10−4 | |
| HE | 3.00 × 10−4 | 2.98 × 10−4 | 2.49 × 10−4 | 3.64 × 10−4 | |
| S | 2.97 × 10−4 | 2.96 × 10−4 | 2.38 × 10−4 | 3.62 × 10−4 | |
| 12 KDa | 3.01 × 10−4 | 2.90 × 10−4 | 2.4 × 10−4 | 4.42 × 10−4 | |
| SE | 4.55 × 10−4 | 4.20 × 10−4 | 2.00 × 10−4 | 8.65 × 10−4 | |
| M | 3.49 × 10−4 | 3.43 × 10−4 | 2.51 × 10−4 | 4.80 × 10−4 | |
| N | 3.75 × 10−4 | 3.71 × 10−4 | 2.90 × 10−4 | 4.78 × 10 | |
| tMRCA | pp1ab | 1947.98 | 1953.40 | 1906.23 | 1971.93 |
| 32 KDa | 1957.34 | 1961.90 | 1921.92 | 1977.44 | |
| HE | 1956.83 | 1960.32 | 1926.97 | 1974.58 | |
| S | 1951.37 | 1953.57 | 1928.94 | 1966.89 | |
| 12 KDa | 1952.13 | 1956.46 | 1912.25 | 1977.13 | |
| SE | 1974.07 | 1979.27 | 1947.92 | 1984.82 | |
| M | 1957.99 | 1962.41 | 1924.42 | 1977.13 | |
| N | 1969.51 | 1972.32 | 1947.62 | 1981.74 |
| FEL | FUBAR | SLAC | ||||
|---|---|---|---|---|---|---|
| Position | dN-dS | p-Value | dN-dS | Posterior Probability | dN-dS | p-Value |
| 113 | 4.714 | 0.011 | 7.060 | 0.993 | 5.711 | 0.013 |
| 174 | - | - | - | - | 4.000 | 0.039 |
| 179 | 9.127 | 0.030 | 17.724 | 0.994 | 10.129477 | 0.012 |
| 257 | 3.009 | 0.039 | 4.547 | 0.937 | - | - |
| 400 | - | - | 3.326 | 0.912 | - | - |
| 499 | 5.817 | 0.013 | 12.690 | 0.997 | 5.816 | 0.021 |
| 501 | 7.518 | 0.001 | 14.566 | 1.000 | 12.412 | 0.000 |
| 509 | 9.840 | 0.001 | 17.373 | 1.000 | 9.071 | 0.002 |
| 510 | 3.790 | 0.024 | 6.358 | 0.984 | 4.722 | 0.030 |
| 525 | - | - | 8.060 | 0.917 | 6.462 | 0.034 |
| 572 | - | - | 4.962 | 0.938 | - | - |
| 579 | 3.456 | 0.032 | 5.916 | 0.972 | 4.219 | 0.046 |
| 966 | - | - | 7.742 | 0.972 | - | - |
| 1298 | 1.307 | 0.026 | 1.403 | 0.918 | - | - |
| Amino Acid 1 | Amino Acid 2 | Position Amino Acid 1 | Position Amino Acid 2 | Distance (Å) |
|---|---|---|---|---|
| 121 | 260 | Internal | Internal | 12 |
| 146 | 148 | External | External | 5.4 |
| 147 | 151 | External | External | 6.8 |
| 244 | 531 | External | External | 29.1 |
| 525 | 546 | External | External | 12.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Drigo, M.; Legnardi, M.; Grassi, L.; Pasotto, D.; Menandro, M.L.; Cecchinato, M.; Tucciarone, C.M. Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus. Viruses 2020, 12, 1285. https://doi.org/10.3390/v12111285
Franzo G, Drigo M, Legnardi M, Grassi L, Pasotto D, Menandro ML, Cecchinato M, Tucciarone CM. Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus. Viruses. 2020; 12(11):1285. https://doi.org/10.3390/v12111285
Chicago/Turabian StyleFranzo, Giovanni, Michele Drigo, Matteo Legnardi, Laura Grassi, Daniela Pasotto, Maria Luisa Menandro, Mattia Cecchinato, and Claudia Maria Tucciarone. 2020. "Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus" Viruses 12, no. 11: 1285. https://doi.org/10.3390/v12111285
APA StyleFranzo, G., Drigo, M., Legnardi, M., Grassi, L., Pasotto, D., Menandro, M. L., Cecchinato, M., & Tucciarone, C. M. (2020). Bovine Coronavirus: Variability, Evolution, and Dispersal Patterns of a No Longer Neglected Betacoronavirus. Viruses, 12(11), 1285. https://doi.org/10.3390/v12111285