Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Live Cell Imaging
2.3. Soft X-ray Tomography
2.4. Immunofluorescence Microscopy
2.5. Transmission Electron Microscopy
2.6. Numerical Simulations of Capsid Motion
3. Results
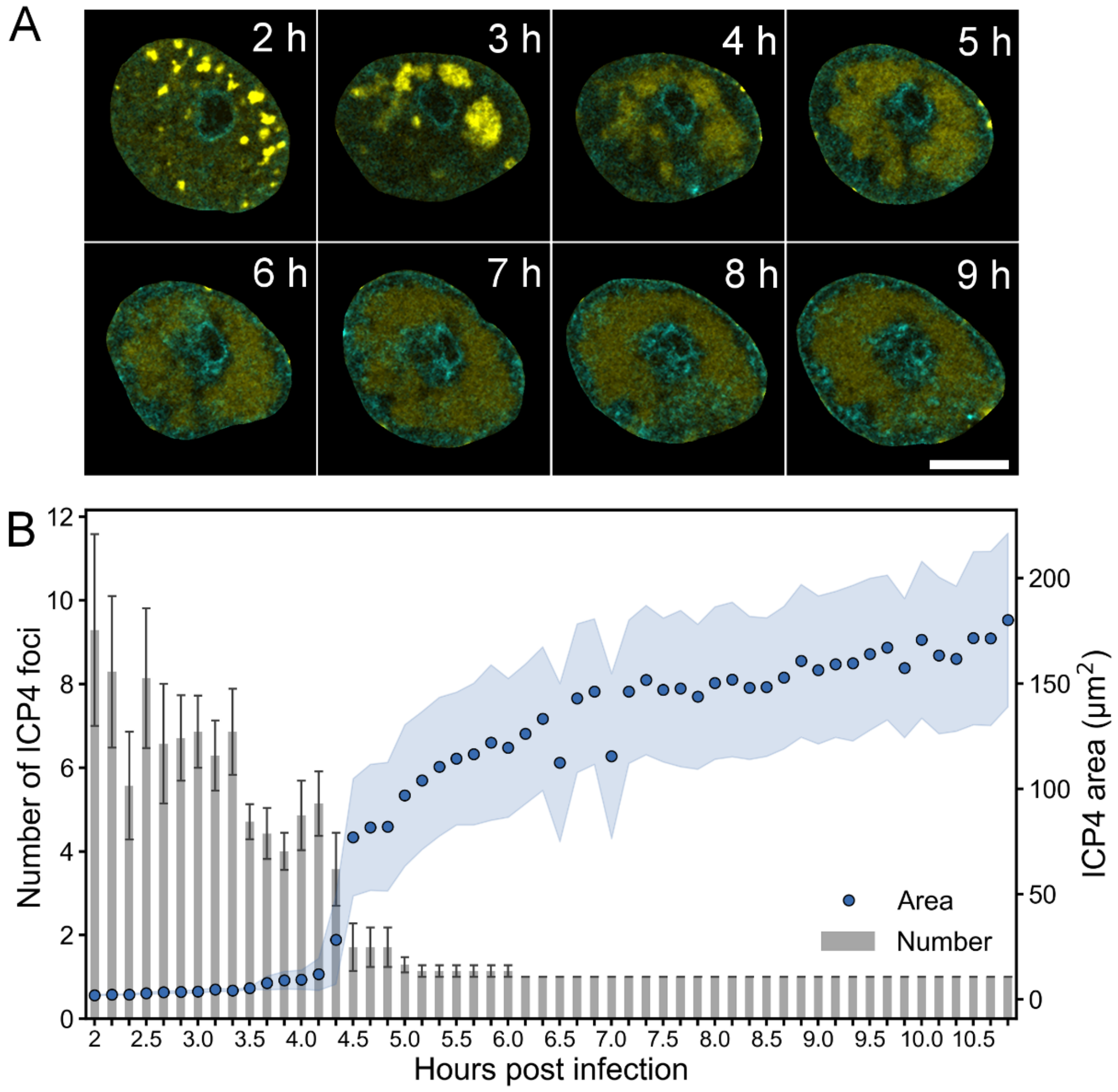
3.1. Formation of Nuclear Replication Compartments
3.2. Infection-Induced Nuclear Swelling and Chromatin Straightening
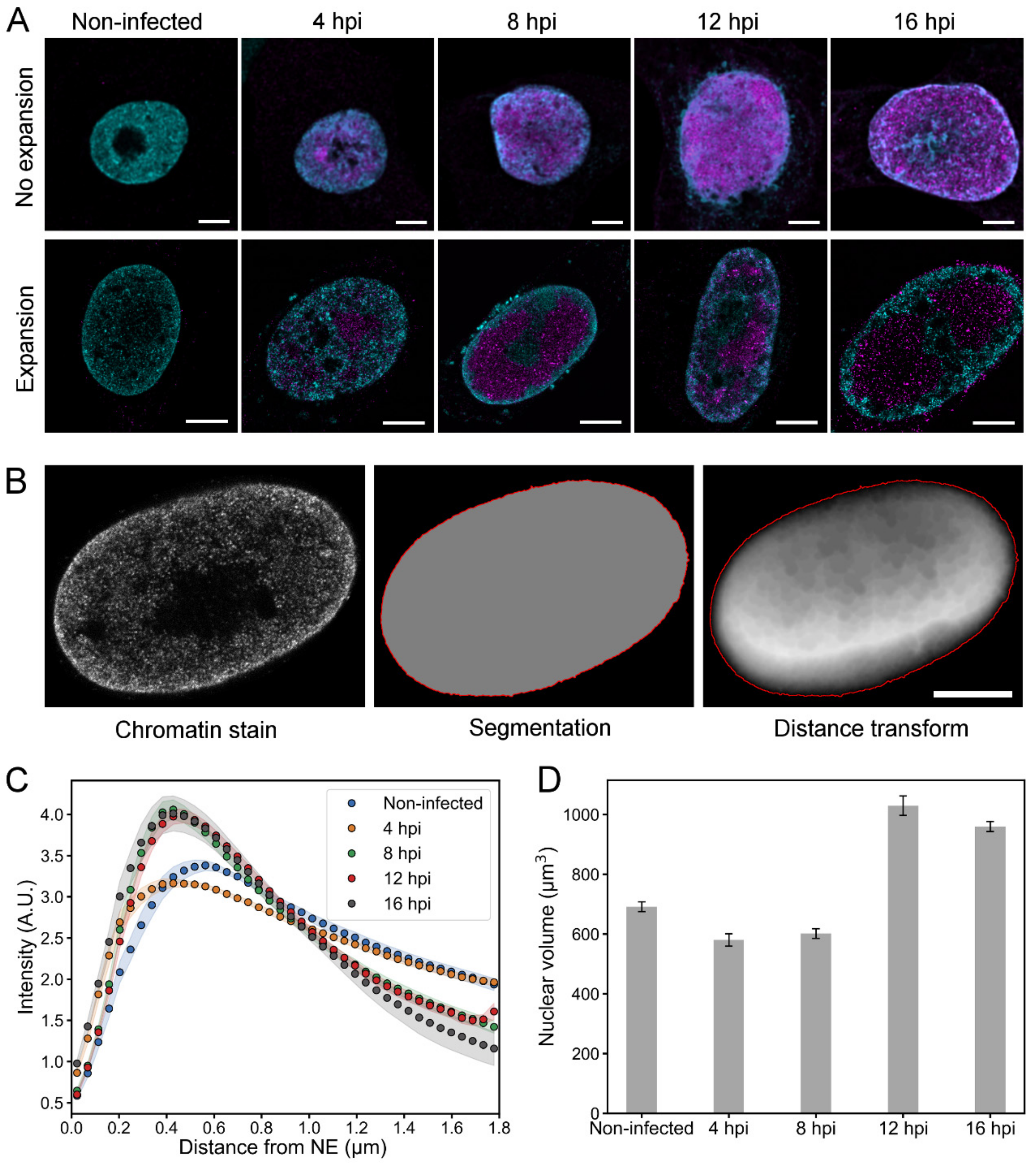
3.3. Virus-Induced Changes in Chromatin Distribution and Nuclear Volume
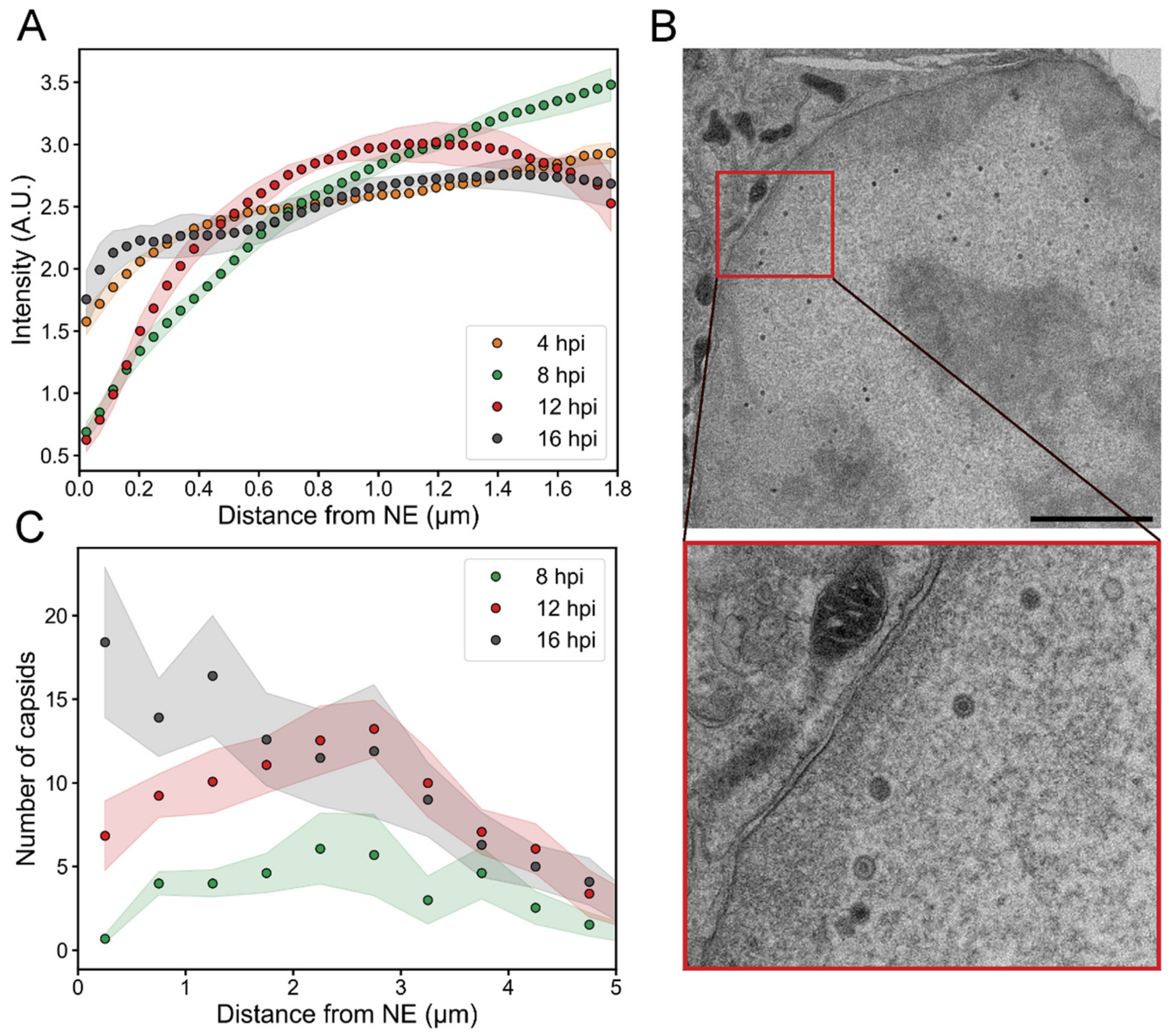
3.4. Intranuclear Distribution of Capsid Proteins and Capsids
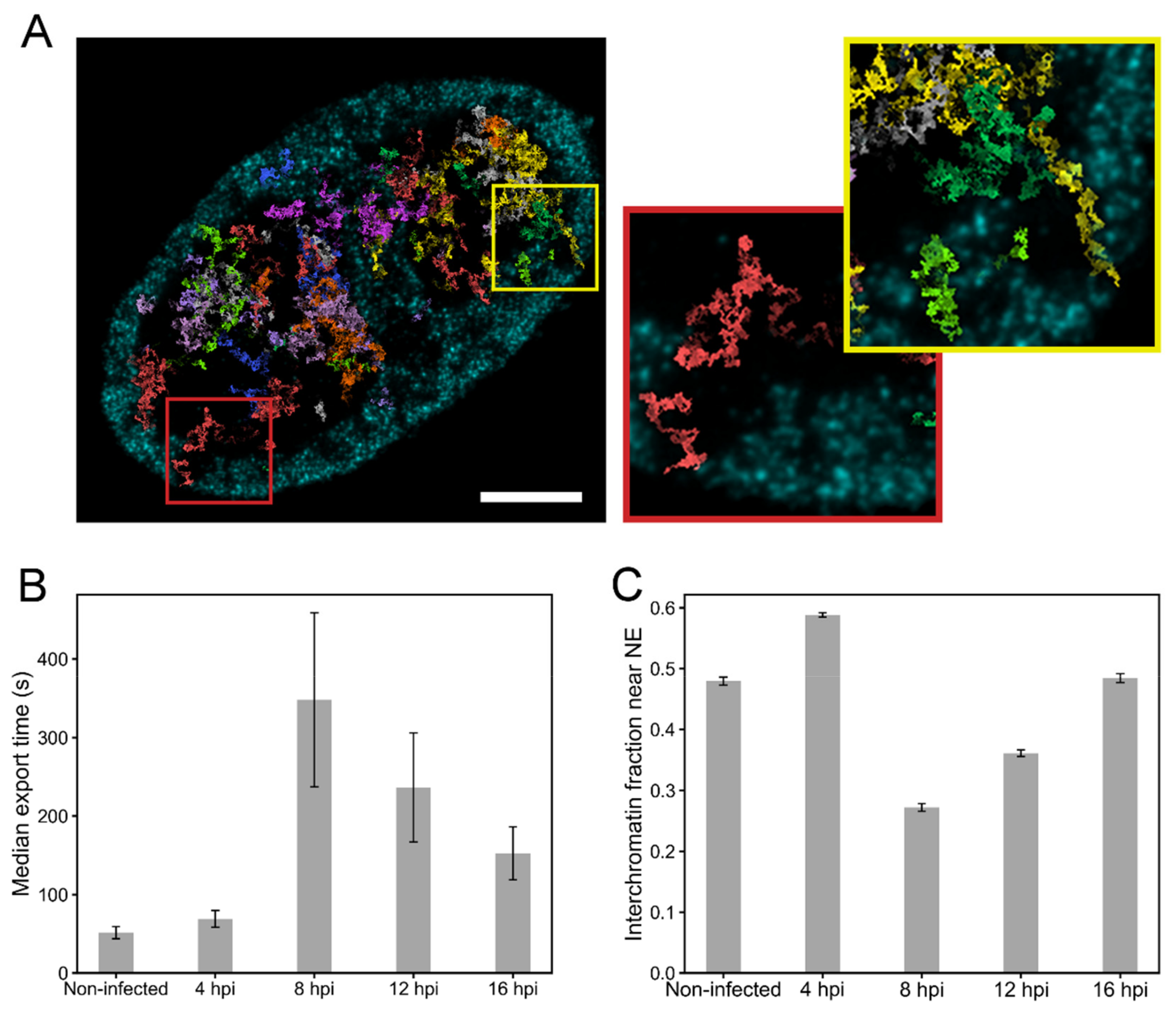
3.5. Simulations of the Nuclear Transport of HSV-1 Capsids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kobiler, O.; Lipman, Y.; Therkelsen, K.; Daubechies, I.; Enquist, L.W. Herpesviruses Carrying a Brainbow Cassette Reveal Replication and Expression of Limited Numbers of Incoming Genomes. Nat. Commun. 2010, 1, 146. [Google Scholar] [CrossRef]
- Kobiler, O.; Brodersen, P.; Taylor, M.P.; Ludmir, E.B.; Enquist, L.W. Herpesvirus Replication Compartments Originate with Single Incoming Viral Genomes. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Monier, K.; Armas, J.C.; Etteldorf, S.; Ghazal, P.; Sullivan, K.F. Annexation of the Interchromosomal Space during Viral Infection. Nat. Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef]
- Myllys, M.; Ruokolainen, V.; Aho, V.; Smith, E.A.; Hakanen, S.; Peri, P.; Salvetti, A.; Timonen, J.; Hukkanen, V.; Larabell, C.A.; et al. Herpes Simplex Virus 1 Induces Egress Channels through Marginalized Host Chromatin. Sci. Rep. 2016, 6, 28844. [Google Scholar] [CrossRef]
- Randall, R.E.; Dinwoodie, N. Intranuclear Localization of Herpes Simplex Virus Immediate-Early and Delayed-Early Proteins: Evidence that ICP 4 is Associated with Progeny Virus DNA. J. Gen. Virol. 1986, 67(Pt. 10), 2163–2177. [Google Scholar] [CrossRef]
- Aho, V.; Myllys, M.; Ruokolainen, V.; Hakanen, S.; Mantyla, E.; Virtanen, J.; Hukkanen, V.; Kuhn, T.; Timonen, J.; Mattila, K.; et al. Chromatin Organization Regulates Viral Egress Dynamics. Sci. Rep. 2017, 7, 3692. [Google Scholar] [CrossRef] [PubMed]
- Booy, F.P.; Newcomb, W.W.; Trus, B.L.; Brown, J.C.; Baker, T.S.; Steven, A.C. Liquid-Crystalline, Phage-Like Packing of Encapsidated DNA in Herpes Simplex Virus. Cell 1991, 64, 1007–1015. [Google Scholar] [CrossRef]
- Lukonis, C.J.; Weller, S.K. Formation of Herpes Simplex Virus Type 1 Replication Compartments by Transfection: Requirements and Localization to Nuclear Domain 10. J. Virol. 1997, 71, 2390–2399. [Google Scholar] [PubMed]
- Newcomb, W.W.; Cockrell, S.K.; Homa, F.L.; Brown, J.C. Polarized DNA Ejection from the Herpesvirus Capsid. J. Mol. Biol. 2009, 392, 885–894. [Google Scholar] [CrossRef]
- Ojala, P.M.; Sodeik, B.; Ebersold, M.W.; Kutay, U.; Helenius, A. Herpes Simplex Virus Type 1 Entry into Host Cells: Reconstitution of Capsid Binding and Uncoating at the Nuclear Pore Complex in Vitro. Mol. Cell. Biol. 2000, 20, 4922–4931. [Google Scholar] [CrossRef]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes Simplex Virus 1 U(L)31 and U(L)34 Gene Products Promote the Late Maturation of Viral Replication Compartments to the Nuclear Periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D. Herpes Simplex Virus Capsid Assembly and DNA Packaging: A Present and Future Antiviral Drug Target. Trends Microbiol. 2011, 19, 606–613. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn Kops, A.; Knipe, D.M. Preexisting Nuclear Architecture Defines the Intranuclear Location of Herpesvirus DNA Replication Structures. J. Virol. 1994, 68, 3512–3526. [Google Scholar] [PubMed]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane Deformation and Scission by the HSV-1 Nuclear Egress Complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.; Ott, M.; Raschbichler, V.; Nagel, C.H.; Binz, A.; Sodeik, B.; Bauerfeind, R.; Bailer, S.M. The Herpes Simplex Virus Protein pUL31 Escorts Nucleocapsids to Sites of Nuclear Egress, a Process Coordinated by its N-Terminal Domain. PLoS Pathog. 2015, 11, e1004957. [Google Scholar] [CrossRef]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Vollmer, B.; Unsay, J.D.; Klupp, B.G.; Garcia-Saez, A.J.; Mettenleiter, T.C.; Antonin, W. A Single Herpesvirus Protein can Mediate Vesicle Formation in the Nuclear Envelope. J. Biol. Chem. 2015, 290, 6962–6974. [Google Scholar] [CrossRef]
- Ungricht, R.; Kutay, U. Mechanisms and Functions of Nuclear Envelope Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 229–245. [Google Scholar] [CrossRef]
- Bosse, J.B.; Hogue, I.B.; Feric, M.; Thiberge, S.Y.; Sodeik, B.; Brangwynne, C.P.; Enquist, L.W. Remodeling Nuclear Architecture Allows Efficient Transport of Herpesvirus Capsids by Diffusion. Proc. Natl. Acad. Sci. USA 2015, 112, 5725. [Google Scholar] [CrossRef]
- Ihalainen, T.O.; Niskanen, E.A.; Jylhava, J.; Paloheimo, O.; Dross, N.; Smolander, H.; Langowski, J.; Timonen, J.; Vihinen-Ranta, M. Parvovirus Induced Alterations in Nuclear Architecture and Dynamics. PLoS ONE 2009, 4, e5948. [Google Scholar] [CrossRef]
- Jiang, M.; Imperiale, M.J. Design Stars: How Small DNA Viruses Remodel the Host Nucleus. Future Virol. 2012, 7, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, T.; Kawasaki, Y.; Abe, A.; Matsumoto, S. Nuclear Marginalization of Host Cell Chromatin Associated with Expansion of Two Discrete Virus-Induced Subnuclear Compartments during Baculovirus Infection. J. Virol. 2008, 82, 6409–6418. [Google Scholar] [CrossRef] [PubMed]
- Dross, N.; Spriet, C.; Zwerger, M.; Muller, G.; Waldeck, W.; Langowski, J. Mapping eGFP Oligomer Mobility in Living Cell Nuclei. PLoS ONE 2009, 4, e5041. [Google Scholar] [CrossRef] [PubMed]
- Gorski, S.A.; Dundr, M.; Misteli, T. The Road Much Traveled: Trafficking in the Cell Nucleus. Curr. Opin. Cell Biol. 2006, 18, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Physiological Importance of RNA and Protein Mobility in the Cell Nucleus. Histochem. Cell Biol. 2008, 129, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Suliman, S.; Ben-Yishay, R.; Yunger, S.; Brody, Y.; Shav-Tal, Y. Dynamics of Single mRNP Nucleocytoplasmic Transport and Export through the Nuclear Pore in Living Cells. Nat. Cell Biol. 2010, 12, 543–552. [Google Scholar] [CrossRef]
- Noble, K.N.; Wente, S.R. Nuclear mRNA on the Move. Nat. Cell Biol. 2010, 12, 525–527. [Google Scholar] [CrossRef]
- Ohkawa, T.; Volkman, L.E.; Welch, M.D. Actin-Based Motility Drives Baculovirus Transit to the Nucleus and Cell Surface. J. Cell Biol. 2010, 190, 187–195. [Google Scholar] [CrossRef]
- Ohkawa, T.; Welch, M.D. Baculovirus Actin-Based Motility Drives Nuclear Envelope Disruption and Nuclear Egress. Curr. Biol. 2018, 28, 2153–2159. [Google Scholar] [CrossRef]
- Forest, T.; Barnard, S.; Baines, J.D. Active Intranuclear Movement of Herpesvirus Capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef]
- Roberts, K.L.; Baines, J.D. Actin in Herpesvirus Infection. Viruses 2011, 3, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Virding, S.; Thiberge, S.Y.; Scherer, J.; Wodrich, H.; Ruzsics, Z.; Koszinowski, U.H.; Enquist, L.W. Nuclear Herpesvirus Capsid Motility is Not Dependent on F-Actin. MBio 2014, 5, 1909. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Enquist, L.W. The Diffusive Way Out: Herpesviruses Remodel the Host Nucleus, Enabling Capsids to Access the Inner Nuclear Membrane. Nucleus 2016, 7, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Sourvinos, G.; Orr, A. Recruitment of Herpes Simplex Virus Type 1 Transcriptional Regulatory Protein ICP4 into Foci Juxtaposed to ND10 in Live, Infected Cells. J. Virol. 2003, 77, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeik, B. The C Terminus of the Large Tegument Protein pUL36 Contains Multiple Capsid Binding Sites that Function Differently during Assembly and Cell Entry of Herpes Simplex Virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-Mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Eling, D.J.; Johnson, P.A.; Sharma, S.; Tufaro, F.; Kipps, T.J. Chronic Lymphocytic Leukemia B Cells are Highly Sensitive to Infection by Herpes Simplex Virus-1 Via Herpesvirus-Entry-Mediator A. Gene Ther. 2000, 7, 1210–1216. [Google Scholar] [CrossRef]
- Spear, P.G.; Longnecker, R. Herpesvirus Entry: An Update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef]
- Le Gros, M.A.; McDermott, G.; Larabell, C.A. X-Ray Tomography of Whole Cells. Curr. Opin. Struct. Biol. 2005, 15, 593–600. [Google Scholar] [CrossRef]
- McDermott, G.; Le Gros, M.A.; Knoechel, C.G.; Uchida, M.; Larabell, C.A. Soft X-Ray Tomography and Cryogenic Light Microscopy: The Cool Combination in Cellular Imaging. Trends Cell Biol. 2009, 19, 587–595. [Google Scholar] [CrossRef]
- Parkinson, D.Y.; Knoechel, C.; Yang, C.; Larabell, C.A.; Le Gros, M.A. Automatic Alignment and Reconstruction of Images for Soft X-Ray Tomography. J. Struct. Biol. 2012, 177, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Farnoosh, R.; Zarpak, B. Image Segmentation using Gaussian Mixture Model. IUST Int. J. Eng. Sci. 2008, 19, 29–32. [Google Scholar]
- Lewiner, T.; Lopes, H.; Vieira, A.W.; Tavares, G. Efficient Implementation of Marching Cubes’ Cases with Topological Guarantees. J. Graph. Tools 2003, 8, 1–15. [Google Scholar] [CrossRef]
- Tillberg, P.W.; Chen, F.; Piatkevich, K.D.; Zhao, Y.; Yu, C.C.; English, B.P.; Gao, L.; Martorell, A.; Suk, H.J.; Yoshida, F.; et al. Protein-Retention Expansion Microscopy of Cells and Tissues Labeled using Standard Fluorescent Proteins and Antibodies. Nat. Biotechnol. 2016, 34, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Magalhães, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Li, C.H.; Lee, C.K. Minimum Cross Entropy Thresholding. Pattern Recognition 1993, 26, 617–625. [Google Scholar] [CrossRef]
- Everett, R.D.; Sourvinos, G.; Leiper, C.; Clements, J.B.; Orr, A. Formation of Nuclear Foci of the Herpes Simplex Virus Type 1 Regulatory Protein ICP4 at Early Times of Infection: Localization, Dynamics, Recruitment of ICP27, and Evidence for the De Novo Induction of ND10-Like Complexes. J. Virol. 2004, 78, 1903–1917. [Google Scholar] [CrossRef]
- Brown, J.C.; Newcomb, W.W. Herpesvirus Capsid Assembly: Insights from Structural Analysis. Curr. Opin. Virol. 2011, 1, 142–149. [Google Scholar] [CrossRef]
- Alazard-Dany, N.; Nicolas, A.; Ploquin, A.; Strasser, R.; Greco, A.; Epstein, A.L.; Fraefel, C.; Salvetti, A. Definition of Herpes Simplex Virus Type 1 Helper Activities for Adeno-Associated Virus Early Replication Events. PLoS Pathog. 2009, 5, e1000340. [Google Scholar] [CrossRef]
- Ihalainen, T.O.; Niskanen, E.A.; Jylhava, J.; Turpeinen, T.; Rinne, J.; Timonen, J.; Vihinen-Ranta, M. Dynamics and Interactions of Parvoviral NS1 Protein in the Nucleus. Cell. Microbiol. 2007, 9, 1946–1959. [Google Scholar] [CrossRef]
- Rubio, M.P.; Guerra, S.; Almendral, J.M. Genome Replication and Postencapsidation Functions Mapping to the Nonstructural Gene Restrict the Host Range of a Murine Parvovirus in Human Cells. J. Virol. 2001, 75, 11573–11582. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sahara, S.; Aoto, M.; Eguchi, Y.; Imamoto, N.; Yoneda, Y.; Tsujimoto, Y. Acinus is a Caspase-3-Activated Protein Required for Apoptotic Chromatin Condensation. Nature 1999, 401, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Tone, S.; Sugimoto, K.; Tanda, K.; Suda, T.; Uehira, K.; Kanouchi, H.; Samejima, K.; Minatogawa, Y.; Earnshaw, W.C. Three Distinct Stages of Apoptotic Nuclear Condensation Revealed by Time-Lapse Imaging, Biochemical and Electron Microscopy Analysis of Cell-Free Apoptosis. Exp. Cell Res. 2007, 313, 3635–3644. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. Condensed Matter in Cell Death. Nature 1999, 401, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. MBio 2016, 7, 16. [Google Scholar] [CrossRef]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes Simplex Virus 1 Infection Induces Activation and Subsequent Inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell. Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Komatsu, T.; Nagata, K.; Wodrich, H. The Role of Nuclear Antiviral Factors Against Invading DNA Viruses: The Immediate Fate of Incoming Viral Genomes. Viruses 2016, 8, 290. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation Modulates Cellular Distribution and DNA Sensing Ability of Interferon-Inducible Protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 Induction of IRF-3 Signaling during Herpesviral Infection and Degradation of IFI16 by the Viral ICP0 Protein. Proc. Natl. Acad. Sci. USA 2012, 109, 3008. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P Granules are Liquid Droplets that Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Burns, L.T.; Wente, S.R. From Hypothesis to Mechanism: Uncovering Nuclear Pore Complex Links to Gene Expression. Mol. Cell. Biol. 2014, 34, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Spector, D.L. Nascent Pre-mRNA Transcripts are Associated with Nuclear Regions Enriched in Splicing Factors. Genes Dev. 1991, 5, 2288–2302. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.B.; Singer, R.H.; Marselle, L.M. Highly Localized Tracks of Specific Transcripts within Interphase Nuclei Visualized by in Situ Hybridization. Cell 1989, 57, 493–502. [Google Scholar] [CrossRef]
- Pederson, T. Diffusional Protein Transport within the Nucleus: A Message in the Medium. Nat. Cell Biol. 2000, 2, 73. [Google Scholar] [CrossRef]
- Politz, J.C.; Tuft, R.A.; Pederson, T.; Singer, R.H. Movement of Nuclear Poly(A) RNA Throughout the Interchromatin Space in Living Cells. Curr. Biol. 1999, 9, 285–291. [Google Scholar] [CrossRef]
- Raices, M.; D’Angelo, M.A. Nuclear Pore Complex Composition: A New Regulator of Tissue-Specific and Developmental Functions. Nat. Rev. Mol. Cell Biol. 2012, 13, 687–699. [Google Scholar] [CrossRef]
- Sheinberger, J.; Shav-Tal, Y. The Dynamic Pathway of Nuclear RNA in Eukaryotes. Nucleus 2013, 4, 195–205. [Google Scholar] [CrossRef]
- Vargas, D.Y.; Raj, A.; Marras, S.A.; Kramer, F.R.; Tyagi, S. Mechanism of mRNA Transport in the Nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 17008–17013. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aho, V.; Mäntylä, E.; Ekman, A.; Hakanen, S.; Mattola, S.; Chen, J.-H.; Weinhardt, V.; Ruokolainen, V.; Sodeik, B.; Larabell, C.; et al. Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection. Viruses 2019, 11, 935. https://doi.org/10.3390/v11100935
Aho V, Mäntylä E, Ekman A, Hakanen S, Mattola S, Chen J-H, Weinhardt V, Ruokolainen V, Sodeik B, Larabell C, et al. Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection. Viruses. 2019; 11(10):935. https://doi.org/10.3390/v11100935
Chicago/Turabian StyleAho, Vesa, Elina Mäntylä, Axel Ekman, Satu Hakanen, Salla Mattola, Jian-Hua Chen, Venera Weinhardt, Visa Ruokolainen, Beate Sodeik, Carolyn Larabell, and et al. 2019. "Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection" Viruses 11, no. 10: 935. https://doi.org/10.3390/v11100935
APA StyleAho, V., Mäntylä, E., Ekman, A., Hakanen, S., Mattola, S., Chen, J.-H., Weinhardt, V., Ruokolainen, V., Sodeik, B., Larabell, C., & Vihinen-Ranta, M. (2019). Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection. Viruses, 11(10), 935. https://doi.org/10.3390/v11100935