
Stage- and Tissue-Specific Expression of MET1 and CMT2 Genes During Germination in Abies koreana E.H.Wilson

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Germination Test
2.2. Germination Stages
2.3. Identification of DNA Methylation-Related Genes
2.4. RT-qPCR Analysis
2.5. Statistical Methods
3. Results
3.1. Developmental Stages of Seed Germination
3.2. Transcriptome Assembly of A. koreana
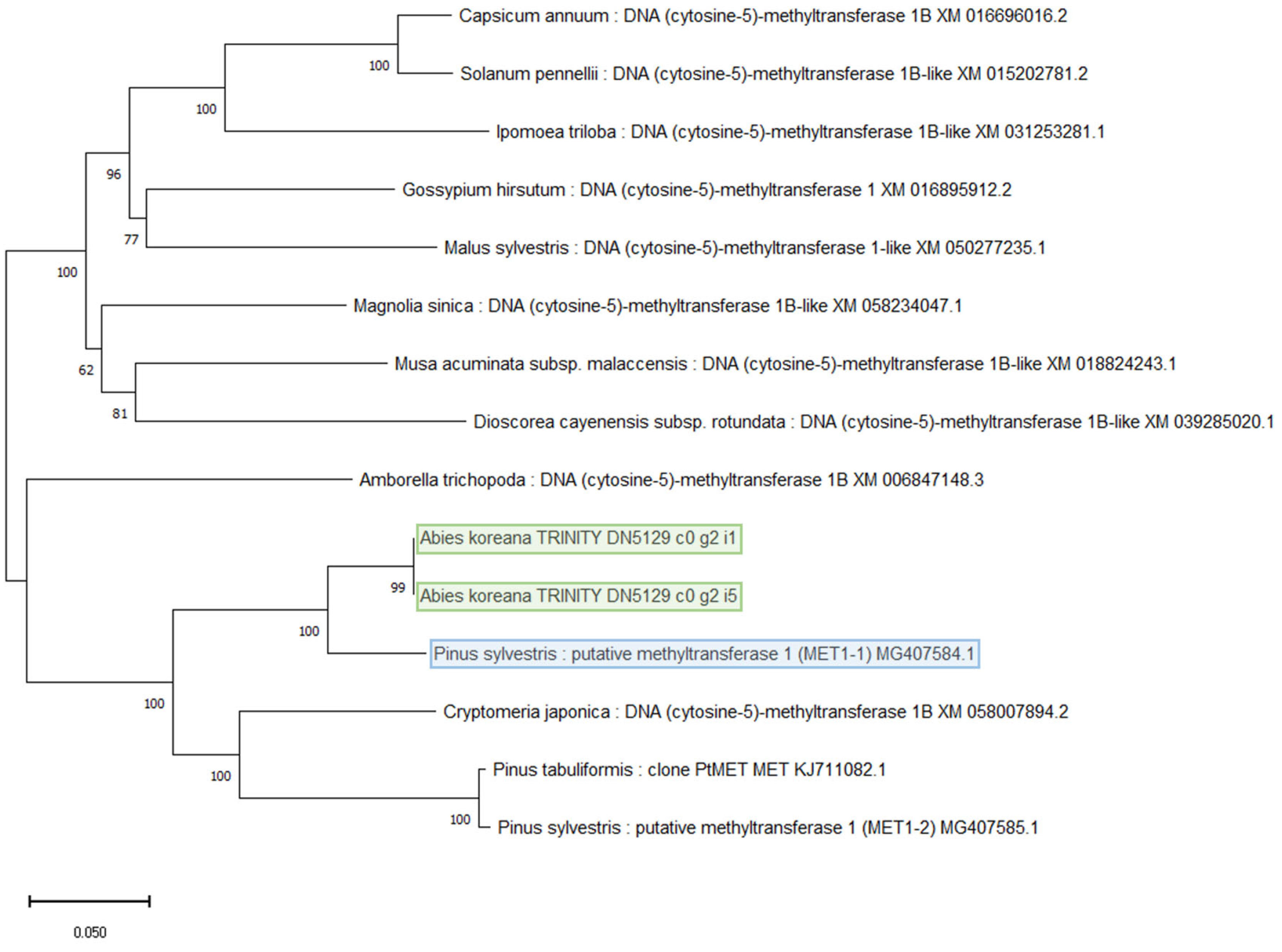
3.3. DNA Methylation Regulatory Genes
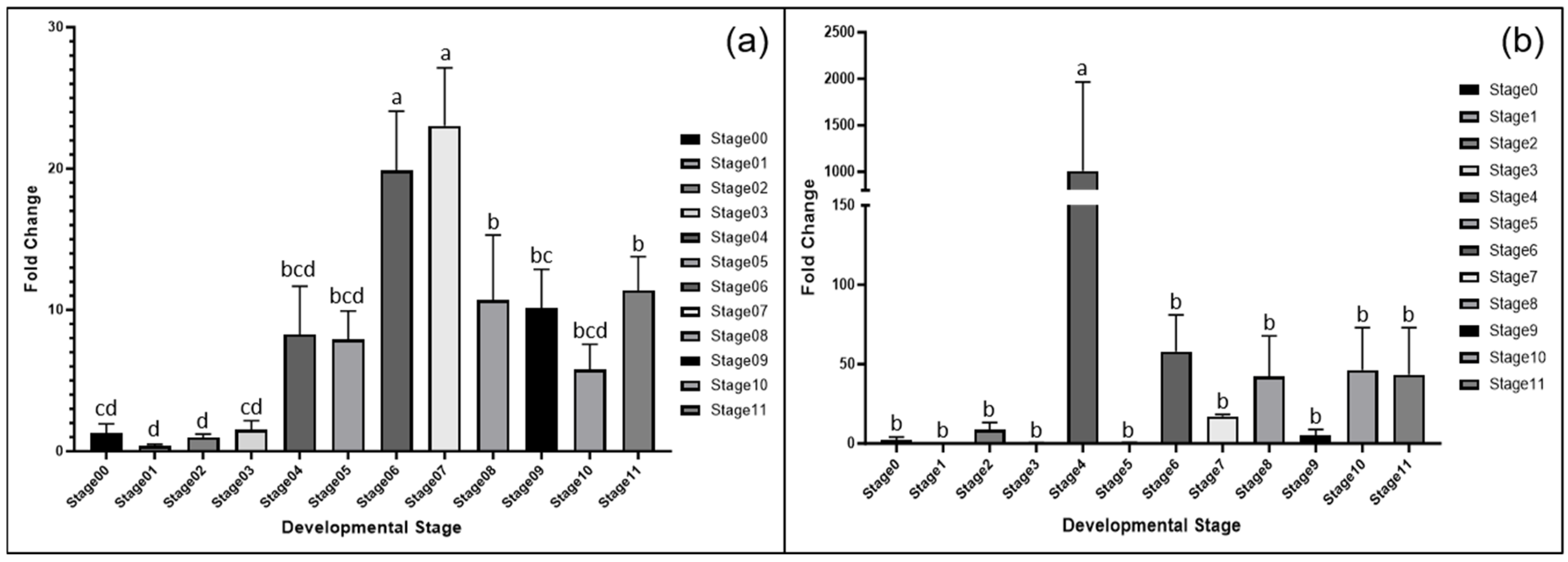
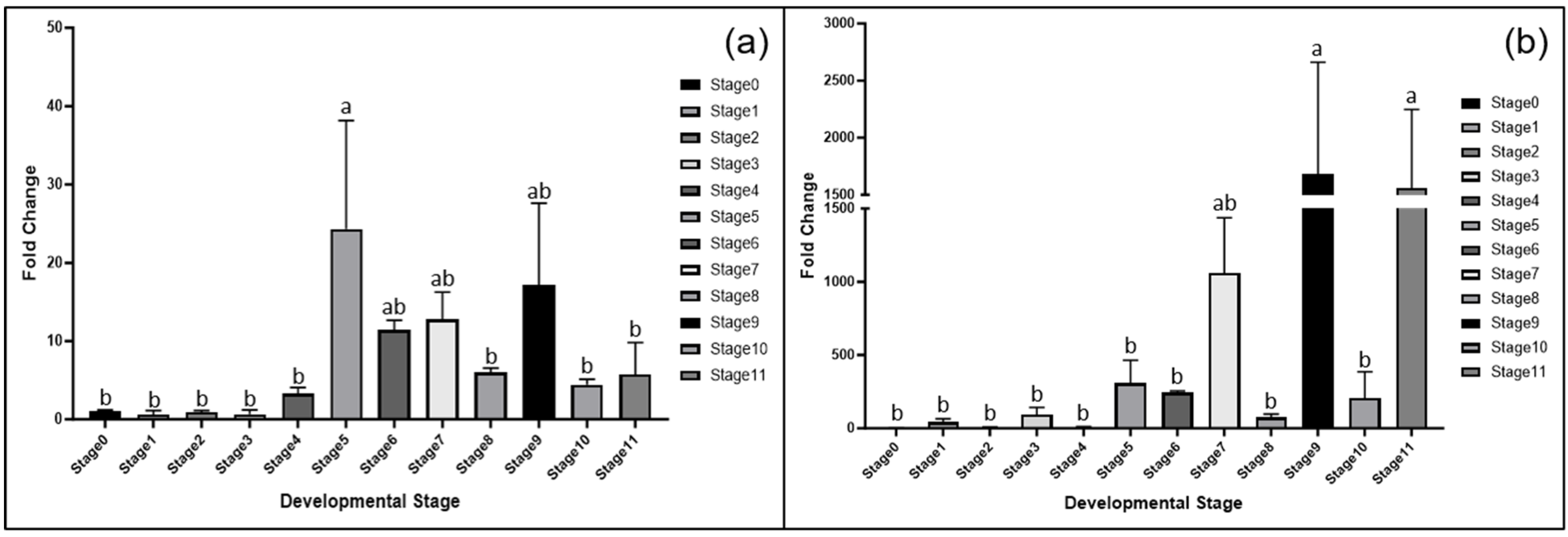
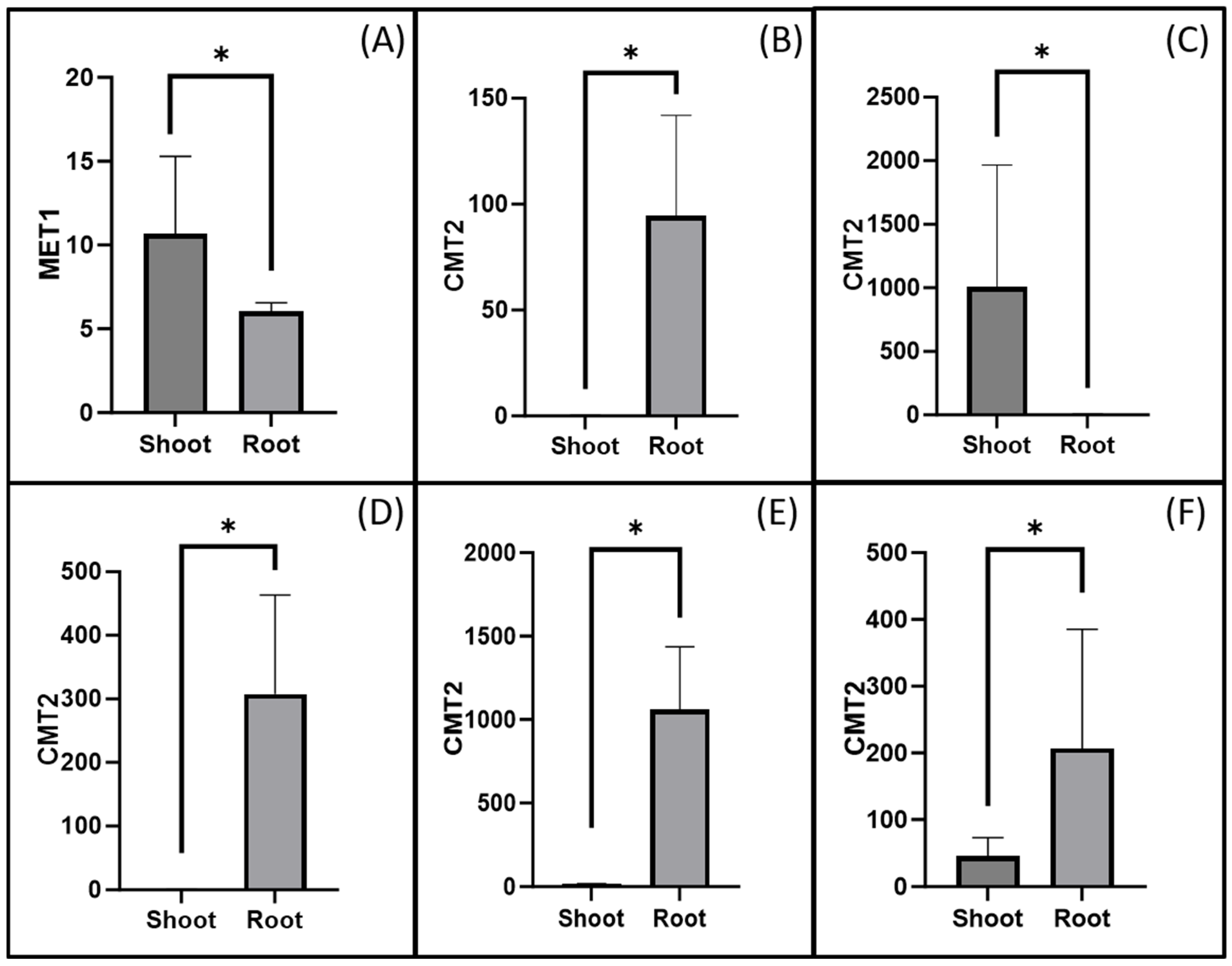
3.4. Gene Expression Analysis
4. Discussion
4.1. Seed Germination and Development
4.2. DNA Methylation and Gene Regulation
4.3. Tissue-Specific Methylation
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Koo, K.A.; Kim, J.; Kong, W.S.; Jung, H.; Kim, G. Projecting the potential distribution of Abies koreana in Korea under the climate change based on RCP scenarios. Korea Soc. Environ. Restor. Technol. 2016, 19, 19–30. [Google Scholar] [CrossRef]
- Ahn, U.S.; Yun, Y.S. Causes of decline in the Korean fir based on spatial distribution in the Mt. Halla region in Korea: A meta-analysis. Forests 2020, 11, 391. [Google Scholar] [CrossRef]
- Choo, M.; Yo, C.; Im, J.; Cho, D.; Kang, Y.; Oh, H.; Lee, J. Trend analysis of vegetation changes of Korean fir (Abies koreana Wilson) in Hallasan and Jirisan using MODIS imagery. Korean Soc. Remote Sens. 2023, 39, 325–338. [Google Scholar]
- Song, J.H.; Han, S.H.; Lee, S.H.; Yun, C.W. Ecological characteristic of Abies koreana stand structure of Mt. Jirisan and Mt. Hallasan. J. Korean Soc. For. Sci. 2021, 110, 590–600. [Google Scholar]
- Kwak, M.; Hong, J.K.; Park, J.H.; Lee, B.Y.; Suh, M.H.; Kim, C.S. Genetic assessment of Abies koreana (Pinaceae), the endangered Korean fir, and conservation implications. Conserv. Genet. Resour. 2017, 18, 1165–1176. [Google Scholar] [CrossRef]
- Koo, K.A.; Kim, D.B. Review forty-year studies of Korean fir (Abies koreana Wilson). J. Ecol. Environ. 2020, 34, 358–371. [Google Scholar] [CrossRef]
- Kim, H.; Kim, E.; Lee, S.; Cho, Y.C. Abnormal winter drought-induced transient dieback of Korean fir in the montane forests of Mt. Jirisan, South Korea. J. Plant. Biol. 2024, 67, 123–136. [Google Scholar] [CrossRef]
- Song, J.H.; Jang, K.H.; Hur, S.D. Variation of seed and germination characteristics of natural populations of Abies koreana Wilson, a Korean endemic species. J. Korean Soc. For. Sci. 2010, 99, 849–854. [Google Scholar]
- Han, B.; Wu, D.; Zhang, Y.; Li, D.Z.; Xu, W.; Liu, A. Epigenetic regulation of seed-specific gene expression by DNA methylation valleys in castor bean. BMC Biol. 2022, 20, 57. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Ecker, J.R. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell. Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Morar, I.M.; Dan, C.; Sestras, R.E.; Stoian-Dod, R.L.; Truta, A.M.; Sestras, A.F.; Sestras, P. Evaluation of different geographic provenances of Silver fir (Abies alba) as seed sources, based on seed traits and germination. Forests 2023, 14, 2186. [Google Scholar] [CrossRef]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18, 172. [Google Scholar] [CrossRef]
- Hwang, J.E.; Kim, Y.J.; Shin, M.H.; Hyun, H.J.; Bohnert, H.J.; Park, H.C. A comprehensive analysis of the Korean fir (Abies koreana) genes expressed under heat stress using transcriptome analysis. Sci. Rep. 2018, 8, 10233. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Shearer, R.C. Proceedings—Conifer Tree Seed in the Inland Mountain West Symposium; Department of Agriculture, Forest Service, Intermountain Research Station: Missoula, MT, USA, 1985; Volume 85, p. 10. [Google Scholar]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055. [Google Scholar] [CrossRef]
- Nonogaki, H. Seed germination and dormancy: The classic story, new puzzles, and evolution. J. Integr. Plant Biol. 2019, 61, 541–563. [Google Scholar] [CrossRef]
- Kermode, A.R. Regulatory mechanisms involved in the transition from seed development to germination. CRC Crit. Rev. Plant Sci. 1990, 9, 155–195. [Google Scholar] [CrossRef]
- Lepiniec, L.; Devic, M.; Roscoe, T.J.; Bouyer, D.; Zhou, D.X.; Boulard, C.; Baud, S.; Dubreucq, B. Molecular and epigenetic regulations and functions of the LAFL transcriptional regulators that control seed development. Plant Reprod. 2018, 31, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; An, L.; Mao, J.; Aluko, O.O.; Ullah, Z.; Xu, F.; Liu, G.; Wang, Q. The CBL-interacting protein kinase NtCIPK23 positively regulates seed germination and early seedling development in tobacco (Nicotiana tabacum L.). Plants 2021, 10, 323. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef]
- Jullien, P.E.; Berger, F. DNA methylation reprogramming during plant sexual reproduction? Trends Genet. 2010, 26, 394–399. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene (Trinity.fasta) | Primer Sequence (5′ → 3′) | Annealing Temperature (°C) | GC Content (%) | Product Size (bp) |
|---|---|---|---|---|
| Actin (ref-gene) (TRINITY_DN6547_c0_g1_i4) | Forward: GCTGACACCATCCCCAGAAT Reverse: CAAGGGCAGTGTTTTCCAGC | 59.7 60 | 55 55 | 389 |
| MET1 (TRINITY_DN5129_c0_g2_i1) | Forward: AGCATCCAACCACACCCTTT Reverse: GACGCCTCTGATGATGCCAA | 59.8 60.5 | 50 55 | 94 |
| CMT2 (TRINITY_DN46777_c0_g1_i5) | Forward: TAACGCCCTAAAAAGCCCCC Reverse: AATTCAAGGTGCGCTGGAGA | 60.3 60 | 55 50 | 343 |
| Transcript | Gene | |
|---|---|---|
| Total number | 1,212,229 | 635,467 |
| Contig N10 | 3955 | 2832 |
| Contig N20 | 2742 | 1701 |
| Contig N30 | 1988 | 1075 |
| Contig N40 | 1394 | 732 |
| Contig N50 | 912 | 534 |
| Median contig length | 329 | 307 |
| Average contig | 604.82 | 478.29 |
| Total assembled bases | 733,181,490 | 303,938,454 |
| Percent GC (%) | 41.00 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-c.; Jeon, K.; Kang, K.-s. Stage- and Tissue-Specific Expression of MET1 and CMT2 Genes During Germination in Abies koreana E.H.Wilson. Forests 2025, 16, 337. https://doi.org/10.3390/f16020337
Hong S-c, Jeon K, Kang K-s. Stage- and Tissue-Specific Expression of MET1 and CMT2 Genes During Germination in Abies koreana E.H.Wilson. Forests. 2025; 16(2):337. https://doi.org/10.3390/f16020337
Chicago/Turabian StyleHong, Sun-cheon, Koeun Jeon, and Kyu-suk Kang. 2025. "Stage- and Tissue-Specific Expression of MET1 and CMT2 Genes During Germination in Abies koreana E.H.Wilson" Forests 16, no. 2: 337. https://doi.org/10.3390/f16020337
APA StyleHong, S.-c., Jeon, K., & Kang, K.-s. (2025). Stage- and Tissue-Specific Expression of MET1 and CMT2 Genes During Germination in Abies koreana E.H.Wilson. Forests, 16(2), 337. https://doi.org/10.3390/f16020337