
Thiamine Deficiency and Neuroinflammation Are Important Contributors to Alcohol Use Disorder

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methodology
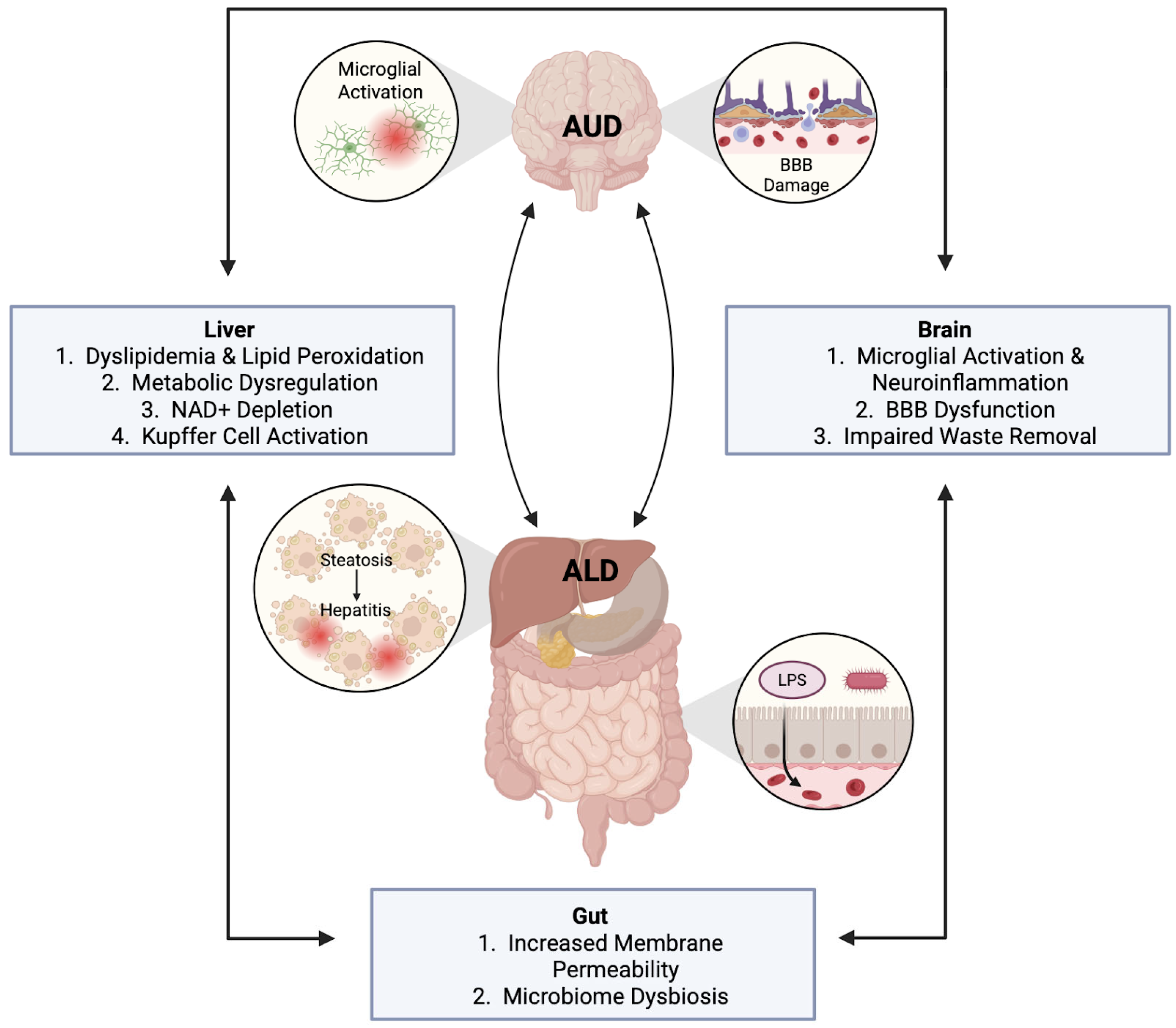
3. Organ Crosstalk in AUD
4. Implications on Thiamine-Related Metabolic Disruptions in AUD
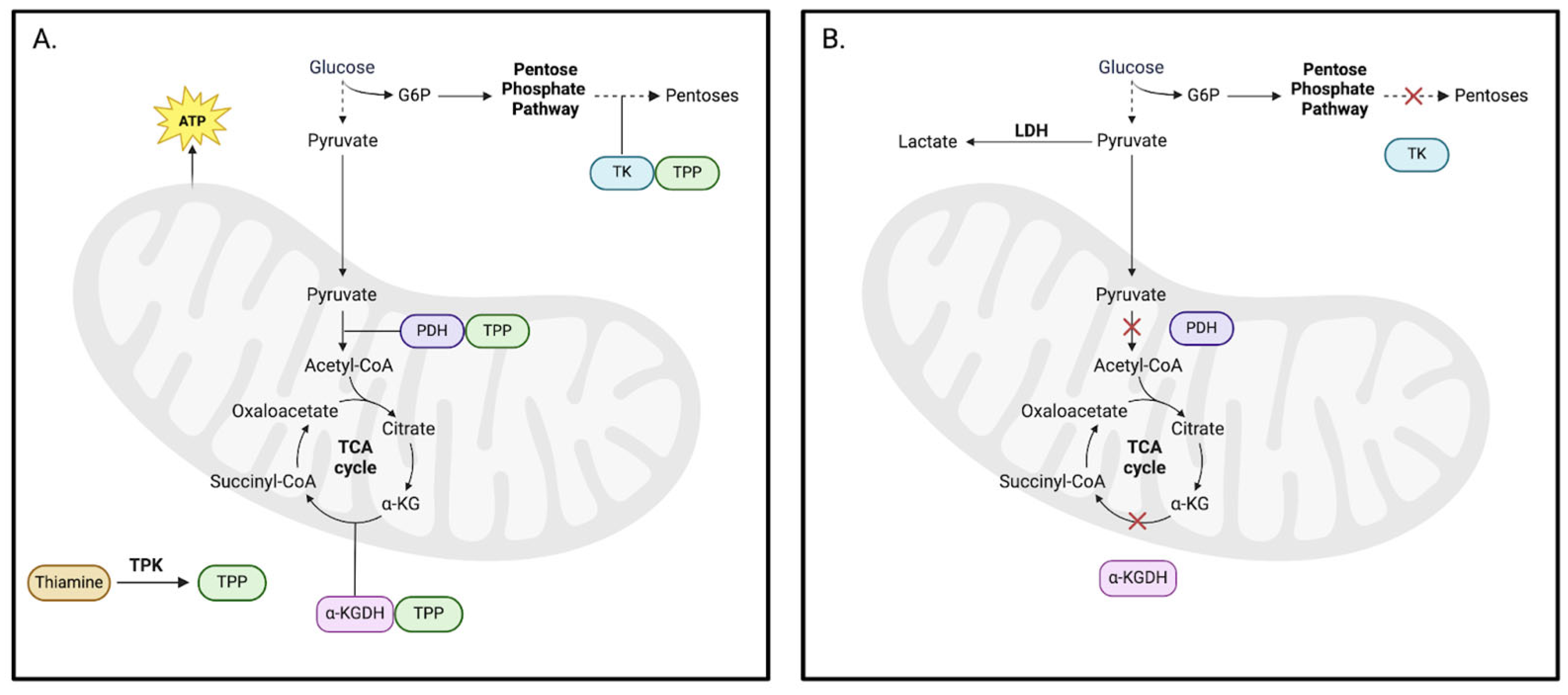
4.1. Thiamine-Related Energy Deficits
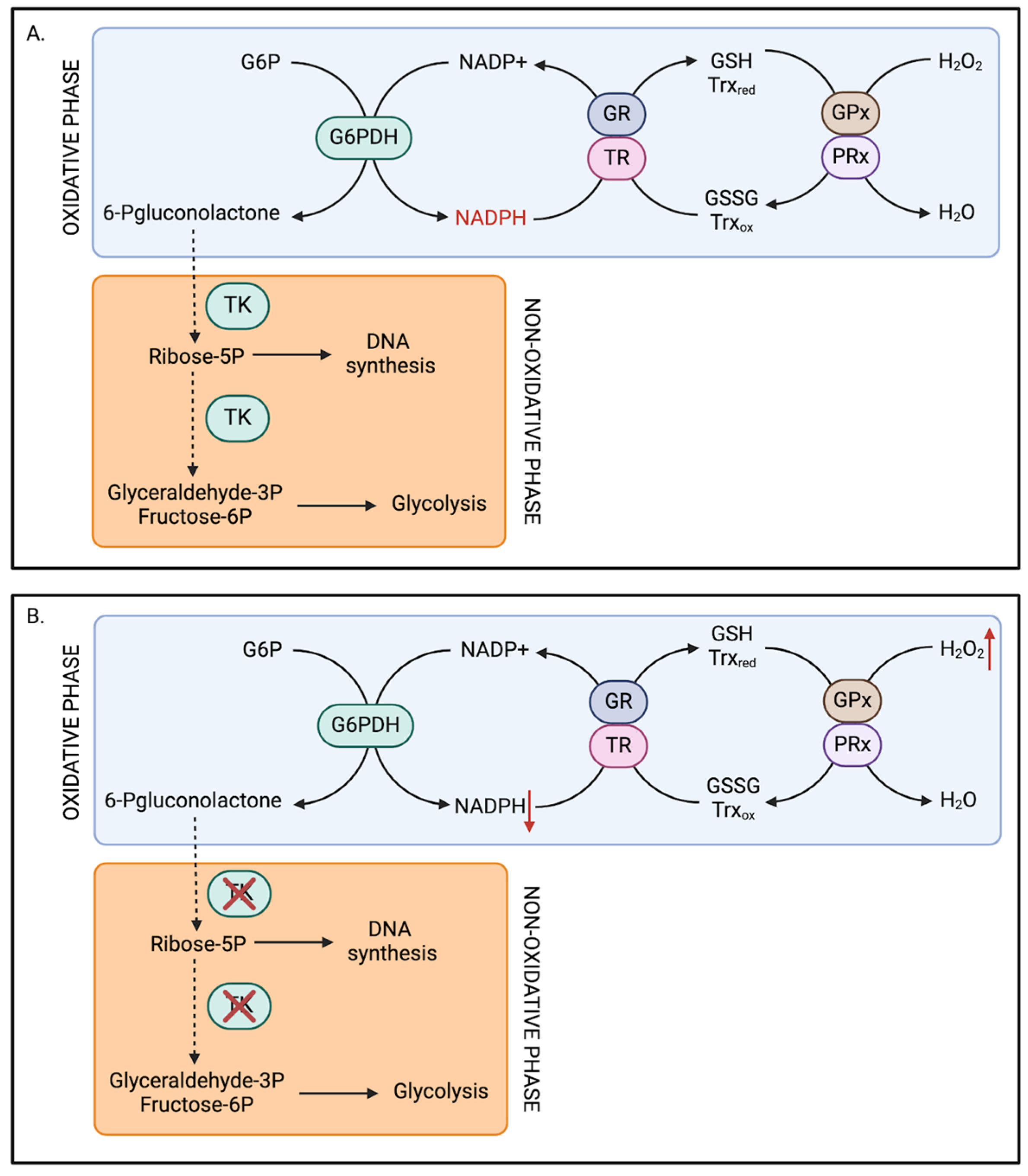
4.2. Thiamine-Related Redox Deficits
4.3. Clinical Implications of Alcohol-Induced Thiamine Deficiency
5. The Role of Neuroinflammation in Alcohol Use Disorder
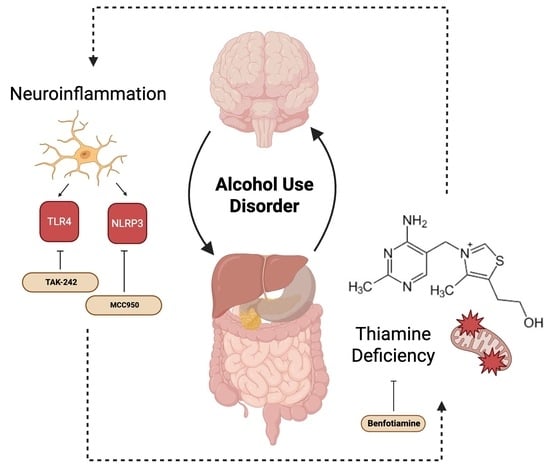
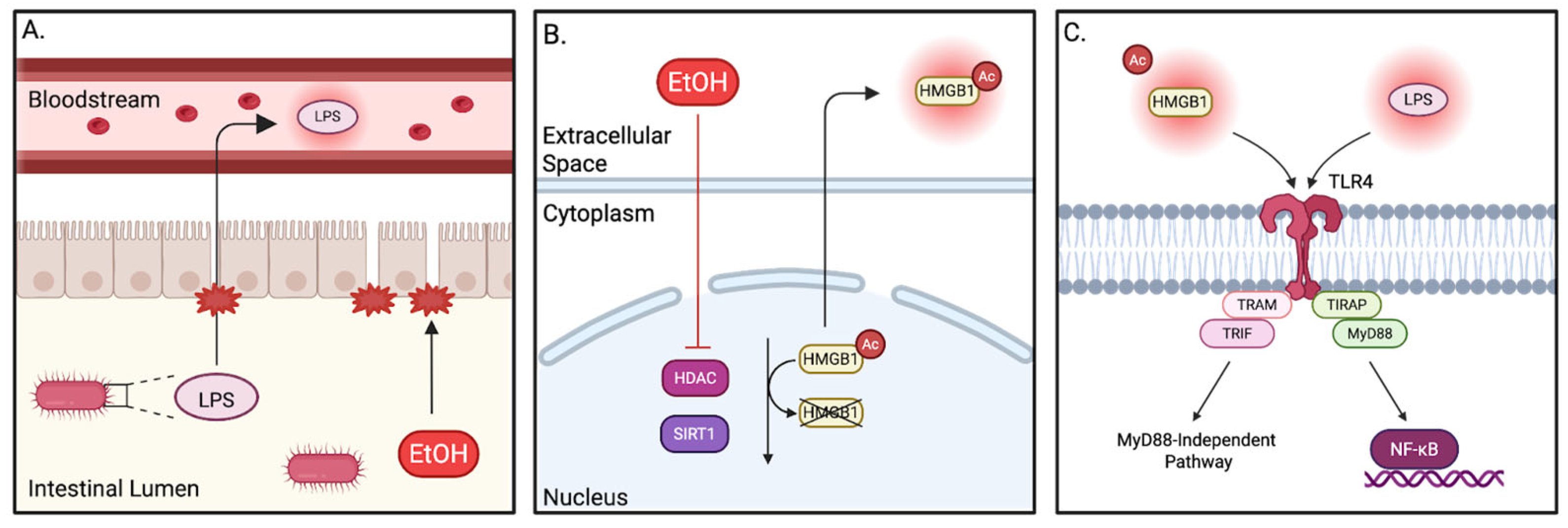
5.1. TLR4
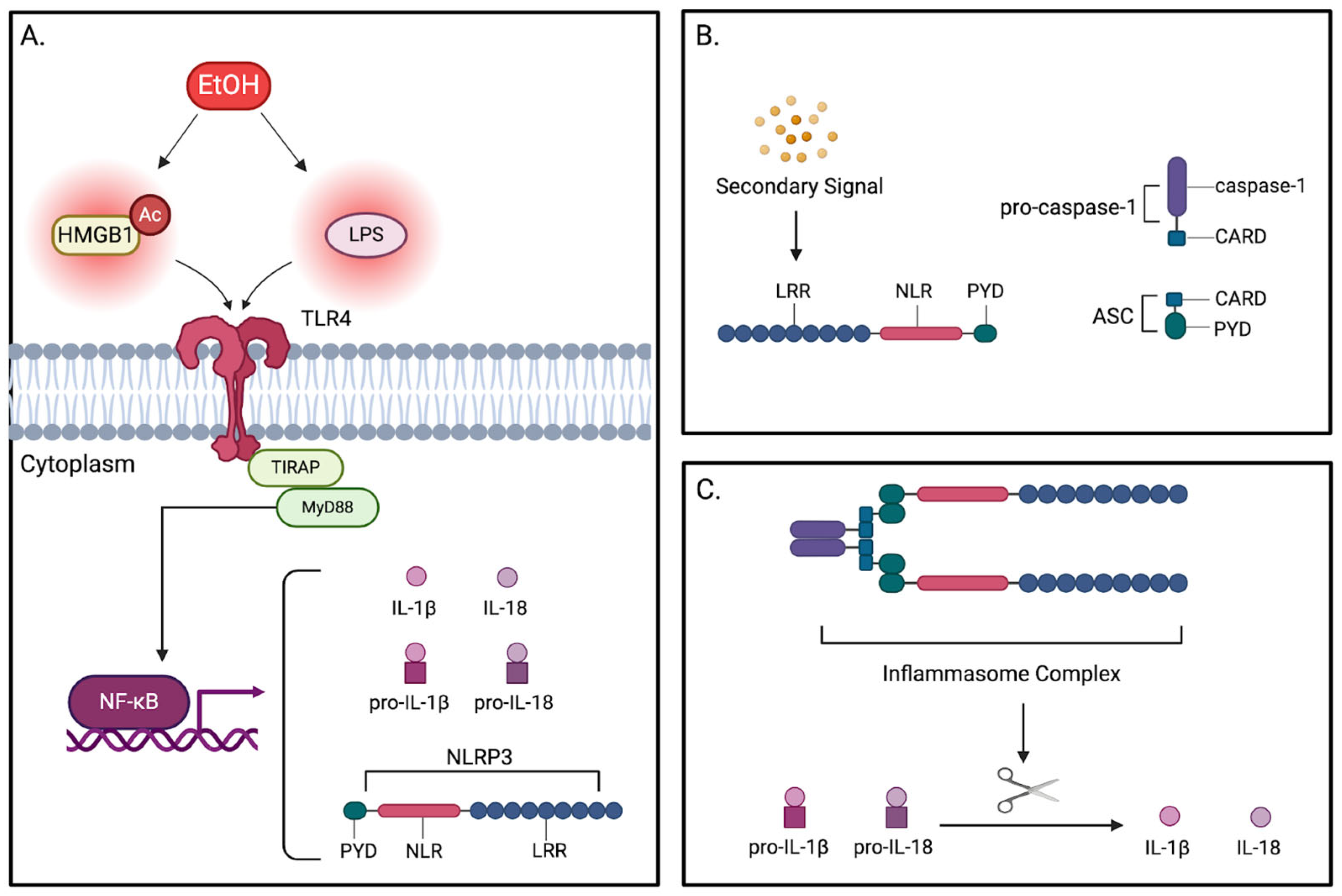
5.2. NLRP3
6. Emerging Connections Between Thiamine Deficiency and Neuroinflammation
7. Future Directions and Research Opportunities
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUD | alcohol use disorder |
| TD | thiamine deficiency |
| CNS | central nervous system |
| GABA | gamma-aminobutyric acid |
| LPS | lipopolysaccharide |
| TNF-α | tumor necrosis factor α |
| ALD | alcohol-associated liver disease |
| EtOH | ethanol |
| TPP | thiamine pyrophosphate |
| TK | transketolase |
| PDH | pyruvate dehydrogenase |
| ɑ-KG | alpha-ketoglutarate |
| ɑ-KGDH | alpha-ketoglutarate dehydrogenase |
| PPP | pentose phosphate pathway |
| TCA cycle | tricarboxylic acid cycle |
| NAD+ | nicotinamide adenine dinucleotide |
| G6P | glucose 6-phosphate |
| TPK | thiamine diphosphokinase |
| ROS | reactive oxygen species |
| GSSG | glutathione disulfide |
| GSH | glutathione |
| BBB | blood–brain barrier |
| WKS | Wernicke–Korsakoff syndrome |
| WE | Wernicke’s encephalopathy |
| KS | Korsakoff syndrome |
| TLR4 | toll-like receptor 4 |
| PRR | pattern recognition receptor |
| PAMPs | pathogen-associated molecular patterns |
| DAMPs | damage-associated molecular patterns |
| TIR | Toll/IL-1 receptor |
| HMGB1 | high-mobility group protein 1 |
| MyD88 | myeloid differentiation primary response gene 88 |
| TIRAP | toll/interleukin-1 receptor domain-containing adaptor protein |
| NF- κB | nuclear factor-κB |
| TRIF | TIR domain-containing adaptor inducing IFN-beta |
| TRAM | TRIF-related adaptor molecule |
| HDACs | histone deacetylases |
| NOD | nucleotide oligomerization domain |
| NLRP3 | NOD-like receptor protein 3 |
| ASC | apoptosis-associated speck-like protein containing a CARD domain |
| AIM2 | absent-in-melanoma 2 |
| PYD | pyrin domain |
| NO | nitric oxide |
| TREM2 | triggering receptor expressed by myeloid cell 2 |
| ADH | alcohol dehydrogenase |
| ALDH2 | aldehyde dehydrogenase 2 |
| CYP2E1 | cytochrome P450 2E1 |
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Bracht, T.; Soravia, L.; Moggi, F.; Stein, M.; Grieder, M.; Federspiel, A.; Tschümperlin, R.; Batschelet, H.M.; Wiest, R.; Denier, N. The role of the orbitofrontal cortex and the nucleus accumbens for craving in alcohol use disorder. Transl. Psychiatry 2021, 11, 267. [Google Scholar] [CrossRef]
- Cooper, S.; Robison, A.J.; Mazei-Robison, M.S. Reward Circuitry in Addiction. Neurotherapeutics 2017, 14, 687–697. [Google Scholar] [CrossRef]
- Galandra, C.; Basso, G.; Manera, M.; Crespi, C.; Giorgi, I.; Vittadini, G.; Poggi, P.; Canessa, N. Salience network structural integrity predicts executive impairment in alcohol use disorders. Sci. Rep. 2018, 8, 14481. [Google Scholar] [CrossRef] [PubMed]
- NIAAA. Alcohol Use Disorder (AUD) in the United States: Age Groups and Demographic Characteristics. Available online: https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics/alcohol-facts-and-statistics/alcohol-use-disorder-aud-united-states-age-groups-and-demographic-characteristics (accessed on 17 January 2025).
- SAMHSA. Racial/Ethnic Differences in Substance Use, Substance Use Disorders, and Substance Use Treatment Utilization Among People Aged 12 or Older (2015–2019); Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2021. [Google Scholar]
- Soto, C.; West, A.E.; Ramos, G.G.; Unger, J.B. Substance and Behavioral Addictions among American Indian and Alaska Native Populations. Int. J. Environ. Res. Public Health 2022, 19, 2974. [Google Scholar] [CrossRef] [PubMed]
- White, A.M. Gender Differences in the Epidemiology of Alcohol Use and Related Harms in the United States. Alcohol Res. 2020, 40, 01. [Google Scholar] [CrossRef]
- Castaldelli-Maia, J.M.; Segura, L.E.; Martins, S.S. The concerning increasing trend of alcohol beverage sales in the U.S. during the COVID-19 pandemic. Alcohol 2021, 96, 37–42. [Google Scholar] [CrossRef]
- White, A.M.; Castle, I.P.; Powell, P.A.; Hingson, R.W.; Koob, G.F. Alcohol-Related Deaths During the COVID-19 Pandemic. JAMA 2022, 327, 1704–1706. [Google Scholar] [CrossRef] [PubMed]
- CDC. Facts About U.S. Deaths from Excessive Alcohol Use. Available online: https://www.cdc.gov/alcohol/facts-stats/index.html (accessed on 19 May 2025).
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef]
- NIAAA. Medications Development Program. Available online: https://www.niaaa.nih.gov/medications-development-program (accessed on 19 May 2025).
- NIAAA. Alcohol Treatment in the United States. Available online: https://www.niaaa.nih.gov/alcohols-effects-health/alcohol-topics-z/alcohol-facts-and-statistics/alcohol-treatment-united-states (accessed on 19 May 2025).
- Venegas, A.; Donato, S.; Meredith, L.R.; Ray, L.A. Understanding low treatment seeking rates for alcohol use disorder: A narrative review of the literature and opportunities for improvement. Am. J. Drug Alcohol Abuse 2021, 47, 664–679. [Google Scholar] [CrossRef]
- Nguyen, L.C.; Durazzo, T.C.; Dwyer, C.L.; Rauch, A.A.; Humphreys, K.; Williams, L.M.; Padula, C.B. Predicting relapse after alcohol use disorder treatment in a high-risk cohort: The roles of anhedonia and smoking. J. Psychiatr. Res. 2020, 126, 1–7. [Google Scholar] [CrossRef]
- WHO. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 92-4-069886-8. [Google Scholar]
- Yan, M.; Man, S.; Sun, B.; Ma, L.; Guo, L.; Huang, L.; Gao, W. Gut liver brain axis in diseases: The implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Bhonchal, S.; Nain, C.K.; Prasad, K.K.; Nada, R.; Sharma, A.K.; Sinha, S.K.; Singh, K. Functional and morphological alterations in small intestine mucosa of chronic alcoholics. J. Gastroenterol. Hepatol. 2008, 23, e43–e48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, J.; Chang, B.; Wang, B.; Zhang, D. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol. Med. Rep. 2014, 9, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Oliviera, J.; Reygaert, W.C. Gram-Negative Bacteria; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar]
- Chen, G.; Shi, F.; Yin, W.; Guo, Y.; Liu, A.; Shuai, J.; Sun, J. Gut microbiota dysbiosis: The potential mechanisms by which alcohol disrupts gut and brain functions. Front. Microbiol. 2022, 13, 916765. [Google Scholar] [CrossRef]
- Rivera, C.A.; Bradford, B.U.; Seabra, V.; Thurman, R.G. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am. J. Physiol. 1998, 275, G1252–G1258. [Google Scholar] [CrossRef]
- Enomoto, N.; Yamashina, S.; Kono, H.; Schemmer, P.; Rivera, C.A.; Enomoto, A.; Nishiura, T.; Nishimura, T.; Brenner, D.A.; Thurman, R.G. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology 1999, 29, 1680–1689. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Toh, E.; Ross, R.A.; Heathers, L.E.; Chandler, K.; Oshodi, A.; McGee, B.; Modlik, E.; Linton, T.; Mangiacarne, D.; et al. Quantity of alcohol drinking positively correlates with serum levels of endotoxin and markers of monocyte activation. Sci. Rep. 2017, 7, 4462. [Google Scholar] [CrossRef]
- Kacimi, R.; Giffard, R.G.; Yenari, M.A. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. 2011, 8, 7. [Google Scholar] [CrossRef]
- Wheeler, M.D. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res. Health 2003, 27, 300–306. [Google Scholar] [PubMed]
- Kirpich, I.A.; McClain, C.J.; Vatsalya, V.; Schwandt, M.; Phillips, M.; Falkner, K.C.; Zhang, L.; Harwell, C.; George, D.T.; Umhau, J.C. Liver Injury and Endotoxemia in Male and Female Alcohol-Dependent Individuals Admitted to an Alcohol Treatment Program. Alcohol Clin. Exp. Res. 2017, 41, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ramkissoon, R.; Shah, V.H. Alcohol Use Disorder and Alcohol-Associated Liver Disease. Alcohol Res. 2022, 42, 13. [Google Scholar] [CrossRef]
- Contreras-Zentella, M.L.; Villalobos-García, D.; Hernández-Muñoz, R. Ethanol Metabolism in the Liver, the Induction of Oxidant Stress, and the Antioxidant Defense System. Antioxidants 2022, 11, 1258. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar]
- Lieber, C.S. Metabolic effects of acetaldehyde. Biochem. Soc. Trans. 1988, 16, 241–247. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161. [Google Scholar] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD + metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Anand, S.K.; Ahmad, M.H.; Sahu, M.R.; Subba, R.; Mondal, A.C. Detrimental Effects of Alcohol-Induced Inflammation on Brain Health: From Neurogenesis to Neurodegeneration. Cell. Mol. Neurobiol. 2023, 43, 1885–1904. [Google Scholar] [CrossRef]
- Khan, M.A.S.; Chang, S.L. Alcohol and the Brain-Gut Axis: The Involvement of Microglia and Enteric Glia in the Process of Neuro-Enteric Inflammation. Cells 2023, 12, 2475. [Google Scholar] [CrossRef]
- Liu, H.; Meng, L.; Wang, J.; Qin, C.; Feng, R.; Chen, Y.; Chen, P.; Zhu, Q.; Ma, M.; Teng, J.; et al. Enlarged perivascular spaces in alcohol-related brain damage induced by dyslipidemia. J. Cereb. Blood Flow Metab. 2024, 44, 1867–1880. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Hepatic encephalopathy. Alcohol Res. Health 2003, 27, 240–246. [Google Scholar]
- Martin, P.R.; Singleton, C.K.; Hiller-Sturmhöfel, S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res. Health 2003, 27, 134–142. [Google Scholar]
- Alexander-Kaufman, K.; Harper, C. Transketolase: Observations in alcohol-related brain damage research. Int. J. Biochem. Cell Biol. 2009, 41, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Polegato, B.F.; Pereira, A.G.; Azevedo, P.S.; Costa, N.A.; Zornoff, L.A.M.; Paiva, S.A.R.; Minicucci, M.F. Role of Thiamin in Health and Disease. Nutr. Clin. Pract. 2019, 34, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Arts, N.J.; Walvoort, S.J.; Kessels, R.P. Korsakoff’s syndrome: A critical review. Neuropsychiatr. Dis. Treat. 2017, 13, 2875–2890. [Google Scholar] [CrossRef]
- Morgan, M.Y. Alcohol and nutrition. Br. Med. Bull. 1982, 38, 21–29. [Google Scholar] [CrossRef]
- Jophlin, L.; Liu, T.Y.; McClain, C.J. Nutritional deficiencies in alcohol use disorder/alcohol-associated liver disease. Curr. Opin. Gastroenterol. 2024, 40, 112–117. [Google Scholar] [CrossRef]
- Langlais, P.J. Alcohol-Related Thiamine Deficiency: Impact on Cognitive and Memory Functioning. Alcohol Health Res. World 1995, 19, 113–121. [Google Scholar] [PubMed]
- Subramanya, S.B.; Subramanian, V.S.; Said, H.M. Chronic alcohol consumption and intestinal thiamin absorption: Effects on physiological and molecular parameters of the uptake process. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G23–G31. [Google Scholar] [CrossRef]
- Xu, H.; Liu, D.; Chen, J.; Li, H.; Xu, M.; Wen, W.; Frank, J.A.; Grahame, N.J.; Zhu, H.; Luo, J. Effects of Chronic Voluntary Alcohol Drinking on Thiamine Concentrations, Endoplasmic Reticulum Stress, and Oxidative Stress in the Brain of Crossed High Alcohol Preferring Mice. Neurotox. Res. 2019, 36, 777–787. [Google Scholar] [CrossRef]
- Laforenza, U.; Patrini, C.; Gastaldi, G.; Rindi, G. Effects of acute and chronic ethanol administration on thiamine metabolizing enzymes in some brain areas and in other organs of the rat. Alcohol Alcohol. 1990, 25, 591–603. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. Focal thalamic degeneration from ethanol and thiamine deficiency is associated with neuroimmune gene induction, microglial activation, and lack of monocarboxylic acid transporters. Alcohol Clin. Exp. Res. 2014, 38, 657–671. [Google Scholar] [CrossRef]
- He, X.; Sullivan, E.V.; Stankovic, R.K.; Harper, C.G.; Pfefferbaum, A. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology 2007, 32, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, P.J.; Self, R.L.; Stepanyan, T.D.; Little, H.J.; Littleton, J.M.; Prendergast, M.A. Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience 2005, 135, 1129–1139. [Google Scholar] [CrossRef]
- Chatterton, B.J.; Nunes, P.T.; Savage, L.M. The Effect of Chronic Ethanol Exposure and Thiamine Deficiency on Myelin-related Genes in the Cortex and the Cerebellum. Alcohol Clin. Exp. Res. 2020, 44, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Baker, H.; Sheu, K.F.; Gibson, G.E. Blood-brain barrier abnormalities in vulnerable brain regions during thiamine deficiency. Exp. Neurol. 1995, 134, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Qian, T.; Fei, G.; Cheng, X.; Zhao, L.; Sang, S.; Zhong, C. Low expression of thiamine pyrophosphokinase-1 contributes to brain susceptibility to thiamine deficiency. Neuroreport 2024, 35, 1000–1009. [Google Scholar] [CrossRef]
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawełczyk, T.; Wróblewska, M.; Szutowicz, A. Acetyl-CoA and acetylcholine metabolism in nerve terminal compartment of thiamine deficient rat brain. J. Neurochem. 2010, 115, 333–342. [Google Scholar] [CrossRef]
- Aikawa, H.; Watanabe, I.S.; Furuse, T.; Iwasaki, Y.; Satoyoshi, E.; Sumi, T.; Moroji, T. Low energy levels in thiamine-deficient encephalopathy. J. Neuropathol. Exp. Neurol. 1984, 43, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Donnino, M.W.; Carney, E.; Cocchi, M.N.; Barbash, I.; Chase, M.; Joyce, N.; Chou, P.P.; Ngo, L. Thiamine deficiency in critically ill patients with sepsis. J. Crit. Care 2010, 25, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Dhir, S.; Tarasenko, M.; Napoli, E.; Giulivi, C. Neurological, Psychiatric, and Biochemical Aspects of Thiamine Deficiency in Children and Adults. Front. Psychiatry 2019, 10, 207. [Google Scholar] [CrossRef]
- Hazell, A.S.; Butterworth, R.F. Update of cell damage mechanisms in thiamine deficiency: Focus on oxidative stress, excitotoxicity and inflammation. Alcohol Alcohol. 2009, 44, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Thiamin deficiency and brain disorders. Nutr. Res. Rev. 2003, 16, 277–284. [Google Scholar] [CrossRef]
- Jakkamsetti, V.; Marin-Valencia, I.; Ma, Q.; Good, L.B.; Terrill, T.; Rajasekaran, K.; Pichumani, K.; Khemtong, C.; Hooshyar, M.A.; Sundarrajan, C.; et al. Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci. Transl. Med. 2019, 11, eaan0457. [Google Scholar] [CrossRef]
- Clergue-Duval, V.; Questel, F.; Azuar, J.; Paquet, C.; Cognat, E.; Amami, J.; Queneau, M.; Dereux, A.; Barré, T.; Bellivier, F.; et al. Brain 18FDG-PET pattern in patients with alcohol-related cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 281–291. [Google Scholar] [CrossRef]
- Ritz, L.; Segobin, S.; Lannuzel, C.; Laniepce, A.; Boudehent, C.; Cabé, N.; Eustache, F.; Vabret, F.; Beaunieux, H.; Pitel, A.L. Cerebellar Hypermetabolism in Alcohol Use Disorder: Compensatory Mechanism or Maladaptive Plasticity? Alcohol Clin. Exp. Res. 2019, 43, 2212–2221. [Google Scholar] [CrossRef]
- Kashyap, B.; Hanson, L.R.; Frey Ii, W.H. Intranasal Insulin: A Treatment Strategy for Addiction. Neurotherapeutics 2020, 17, 105–115. [Google Scholar] [CrossRef]
- Gibson, G.E.; Ksiezak-Reding, H.; Sheu, K.F.; Mykytyn, V.; Blass, J.P. Correlation of enzymatic, metabolic, and behavioral deficits in thiamin deficiency and its reversal. Neurochem. Res. 1984, 9, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.; Butterworth, R.F. Reduced activities of thiamine-dependent enzymes in brains of alcoholics in the absence of Wernicke’s encephalopathy. Alcohol Clin. Exp. Res. 1995, 19, 1073–1077. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Kril, J.J.; Harper, C.G. Thiamine-dependent enzyme changes in the brains of alcoholics: Relationship to the Wernicke-Korsakoff syndrome. Alcohol Clin. Exp. Res. 1993, 17, 1084–1088. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, X.; Zhao, J.; Wang, Y.; Peng, Y.; Zhong, C. Decreased transketolase activity contributes to impaired hippocampal neurogenesis induced by thiamine deficiency. J. Neurochem. 2009, 111, 537–546. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bist, R.; Bubber, P. Thiamine deficiency induces oxidative stress in brain mitochondria of Mus musculus. J. Physiol. Biochem. 2013, 69, 539–546. [Google Scholar] [CrossRef]
- Langlais, P.J.; Anderson, G.; Guo, S.X.; Bondy, S.C. Increased cerebral free radical production during thiamine deficiency. Metab. Brain Dis. 1997, 12, 137–143. [Google Scholar] [CrossRef]
- Viña, J.; Estrela, J.M.; Guerri, C.; Romero, F.J. Effect of ethanol on glutathione concentration in isolated hepatocytes. Biochem. J. 1980, 188, 549–552. [Google Scholar] [CrossRef]
- Vasan, S.; Kumar, A. Wernicke Encephalopathy; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ciccia, R.M.; Langlais, P.J. An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcohol Clin. Exp. Res. 2000, 24, 622–634. [Google Scholar]
- Vedder, L.C.; Hall, J.M.; Jabrouin, K.R.; Savage, L.M. Interactions between chronic ethanol consumption and thiamine deficiency on neural plasticity, spatial memory, and cognitive flexibility. Alcohol Clin. Exp. Res. 2015, 39, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Listabarth, S.; Vyssoki, B.; Marculescu, R.; Gleiss, A.; Groemer, M.; Trojer, A.; Harrer, C.; Weber, S.; König, D. Can thiamine substitution restore cognitive function in alcohol use disorder? Alcohol Alcohol. 2023, 58, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.L.; Bowden, S.C.; Whelan, G. Thiamin treatment and working memory function of alcohol-dependent people: Preliminary findings. Alcohol Clin. Exp. Res. 2001, 25, 112–116. [Google Scholar]
- Kopelman, M.D.; Thomson, A.D.; Guerrini, I.; Marshall, E.J. The Korsakoff syndrome: Clinical aspects, psychology and treatment. Alcohol Alcohol. 2009, 44, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Fama, R. Wernicke’s encephalopathy and Korsakoff’s syndrome revisited. Neuropsychol. Rev. 2012, 22, 69–71. [Google Scholar] [CrossRef]
- Eva, L.; Brehar, F.M.; Florian, I.A.; Covache-Busuioc, R.A.; Costin, H.P.; Dumitrascu, D.I.; Bratu, B.G.; Glavan, L.A.; Ciurea, A.V. Neuropsychiatric and Neuropsychological Aspects of Alcohol-Related Cognitive Disorders: An In-Depth Review of Wernicke’s Encephalopathy and Korsakoff’s Syndrome. J. Clin. Med. 2023, 12, 6101. [Google Scholar] [CrossRef]
- Szabo, G.; Saha, B. Alcohol’s Effect on Host Defense. Alcohol Res. 2015, 37, 159–170. [Google Scholar]
- Afshar, M.; Richards, S.; Mann, D.; Cross, A.; Smith, G.B.; Netzer, G.; Kovacs, E.; Hasday, J. Acute immunomodulatory effects of binge alcohol ingestion. Alcohol 2015, 49, 57–64. [Google Scholar] [CrossRef]
- Wang, H.J.; Zakhari, S.; Jung, M.K. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J. Gastroenterol. 2010, 16, 1304–1313. [Google Scholar] [CrossRef]
- Zillich, L.; Poisel, E.; Frank, J.; Foo, J.C.; Friske, M.M.; Streit, F.; Sirignano, L.; Heilmann-Heimbach, S.; Heimbach, A.; Hoffmann, P.; et al. Multi-omics signatures of alcohol use disorder in the dorsal and ventral striatum. Transl. Psychiatry 2022, 12, 190. [Google Scholar] [CrossRef]
- Adams, C.; Conigrave, J.H.; Lewohl, J.; Haber, P.; Morley, K.C. Alcohol use disorder and circulating cytokines: A systematic review and meta-analysis. Brain Behav. Immun. 2020, 89, 501–512. [Google Scholar] [CrossRef]
- Moura, H.F.; Hansen, F.; Galland, F.; Silvelo, D.; Rebelatto, F.P.; Ornell, F.; Massuda, R.; Scherer, J.N.; Schuch, F.; Kessler, F.H.; et al. Inflammatory cytokines and alcohol use disorder: Systematic review and meta-analysis. Braz. J. Psychiatry 2022, 44, 548–556. [Google Scholar] [CrossRef]
- Torres, S.; Segalés, P.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells 2022, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Meredith, L.R.; Burnette, E.M.; Grodin, E.N.; Irwin, M.R.; Ray, L.A. Immune treatments for alcohol use disorder: A translational framework. Brain Behav. Immun. 2021, 97, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, W.; Li, Y.; Zhai, J. The SIRT1-HMGB1 axis: Therapeutic potential to ameliorate inflammatory responses and tumor occurrence. Front. Cell Dev. Biol. 2022, 10, 986511. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef]
- Crews, F.T.; Qin, L.; Sheedy, D.; Vetreno, R.P.; Zou, J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatry 2013, 73, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Vetreno, R.P.; Crews, F.T. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience 2012, 226, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Antón, M.; Alén, F.; Gómez de Heras, R.; Serrano, A.; Pavón, F.J.; Leza, J.C.; García-Bueno, B.; Rodríguez de Fonseca, F.; Orio, L. Oleoylethanolamide prevents neuroimmune HMGB1/TLR4/NF-kB danger signaling in rat frontal cortex and depressive-like behavior induced by ethanol binge administration. Addict. Biol. 2017, 22, 724–741. [Google Scholar] [CrossRef]
- Orio, L.; Antón, M.; Rodríguez-Rojo, I.C.; Correas, Á.; García-Bueno, B.; Corral, M.; de Fonseca, F.R.; García-Moreno, L.M.; Maestú, F.; Cadaveira, F. Young alcohol binge drinkers have elevated blood endotoxin, peripheral inflammation and low cortisol levels: Neuropsychological correlations in women. Addict. Biol. 2018, 23, 1130–1144. [Google Scholar] [CrossRef]
- Farhana, A.; Khan, Y.S. Biochemistry, Lipopolysaccharide; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Zou, J.Y.; Crews, F.T. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef]
- Mu, S.; Li, Z.; Lin, L.; Wang, D.; Yang, F.; Chen, L.; Xian, L.; Lin, K.; Lin, Y.; Ye, D.; et al. SIRT1-Mediated HMGB1 Deacetylation Suppresses Neutrophil Extracellular Traps Related to Blood-Brain Barrier Impairment After Cerebral Venous Thrombosis. Mol. Neurobiol. 2024, 61, 6060–6076. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Leo, M.A.; Wang, X.; Decarli, L.M. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem. Biophys. Res. Commun. 2008, 370, 44–48. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Czerwińska-Błaszczyk, A.; Pawlak, E.; Pawłowski, T. The Significance of Toll-Like Receptors in the Neuroimmunologic Background of Alcohol Dependence. Front. Psychiatry 2021, 12, 797123. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef]
- Holloway, K.N.; Douglas, J.C.; Rafferty, T.M.; Kane, C.J.M.; Drew, P.D. Ethanol Induces Neuroinflammation in a Chronic Plus Binge Mouse Model of Alcohol Use Disorder via TLR4 and MyD88-Dependent Signaling. Cells 2023, 12, 2109. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Baliño, P.; Aragón, C.M.; Guerri, C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of TLR4 and TLR2. Neuropharmacology 2015, 89, 352–359. [Google Scholar] [CrossRef]
- Blanco, A.M.; Vallés, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Baliño, P.; Alfonso-Loeches, S.; Aragón, C.M.; Guerri, C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav. Immun. 2011, 25 (Suppl. S1), S80–S91. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.; Pascual, M.; Pla, A.; Maldonado, C.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. TLR4 elimination prevents synaptic and myelin alterations and long-term cognitive dysfunctions in adolescent mice with intermittent ethanol treatment. Brain Behav. Immun. 2015, 45, 233–244. [Google Scholar] [CrossRef]
- Wu, Y.; Lousberg, E.L.; Moldenhauer, L.M.; Hayball, J.D.; Coller, J.K.; Rice, K.C.; Watkins, L.R.; Somogyi, A.A.; Hutchinson, M.R. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br. J. Pharmacol. 2012, 165, 1319–1329. [Google Scholar] [CrossRef]
- Vore, A.S.; Deak, T. Alcohol, inflammation, and blood-brain barrier function in health and disease across development. Int. Rev. Neurobiol. 2022, 161, 209–249. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Pascual, M.; Gómez-Pinedo, U.; Pascual-Lucas, M.; Renau-Piqueras, J.; Guerri, C. Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia 2012, 60, 948–964. [Google Scholar] [CrossRef]
- Pascual, M.; Montesinos, J.; Montagud-Romero, S.; Forteza, J.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J. Neuroinflamm. 2017, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of blood-brain barrier integrity in postmortem alcoholic brain: Preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef]
- June, H.L.; Liu, J.; Warnock, K.T.; Bell, K.A.; Balan, I.; Bollino, D.; Puche, A.; Aurelian, L. CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 2015, 40, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, A.R.; Kelly, T.; Puche, A.; Esoga, C.; June, H.L.; Elnabawi, A.; Merchenthaler, I.; Sieghart, W.; Aurelian, L. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc. Natl. Acad. Sci. USA 2011, 108, 4465–4470. [Google Scholar] [CrossRef]
- Balan, I.; Warnock, K.T.; Puche, A.; Gondre-Lewis, M.C.; June, H.; Aurelian, L. The GABA A Receptor α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and Nicotine Abuse. Brain Sci. 2018, 8, 72. [Google Scholar] [CrossRef]
- Lippai, D.; Bala, S.; Petrasek, J.; Csak, T.; Levin, I.; Kurt-Jones, E.A.; Szabo, G. Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J. Leukoc. Biol. 2013, 94, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef]
- Rosenberger, K.; Derkow, K.; Dembny, P.; Krüger, C.; Schott, E.; Lehnardt, S. The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J. Neuroinflamm. 2014, 11, 166. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Chernis, J.; Da Costa, A.; Mayfield, J.; Harris, R.A. Ethanol Consumption in Mice Lacking CD14, TLR2, TLR4, or MyD88. Alcohol Clin. Exp. Res. 2017, 41, 516–530. [Google Scholar] [CrossRef]
- Pang, M.; Bala, S.; Kodys, K.; Catalano, D.; Szabo, G. Inhibition of TLR8- and TLR4-induced Type I IFN induction by alcohol is different from its effects on inflammatory cytokine production in monocytes. BMC Immunol. 2011, 12, 55. [Google Scholar] [CrossRef]
- Gustot, T.; Lemmers, A.; Moreno, C.; Nagy, N.; Quertinmont, E.; Nicaise, C.; Franchimont, D.; Louis, H.; Devière, J.; Le Moine, O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006, 43, 989–1000. [Google Scholar] [CrossRef]
- Makoni, N.J.; Nichols, M.R. The intricate biophysical puzzle of caspase-1 activation. Arch. Biochem. Biophys. 2021, 699, 108753. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.L.; Roodsari, S.K.; Cheng, Y.; Dempsey, R.E.; Hu, W. Microglia NLRP3 Inflammasome and Neuroimmune Signaling in Substance Use Disorders. Biomolecules 2023, 13, 922. [Google Scholar] [CrossRef]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Anton, P.E.; Rutt, L.N.; Kaufman, M.L.; Busquet, N.; Kovacs, E.J.; McCullough, R.L. Binge ethanol exposure in advanced age elevates neuroinflammation and early indicators of neurodegeneration and cognitive impairment in female mice. Brain Behav. Immun. 2024, 116, 303–316. [Google Scholar] [CrossRef]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Ureña-Peralta, J.R.; Morillo-Bargues, M.J.; Oliver-De La Cruz, J.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell. Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Ureña-Peralta, J.; Morillo-Bargues, M.J.; Gómez-Pinedo, U.; Guerri, C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem. Res. 2016, 41, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.P.; Cho, Y.; Tornai, D.; Coban, S.; Catalano, D.; Szabo, G. Inhibition of the Inflammasome Signaling Cascade Reduces Alcohol Consumption in Female But Not Male Mice. Alcohol Clin. Exp. Res. 2020, 44, 567–578. [Google Scholar] [CrossRef]
- Li, Z.; Vidjro, O.E.; Guo, G.; Du, Y.; Zhou, Y.; Xie, Q.; Li, J.; Gao, K.; Zhou, L.; Ma, T. NLRP3 deficiency decreases alcohol intake controlling anxiety-like behavior via modification of glutamatergic transmission in corticostriatal circuits. J. Neuroinflamm. 2022, 19, 308. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Kellar, D.; Lake, A.; Finn, P.; Rebec, G.V.; Dharmadhikari, S.; Dydak, U.; Newman, S. Effects of Alcohol Cues on MRS Glutamate Levels in the Anterior Cingulate. Alcohol Alcohol. 2018, 53, 209–215. [Google Scholar] [CrossRef]
- Nurmi, K.; Virkanen, J.; Rajamäki, K.; Niemi, K.; Kovanen, P.T.; Eklund, K.K. Ethanol inhibits activation of NLRP3 and AIM2 inflammasomes in human macrophages--a novel anti-inflammatory action of alcohol. PLoS ONE 2013, 8, e78537. [Google Scholar] [CrossRef]
- Vemuganti, R.; Kalluri, H.; Yi, J.H.; Bowen, K.K.; Hazell, A.S. Gene expression changes in thalamus and inferior colliculus associated with inflammation, cellular stress, metabolism and structural damage in thiamine deficiency. Eur. J. Neurosci. 2006, 23, 1172–1188. [Google Scholar] [CrossRef]
- Wang, D.; Hazell, A.S. Microglial activation is a major contributor to neurologic dysfunction in thiamine deficiency. Biochem. Biophys. Res. Commun. 2010, 402, 123–128. [Google Scholar] [CrossRef]
- Toledo Nunes, P.; Vedder, L.C.; Deak, T.; Savage, L.M. A Pivotal Role for Thiamine Deficiency in the Expression of Neuroinflammation Markers in Models of Alcohol-Related Brain Damage. Alcohol Clin. Exp. Res. 2019, 43, 425–438. [Google Scholar] [CrossRef]
- Ke, Z.J.; Calingasan, N.Y.; DeGiorgio, L.A.; Volpe, B.T.; Gibson, G.E. CD40-CD40L interactions promote neuronal death in a model of neurodegeneration due to mild impairment of oxidative metabolism. Neurochem. Int. 2005, 47, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, G.; Li, Y.; Pan, Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front. Immunol. 2022, 13, 840029. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.; San Felipe, D.; Ballesta, A.; Alén, F.; Rodríguez de Fonseca, F.; García-Bueno, B.; Marco, E.M.; Orio, L. Cerebellar and cortical TLR4 activation and behavioral impairments in Wernicke-Korsakoff Syndrome: Pharmacological effects of oleoylethanolamide. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 108, 110190. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.; Escudero, B.; Gómez-Blázquez, E.; Rebolledo-Poves, A.B.; López-Gallardo, M.; Guerrero, C.; Marco, E.M.; Orio, L. Upregulation of TLR4/MyD88 pathway in alcohol-induced Wernicke’s encephalopathy: Findings in preclinical models and in a postmortem human case. Front. Pharmacol. 2022, 13, 866574. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, L.; Qiu, H.; Qian, T.; Sang, S.; Zhong, C. The impact of thiamine deficiency and benfotiamine treatment on Nod-like receptor protein-3 inflammasome in microglia. Neuroreport 2021, 32, 1041–1048. [Google Scholar] [CrossRef]
- Page, M.G.; Ankoma-Sey, V.; Coulson, W.F.; Bender, D.A. Brain glutamate and gamma-aminobutyrate (GABA) metabolism in thiamin-deficient rats. Br. J. Nutr. 1989, 62, 245–253. [Google Scholar] [CrossRef]
- Mayfield, J.; Arends, M.A.; Harris, R.A.; Blednov, Y.A. Genes and Alcohol Consumption: Studies with Mutant Mice. Int. Rev. Neurobiol. 2016, 126, 293–355. [Google Scholar] [CrossRef]
- Warden, A.S.; Triplett, T.A.; Lyu, A.; Grantham, E.K.; Azzam, M.M.; DaCosta, A.; Mason, S.; Blednov, Y.A.; Ehrlich, L.I.R.; Mayfield, R.D.; et al. Microglia depletion and alcohol: Transcriptome and behavioral profiles. Addict. Biol. 2021, 26, e12889. [Google Scholar] [CrossRef]
- Hazell, A.S.; Sheedy, D.; Oanea, R.; Aghourian, M.; Sun, S.; Jung, J.Y.; Wang, D.; Wang, C. Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 2010, 58, 148–156. [Google Scholar] [CrossRef]
- Wang, K.; Yang, L.; Li, Q.; Yang, X.; Chen, Z.; Zhou, Y.; Jia, Y.; Gong, Z. Long-Term Alcohol Exposure Aggravates Ischemic Stroke-Induced Damage by Promoting Pericyte NLRP3 Inflammasome Activation via Pre-Activating the TLR4/NF-κB Pathway in Rats. J. Inflamm. Res. 2024, 17, 4791–4810. [Google Scholar] [CrossRef]
- Liu, S.; Wu, J.; Chen, P.; Mohammed, S.A.D.; Zhang, J. TAK-242 Ameliorates Hepatic Fibrosis by Regulating the Liver-Gut Axis. Biomed. Res. Int. 2022, 2022, 4949148. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ju, Y.; Wu, Q.; Sun, G.; Yan, Z. TAK-242, a toll-like receptor 4 antagonist, against brain injury by alleviates autophagy and inflammation in rats. Open Life Sci. 2023, 18, 20220662. [Google Scholar] [CrossRef]
- Deschamps, C.; Uyttersprot, F.; Debris, M.; Marié, C.; Fouquet, G.; Marcq, I.; Vilpoux, C.; Naassila, M.; Pierrefiche, O. Anti-inflammatory drugs prevent memory and hippocampal plasticity deficits following initial binge-like alcohol exposure in adolescent male rats. Psychopharmacology 2022, 239, 2245–2262. [Google Scholar] [CrossRef]
- Kasanetz, F.; Deroche-Gamonet, V.; Berson, N.; Balado, E.; Lafourcade, M.; Manzoni, O.; Piazza, P.V. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 2010, 328, 1709–1712. [Google Scholar] [CrossRef]
- Fu, Q.; Li, J.; Qiu, L.; Ruan, J.; Mao, M.; Li, S.; Mao, Q. Inhibiting NLRP3 inflammasome with MCC950 ameliorates perioperative neurocognitive disorders, suppressing neuroinflammation in the hippocampus in aged mice. Int. Immunopharmacol. 2020, 82, 106317. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Savic, D.; Laketa, D.; Bjelobaba, I.; Milenkovic, I.; Pekovic, S.; Nedeljkovic, N.; Lavrnja, I. Benfotiamine attenuates inflammatory response in LPS stimulated BV-2 microglia. PLoS ONE 2015, 10, e0118372. [Google Scholar] [CrossRef] [PubMed]
- Portari, G.V.; Ovidio, P.P.; Deminice, R.; Jordão, A.A. Protective effect of treatment with thiamine or benfotiamine on liver oxidative damage in rat model of acute ethanol intoxication. Life Sci. 2016, 162, 21–24. [Google Scholar] [CrossRef]
- Manzardo, A.M.; He, J.; Poje, A.; Penick, E.C.; Campbell, J.; Butler, M.G. Double-blind, randomized placebo-controlled clinical trial of benfotiamine for severe alcohol dependence. Drug Alcohol Depend. 2013, 133, 562–570. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Zhang, L.; Sun, Y.; Liang, Y.; Li, H.; Zhang, Y. Role of trigger receptor 2 expressed on myeloid cells in neuroinflammation-neglected multidimensional regulation of microglia. Neurochem. Int. 2023, 171, 105639. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef]
- Cantoni, C.; Bollman, B.; Licastro, D.; Xie, M.; Mikesell, R.; Schmidt, R.; Yuede, C.M.; Galimberti, D.; Olivecrona, G.; Klein, R.S.; et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015, 129, 429–447. [Google Scholar] [CrossRef] [PubMed]
- van Lengerich, B.; Zhan, L.; Xia, D.; Chan, D.; Joy, D.; Park, J.I.; Tatarakis, D.; Calvert, M.; Hummel, S.; Lianoglou, S.; et al. A TREM2-activating antibody with a blood-brain barrier transport vehicle enhances microglial metabolism in Alzheimer’s disease models. Nat. Neurosci. 2023, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Mirescu, C.; Dejanovic, B.; Larson, K.C.; Kiragasi, B.; Gergits, F.W.; Figley, M.D.; Renoux, A.J.; Huang, L.; Pandya, B.A.; Houze, J.B.; et al. Pharmacological and functional characterization of the first small molecule TREM2 agonist, VG-3927, for the treatment of Alzheimer’s disease. Alzheimer’s Dement. 2025, 20, e089318. [Google Scholar] [CrossRef]
- Crews, F.T.; Zou, J.; Coleman, L.G. Extracellular microvesicles promote microglia-mediated pro-inflammatory responses to ethanol. J. Neurosci. Res. 2021, 99, 1940–1956. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.E.; Alicea Pauneto, C.D.M.; Barnett, A.M.; Coleman, L.G. Chronic Ethanol Causes Persistent Increases in Alzheimer’s Tau Pathology in Female 3xTg-AD Mice: A Potential Role for Lysosomal Impairment. Front. Behav. Neurosci. 2022, 16, 886634. [Google Scholar] [CrossRef]
- Lan, L.; Wang, H.; Zhang, X.; Shen, Q.; Li, X.; He, L.; Rong, X.; Peng, J.; Mo, J.; Peng, Y. Chronic exposure of alcohol triggers microglia-mediated synaptic elimination inducing cognitive impairment. Exp. Neurol. 2022, 353, 114061. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalapatapu, N.; Skinner, S.G.; D’Addezio, E.G.; Ponna, S.; Cadenas, E.; Davies, D.L. Thiamine Deficiency and Neuroinflammation Are Important Contributors to Alcohol Use Disorder. Pathophysiology 2025, 32, 34. https://doi.org/10.3390/pathophysiology32030034
Kalapatapu N, Skinner SG, D’Addezio EG, Ponna S, Cadenas E, Davies DL. Thiamine Deficiency and Neuroinflammation Are Important Contributors to Alcohol Use Disorder. Pathophysiology. 2025; 32(3):34. https://doi.org/10.3390/pathophysiology32030034
Chicago/Turabian StyleKalapatapu, Nikhila, Samantha G. Skinner, Emma G. D’Addezio, Srija Ponna, Enrique Cadenas, and Daryl L. Davies. 2025. "Thiamine Deficiency and Neuroinflammation Are Important Contributors to Alcohol Use Disorder" Pathophysiology 32, no. 3: 34. https://doi.org/10.3390/pathophysiology32030034
APA StyleKalapatapu, N., Skinner, S. G., D’Addezio, E. G., Ponna, S., Cadenas, E., & Davies, D. L. (2025). Thiamine Deficiency and Neuroinflammation Are Important Contributors to Alcohol Use Disorder. Pathophysiology, 32(3), 34. https://doi.org/10.3390/pathophysiology32030034