Mutation and Microsatellite Instability (MSI) Affect the Differential Gene Expression of Folic Acid and 5-Flourouracil Metabolism-Related Genes in Colorectal Carcinoma

 , , , and
, , , and
Simple Summary
Abstract
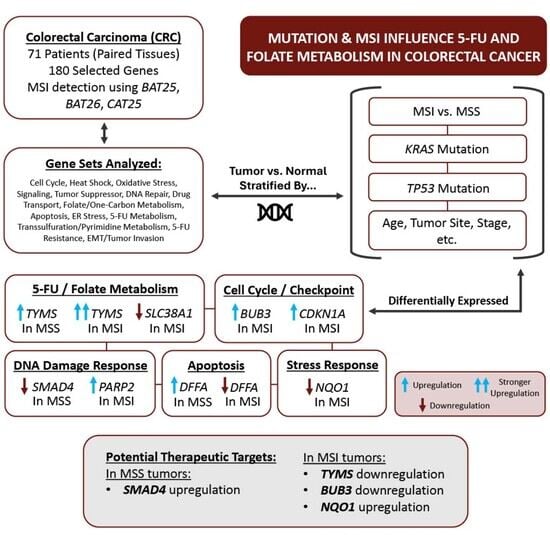
1. Introduction
2. Materials and Methods
2.1. Gene Selection and Functional Relevance
2.2. MSI Detection
2.3. Statistical Analysis
3. Results
3.1. Association of MSI Status on Differential Expression of Genes
3.2. Association of MSI Status and KRAS Mutation Status on Differential Expression of Genes
3.3. Association of Age of Onset (<40 Years and >40 Years) on Differential Expression of Genes
4. Discussion
4.1. Folate and One-Carbon Metabolism
4.2. Cell Cycle Regulation and Checkpoint Control
4.3. DNA Damage Response and Repair
4.4. Apoptosis Regulation
4.5. Stress Response Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| CRC | Colorectal Cancer |
| MSI | Microsatellite Instability |
| MSS | Microsatellite Stable |
| FA | Folic Acid |
| CI | Confidence Interval |
| ACS | American Cancer Society |
| mCRC | Metastatic Colorectal Cancer |
| DNA | Deoxyribonucleic Acid |
| RNA | Ribonucleic Acid |
| EMT | Epithelial–Mesenchymal Transition |
| TIL | Tumor-Infiltrating Lymphocyte |
| MMR | Mismatch Repair |
| MMR-D | Mismatch Repair Deficiency |
| MSI-H | MSI-High |
| BSMMU | Bangabandhu Sheikh Mujib Medical University |
| ER | Endoplasmic Reticulum |
| HRM | High Resolution Melt |
| PCR | Polymerase Chain Reaction |
| ANOVA | Analysis of Variance |
| FC | Fold Change |
| GO | Gene Ontology |
| FDR | False Discovery Rate |
| KRAS | Kristen Rat Sarcoma Virus |
| EOCRC | Early-Onset CRC |
| LOCRC | Late-Onset CRC |
| PEMT | Phosphatidylethanolamine Methyltransferase |
| FdUMP | Fluorodeoxyuridylate |
| TYMS | Thymidylate Synthase |
| SHMT1 | Serine Hydroxymethyltransferase 1 |
| SLC38A1 | Solute Carrier Family 38 Member 1 |
| BER | Base Excision Repair |
| DFF | DNA Fragmentation Factor Complex |
References
- Kandel, A.; Thida, A.M.; Preet, M. A Review of the Early Detection of Colon Cancer and the Role of Circulating Tumor DNA. Cureus 2025, 17, e84394. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdelgadir, O.; Kuo, Y.F.; Khan, M.F.; Okorodudu, A.O.; Cheng, Y.W.; Dong, J. Mortality Outcome Associated with Specific KRAS, NRAS, and BRAF Hot-Spot Mutations in Metastatic Colorectal Cancer Patients: A Retrospective Cohort Study. Diagnostics 2025, 15, 590. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chan, G.H.J.; Chee, C.E. Making sense of adjuvant chemotherapy in colorectal cancer. J. Gastrointest. Oncol. 2019, 10, 1183–1192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gmeiner, W.H.; Okechukwu, C.C. Review of 5-FU resistance mechanisms in colorectal cancer: Clinical significance of attenuated on-target effects. Cancer Drug Resist. 2023, 6, 257–272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Długosz-Pokorska, A.; Pięta, M.; Janecki, T.; Janecka, A. New uracil analogs as downregulators of ABC transporters in 5-fluorouracil-resistant human leukemia HL-60 cell line. Mol. Biol. Rep. 2019, 46, 5831–5839. [Google Scholar] [CrossRef] [PubMed]
- Sethy, C.; Kundu, C.N. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomed. Pharmacother. 2021, 137, 111285. [Google Scholar] [CrossRef] [PubMed]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barathan, M.; Zulpa, A.K.; Ng, S.L.; Lokanathan, Y.; Ng, M.H.; Law, J.X. Innovative Strategies to Combat 5-Fluorouracil Resistance in Colorectal Cancer: The Role of Phytochemicals and Extracellular Vesicles. Int J Mol Sci. 2024, 25, 7470. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ghafouri-Fard, S.; Abak, A.; Tondro Anamag, F.; Shoorei, H.; Fattahi, F.; Javadinia, S.A.; Basiri, A.; Taheri, M. 5-Fluorouracil: A Narrative Review on the Role of Regulatory Mechanisms in Driving Resistance to This Chemotherapeutic Agent. Front. Oncol. 2021, 11, 658636. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- LaCourse, K.D.; Zepeda-Rivera, M.; Kempchinsky, A.G.; Baryiames, A.; Minot, S.S.; Johnston, C.D.; Bullman, S. The cancer chemotherapeutic 5-fluorouracil is a potent Fusobacterium nucleatum inhibitor and its activity is modified by intratumoral microbiota. Cell Rep. 2022, 41, 111625. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seiple, L.; Jaruga, P.; Dizdaroglu, M.; Stivers, J.T. Linking uracil base excision repair and 5-fluorouracil toxicity in yeast. Nucleic Acids Res. 2006, 34, 140–151. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Showalter, S.L.; Showalter, T.N.; Witkiewicz, A.; Havens, R.; Kennedy, E.P.; Hucl, T.; Kern, S.E.; Yeo, C.J.; Brody, J.R. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol. Ther. 2008, 7, 986–994. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Azwar, S.; Seow, H.F.; Abdullah, M.; Faisal Jabar, M.; Mohtarrudin, N. Recent Updates on Mechanisms of Resistance to 5-Fluorouracil and Reversal Strategies in Colon Cancer Treatment. Biology 2021, 10, 854. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zsigrai, S.; Kalmár, A.; Barták, B.K.; Nagy, Z.B.; Szigeti, K.A.; Valcz, G.; Kothalawala, W.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. Folic Acid Treatment Directly Influences the Genetic and Epigenetic Regulation along with the Associated Cellular Maintenance Processes of HT-29 and SW480 Colorectal Cancer Cell Lines. Cancers 2022, 14, 1820. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Danenberg, P.V.; Gustavsson, B.; Johnston, P.; Lindberg, P.; Moser, R.; Odin, E.; Peters, G.J.; Petrelli, N. Folates as adjuvants to anticancer agents: Chemical rationale and mechanism of action. Crit. Rev. Oncol. Hematol. 2016, 106, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.B.; Lincz, L.F.; Adler, K.; Scorgie, F.E.; Ackland, S.P.; Sakoff, J.A. Predicting 5-fluorouracil toxicity in colorectal cancer patients from peripheral blood cell telomere length: A multivariate analysis. Br. J. Cancer 2012, 107, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gmeiner, W.H. Fluoropyrimidine Modulation of the Anti-Tumor Immune Response-Prospects for Improved Colorectal Cancer Treatment. Cancers 2020, 12, 1641. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jang, H.Y.; Kim, D.H.; Lee, H.J.; Kim, W.D.; Kim, S.Y.; Hwang, J.J.; Lee, S.J.; Moon, D.H. Schedule-dependent synergistic effects of 5-fluorouracil and selumetinib in KRAS or BRAF mutant colon cancer models. Biochem. Pharmacol. 2019, 160, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Klingbiel, D.; Saridaki, Z.; Roth, A.D.; Bosman, F.T.; Delorenzi, M.; Tejpar, S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: Results of the PETACC-3 trial. Ann. Oncol. 2015, 26, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Thrall, M.M.; Wood, P.; King, V.; Rivera, W.; Hrushesky, W. Investigation of the comparative toxicity of 5-FU bolus versus 5-FU continuous infusion circadian chemotherapy with concurrent radiation therapy in locally advanced rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Zalcberg, J.; Kerr, D.; Seymour, L.; Palmer, M. Haematological and non-haematological toxicity after 5-fluorouracil and leucovorin in patients with advanced colorectal cancer is significantly associated with gender, increasing age and cycle number. Tomudex International Study Group. Eur. J. Cancer 1998, 34, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Richman, S. Deficient mismatch repair: Read all about it (Review). Int. J. Oncol. 2015, 47, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, W.; Frankel, W.L. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod. Pathol. 2019, 32 (Suppl. S1), 1–15. [Google Scholar] [CrossRef] [PubMed]
- Battaglin, F.; Naseem, M.; Lenz, H.J.; Salem, M.E. Microsatellite instability in colorectal cancer: Overview of its clinical significance and novel perspectives. Clin. Adv. Hematol. Oncol. 2018, 16, 735–745. [Google Scholar] [PubMed] [PubMed Central]
- Gutierrez, C.; Ogino, S.; Meyerhardt, J.A.; Iorgulescu, J.B. The Prevalence and Prognosis of Microsatellite Instability-High/Mismatch Repair-Deficient Colorectal Adenocarcinomas in the United States. JCO Precis. Oncol. 2023, 7, e2200179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tomasello, G.; Ghidini, M.; Galassi, B.; Grossi, F.; Luciani, A.; Petrelli, F. Survival benefit with adjuvant chemotherapy in stage III microsatellite-high/deficient mismatch repair colon cancer: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 1055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jasmine, F.; Rahaman, R.; Dodsworth, C.; Roy, S.; Paul, R.; Raza, M.; Paul-Brutus, R.; Kamal, M.; Ahsan, H.; Kibriya, M.G. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: Opportunities for future personalized treatment. PLoS ONE 2012, 7, e31968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Pekow, J.; Munoz, A.; Weber, C.; Raza, M.; Kamal, M.; Ahsan, H.; Bissonnette, M. Pathways Related to Colon Inflammation Are Associated with Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study. Cancers 2023, 15, 2921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Raza, M.; Jasmine, F.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Dodsworth, C.; Rakibuz-Zaman, M.; Kamal, M.; Ahsan, H. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med. Genom. 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Raza, M.; Kamal, M.; Haq, Z.; Paul, R.; Mareczko, A.; Pierce, B.L.; Ahsan, H.; Jasmine, F. Relative Telomere Length Change in Colorectal Carcinoma and Its Association with Tumor Characteristics, Gene Expression and Microsatellite Instability. Cancers 2022, 14, 2250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Wei, Q.; Chen, Y.; Long, S.; Yao, Y.; Fu, K. Identification of Hub Genes Associated with Sensitivity of 5-Fluorouracil Based Chemotherapy for Colorectal Cancer by Integrated Bioinformatics Analysis. Front. Oncol. 2021, 11, 604315. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fulcher, L.J.; Batley, C.; Sobajima, T.; Barr, F.A. Time as a danger signal promoting G1 arrest after mitosis. Trends Cell Biol. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S phase cell-cycle genomic alterations and accompanying co-alterations with individualized CDK4/6 inhibitor-based regimens. JCI Insight 2021, 6, e142547. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manandhar, M.; Boulware, K.S.; Wood, R.D. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene 2015, 569, 153–161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miyashita, K.; Shioi, S.; Kajitani, T.; Koi, Y.; Shimokawa, M.; Makiyama, A.; Oda, S.; Esaki, T. More subtle microsatellite instability better predicts fluorouracil insensitivity in colorectal cancer patients. Sci. Rep. 2024, 14, 27257. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Rohde, L.H.; Wu, H. Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair. Curr. Genom. 2009, 10, 250–258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, Y.K.; Kim, M.J.; Kim, M.S.; Lee, J.H.; Chung, S.J.; Song, J.S.; Chae, Y.J.; Lee, K.R. Role of the efflux transporters Abcb1 and Abcg2 in the brain distribution of olaparib in mice. Eur. J. Pharm. Sci. 2022, 173, 106177. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.; Sun, X.; Sun, J.; Zhang, J.; Wang, Y. Epithelial-mesenchymal transition in colorectal cancer metastasis and progression: Molecular mechanisms and therapeutic strategies. Cell Death Discov. 2025, 11, 336. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Somu, P.; Mohanty, S.; Basavegowda, N.; Yadav, A.K.; Paul, S.; Baek, K.H. The Interplay between Heat Shock Proteins and Cancer Pathogenesis: A Novel Strategy for Cancer Therapeutics. Cancers 2024, 16, 638. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Afzal, S.; Jensen, S.A.; Vainer, B.; Vogel, U.; Matsen, J.P.; Sørensen, J.B.; Andersen, P.K.; Poulsen, H.E. MTHFR polymorphisms and 5-FU-based adjuvant chemotherapy in colorectal cancer. Ann. Oncol. 2009, 20, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thorn, C.F.; Marsh, S.; Carrillo, M.W.; McLeod, H.L.; Klein, T.E.; Altman, R.B. PharmGKB summary: Fluoropyrimidine pathways. Pharmacogenet Genom. 2011, 21, 237–242. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Feng, H.; Yu, J.; Xu, Z.; Sang, Q.; Li, F.; Chen, M.; Chen, Y.; Yu, B.; Zhu, N.; Xia, J.; et al. SLC7A9 suppression increases chemosensitivity by inducing ferroptosis via the inhibition of cystine transport in gastric cancer. eBioMedicine 2024, 109, 105375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, D.; Kang, R. NFE2L2 and ferroptosis resistance in cancer therapy. Cancer Drug Resist. 2024, 7, 41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, Y.; Xu, Z.; Zhou, H.; Xu, R.; Xu, J.; Liu, W.; Wu, Y.; Qiu, Y.; Zhang, G.; Huang, X.; et al. RBP7 functions as a tumor suppressor in HR + breast cancer by inhibiting the AKT/SREBP1 pathway and reducing fatty acid. Cancer Cell Int. 2024, 24, 118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhan, Y.; Jiang, L.; Jin, X.; Ying, S.; Wu, Z.; Wang, L.; Yu, W.; Tong, J.; Zhang, L.; Lou, Y.; et al. Inhibiting RRM2 to enhance the anticancer activity of chemotherapy. Biomed. Pharmacother. 2021, 133, 110996. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/β-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. Biomed. Res. Int. 2020, 2020, 9390878. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsu, C.P.; Kao, T.Y.; Chang, W.L.; Nieh, S.; Wang, H.L.; Chung, Y.C. Clinical significance of tumor suppressor PTEN in colorectal carcinoma. Eur. J. Surg. Oncol. 2011, 37, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Zhou, J.; Chen, Z.R.; Chng, W.J. P53 mutations in colorectal cancer—Molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Y.; Xue, J.; Li, Y.; Wu, X.; Qu, M.; Xu, D.; Shi, Y. Expression, clinical significance and correlation of RUNX3 and HER2 in colorectal cancer. J. Gastrointest. Oncol. 2021, 12, 1577–1589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, X.; Ke, K.; Jin, W.; Zhu, Q.; Zhu, Q.; Mei, R.; Zhang, R.; Yu, S.; Shou, L.; Sun, X.; et al. Identification of Genes Related to 5-Fluorouracil Based Chemotherapy for Colorectal Cancer. Front. Immunol. 2022, 13, 887048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Khamkevych, Y.; Raza, M.; Kamal, M.; Bissonnette, M.; Ahsan, H. Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy. Medicina 2024, 60, 348. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morifuji, M.; Hiyama, E.; Murakami, Y.; Imamura, Y.; Sueda, T.; Yokoyama, T. Fluorescent-based BAT-26 analysis for distinct screening of microsatellite instability in colorectal cancers. Int. J. Oncol. 2003, 22, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Deschoolmeester, V.; Baay, M.; Wuyts, W.; Van Marck, E.; Van Damme, N.; Vermeulen, P.; Lukaszuk, K.; Lardon, F.; Vermorken, J.B. Detection of microsatellite instability in colorectal cancer using an alternative multiplex assay of quasi-monomorphic mononucleotide markers. J. Mol. Diagn. 2008, 10, 154–159. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Janavicius, R.; Matiukaite, D.; Jakubauskas, A.; Griskevicius, L. Microsatellite instability detection by high-resolution melting analysis. Clin. Chem. 2010, 56, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Downey, T. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 2006, 411, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.M.; Rankin, C.; Toriola, A.T.; Makar, K.W.; Altug-Teber, Ö.; Benedetti, J.K.; Holmes, R.S.; Smalley, S.R.; Blanke, C.D.; Lenz, H.J. Polymorphisms in folate-metabolizing enzymes and response to 5-fluorouracil among patients with stage II or III rectal cancer (INT-0144; SWOG 9304). Cancer 2014, 120, 3329–3337. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jasmine, F.; Haq, Z.; Kamal, M.; Raza, M.; da Silva, G.; Gorospe, K.; Paul, R.; Strzempek, P.; Ahsan, H.; Kibriya, M.G. Interaction between Microsatellite Instability (MSI) and Tumor DNA Methylation in the Pathogenesis of Colorectal Carcinoma. Cancers 2021, 13, 4956. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate synthase: A critical target for cancer chemotherapy. Clin. Color. Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Lee, J.S.; Min, H.Y.; Lee, H.Y. Acquired resistance to 5-fluorouracil via HSP90/Src-mediated increase in thymidylate synthase expression in colon cancer. Oncotarget 2015, 6, 32622–32633. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nishizawa, N.; Kurasaka, C.; Ogino, Y.; Sato, A. Regulation of 5-fluorodeoxyuridine monophosphate-thymidylate synthase ternary complex levels by autophagy confers resistance to 5-fluorouracil. FASEB Bioadv. 2023, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jo, W.S.; Carethers, J.M. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006, 2, 51–60. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vance, D.E. Physiological roles of phosphatidylethanolamine N-methyltransferase. Biochim. Biophys Acta 2013, 1831, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, F.F.; Xie, W.; Chen, S.Q.; Wang, X.K.; Liu, Q.; Pan, X.K.; Su, F.; Feng, M.H. SLC38A1 promotes proliferation and migration of human colorectal cancer cells. J. Huazhong Univ. Sci. Technolog Med. Sci. 2017, 37, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Guiducci, G.; Paone, A.; Rinaldo, S.; Giardina, G.; Liberati, F.R.; Cutruzzolá, F.; Tartaglia, G.G. Modelling of SHMT1 riboregulation predicts dynamic changes of serine and glycine levels across cellular compartments. Comput. Struct. Biotechnol. J. 2021, 19, 3034–3041. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macfarlane, A.J.; Perry, C.A.; McEntee, M.F.; Lin, D.M.; Stover, P.J. Shmt1 heterozygosity impairs folate-dependent thymidylate synthesis capacity and modifies risk of Apc(min)-mediated intestinal cancer risk. Cancer Res. 2011, 71, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de la Chapelle, A.; Hampel, H. Clinical relevance of microsatellite instability in colorectal cancer. J. Clin. Oncol. 2010, 28, 3380–3387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Löhr, K.; Möritz, C.; Contente, A.; Dobbelstein, M. p21/CDKN1A mediates negative regulation of transcription by p53. J. Biol. Chem. 2003, 278, 32507–32516. [Google Scholar] [CrossRef] [PubMed]
- Kho, P.S.; Wang, Z.; Zhuang, L.; Li, Y.; Chew, J.L.; Ng, H.H.; Liu, E.T.; Yu, Q. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J. Biol. Chem. 2004, 279, 21183–21192. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, P.M.; Svendsrud, D.H.; Kravik, K.L.; Stokke, T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol. Cancer 2006, 5, 20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, B.; Yang, P.; Zhang, S. Cell fate regulation governed by p53: Friends or reversible foes in cancer therapy. Cancer Commun. 2024, 44, 297–360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zuo, Z.; Zhou, Z.; Chang, Y.; Liu, Y.; Shen, Y.; Li, Q.; Zhang, L. Ribonucleotide reductase M2 (RRM2): Regulation, function and targeting strategy in human cancer. Genes. Dis. 2024, 11, 218–233. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hosea, R.; Duan, W.; Meliala, I.T.S.; Li, W.; Wei, M.; Hillary, S.; Zhao, H.; Miyagishi, M.; Wu, S.; Kasim, V. YY2/BUB3 Axis promotes SAC Hyperactivation and Inhibits Colorectal Cancer Progression via Regulating Chromosomal Instability. Adv. Sci. 2024, 11, e2308690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, B.; Leng, C.; Wu, C.; Zhang, Z.; Dou, L.; Luo, X.; Zhang, B.; Chen, X. Smad4 sensitizes colorectal cancer to 5-fluorouracil through cell cycle arrest by inhibiting the PI3K/Akt/CDC2/survivin cascade. Oncol. Rep. 2016, 35, 1807–1815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, B.; Chen, X.; Bae, S.; Singh, K.; Washington, M.K.; Datta, P.K. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br. J. Cancer 2014, 110, 946–957. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shin, K.H.; Park, Y.J.; Park, J.G. PTEN gene mutations in colorectal cancers displaying microsatellite instability. Cancer Lett. 2001, 174, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X. circPTEN suppresses colorectal cancer progression through regulating PTEN/AKT pathway. Mol. Ther. Nucleic Acids. 2021, 26, 1418–1432. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fang, L.; Li, H.; Wang, L.; Hu, J.; Jin, T.; Wang, J.; Yang, B.B. MicroRNA-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing PTEN expression. Oncotarget 2014, 5, 2974–2987. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jian, B.; Li, Z.; Xiao, D.; He, G.; Bai, L.; Yang, Q. Downregulation of microRNA-193-3p inhibits tumor proliferation migration and chemoresistance in human gastric cancer by regulating PTEN gene. Tumour Biol. 2016, 37, 8941–8949. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.Y.; Chen, Q.J.; Xu, K.; Ren, H.T.; Bao, X.; Ma, Y.N.; Wei, Y.; Ma, H.B. Involvement of microRNA-141-3p in 5-fluorouracil and oxaliplatin chemo-resistance in esophageal cancer cells via regulation of PTEN. Mol. Cell Biochem. 2016, 422, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nyiraneza, C.; Jouret-Mourin, A.; Kartheuser, A.; Camby, P.; Plomteux, O.; Detry, R.; Dahan, K.; Sempoux, C. Distinctive patterns of p53 protein expression and microsatellite instability in human colorectal cancer. Hum. Pathol. 2011, 42, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.; Bracht, E.V.; Reijnders, D.; Safarikova, B.; Jelinkova, I.; Grandien, A.; Vaculova, A.H.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil-induced RNA stress engages a TRAIL-DISC-dependent apoptosis axis facilitated by p53. Oncotarget 2015, 6, 43679–43697. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Babinčák, M.; Jendželovský, R.; Košuth, J.; Majernik, M.; Vargova, J.; Mikulášek, K.; Zdráhal, Z.; Fedoročko, P. Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp. Cancers 2021, 13, 1646. Cancers 2021, 13, 1646. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nazim, U.M.; Rasheduzzaman, M.; Lee, Y.J.; Seol, D.W.; Park, S.Y. Enhancement of TRAIL-induced apoptosis by 5-fluorouracil requires activating Bax and p53 pathways in TRAIL-resistant lung cancers. Oncotarget 2017, 8, 18095–18105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maecker, H.L.; Koumenis, C.; Giaccia, A.J. p53 promotes selection for Fas-mediated apoptotic resistance. Cancer Res. 2000, 60, 4638–4644. [Google Scholar] [PubMed]
- Xiao, W.; Ibrahim, M.L.; Redd, P.S.; Klement, J.D.; Lu, C.; Yang, D.; Savage, N.M.; Liu, K. Loss of Fas Expression and Function Is Coupled with Colon Cancer Resistance to Immune Checkpoint Inhibitor Immunotherapy. Mol. Cancer Res. 2019, 17, 420–430. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, H.; Zuo, J.; Li, B.; Chen, R.; Luo, K.; Xiang, X.; Lu, S.; Huang, C.; Liu, L.; Tang, J.; et al. Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. 2023, 63, 102754. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chun, K.S.; Joo, S.H. Modulation of Reactive Oxygen Species to Overcome 5-Fluorouracil Resistance. Biomol. Ther. 2022, 30, 479–489. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Sci. Rep. 2019, 9, 3210. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irawan, B.; Labeda, I.; Lusikooy, R.E.; Sampetoding, S.; Kusuma, M.I.; Uwuratuw, J.A.; Syarifuddin, E.; Faruk, M. Association of superoxide dismutase enzyme with staging and grade of differentiation colorectal cancer: A cross-sectional study. Ann. Med. Surg. 2020, 58, 194–199. [Google Scholar] [CrossRef]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. 2021, 48, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, C.; Zhang, P.; Gao, X.; Guan, W.; Wang, F.; Li, X.; Yuan, J.; Dou, H.; Xu, G. Enhanced Intracellular Reactive Oxygen Species by Photodynamic Therapy Effectively Promotes Chemoresistant Cell Death. Int. J. Biol. Sci. 2022, 18, 374–385. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Description | p-Value | FDR (p-Value) | Fold-Change | (95% CI) |
|---|---|---|---|---|
| Cell Cycle Checkpoint Genes | 8.94 × 10−217 | 1.25 × 10−215 | 1.27 | (1.25–1.29) |
| Heat Shock Response Genes | 3.04 × 10−53 | 2.13 × 10−52 | 1.32 | (1.27–1.37) |
| Oxidative Stress Response Genes | 5.43 × 10−33 | 2.54 × 10−32 | 1.11 | (1.09–1.13) |
| Signaling pathway | 4.09 × 10−23 | 1.43 × 10−22 | 1.08 | (1.06–1.10) |
| Tumor suppressor gene | 5.08 × 10−21 | 1.42 × 10−20 | −1.16 | (−1.20–−1.13) |
| DNA Repair Mechanisms | 1.17 × 10−15 | 2.73 × 10−15 | 1.04 | (1.03–1.05) |
| Drug Transport | 3.46 × 10−10 | 6.91 × 10−10 | 1.07 | (1.05–1.10) |
| Folate & One carbon metabolism | 6.65 × 10−8 | 1.16 × 10−7 | 1.04 | (1.02–1.05) |
| Apoptotic gene | 1.78 × 10−6 | 2.77 × 10−6 | −1.02 | (−1.03–−1.01) |
| Endoplasmic Reticulum (ER) Stress | 0.010382 | 0.014534 | −1.03 | (−1.06–−1.01) |
| 5FU Metabolism | 0.016943 | 0.021564 | 1.04 | (1.01–1.08) |
| Transsulfuration & Pyrimidine metabolism | 0.05517 | 0.064365 | 1.06 | (−1.00–1.13) |
| known 5FU-resistance genes | 0.363536 | 0.3915 | 1.01 | (−1.01–1.03) |
| EMT & tumor invasion | 0.578479 | 0.578479 | 1.01 | (−1.03–1.05) |
| Gene Set | Interaction p-Value | FDR p-Value | KRAS Mutant | KRAS Wild | ||
|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | Fold Change | (95% CI) | |||
| Cell Cycle Checkpoint Genes | 1.09 × 10−8 | 1.53 × 10−7 | 1.33 | (1.30 to 1.37) | 1.24 | (1.22 to 1.26) |
| Heat Shock Response Genes | 4.57 × 10−4 | 3.20 × 10−3 | 1.45 | (1.36 to 1.55) | 1.26 | (1.21 to 1.31) |
| Oxidative Stress Response Genes | 2.65 × 10−2 | 1.24 × 10−1 | 1.14 | (1.11 to 1.18) | 1.09 | (1.07 to 1.11) |
| Gene Set | Interaction p-Value | FDR p-Value | TP53 Mutant | TP53 Wild | ||
|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | Fold Change | (95% CI) | |||
| Oxidative Stress Response Genes | 1.69 × 10−11 | 2.36 × 10−10 | 1.21 | (1.17 to 1.25) | 1.05 | (1.03 to 1.08) |
| Endoplasmic Reticulum (ER) Stress | 1.28 × 10−5 | 8.98 × 10−5 | 1.05 | (1.00 to 1.10) | −1.07 | (−1.11 to −1.04) |
| Signaling pathway | 2.61 × 10−3 | 1.22 × 10−2 | 1.12 | (1.09 to 1.15) | 1.06 | (1.04 to 1.08) |
| Gene Set | Interaction p-Value | FDR p-Value | CRC with MSS | CRC with MSI | ||
|---|---|---|---|---|---|---|
| Fold Change | (95% CI) | Fold Change | (95% CI) | |||
| Cell Cycle Checkpoint Genes | 3.72 × 10−15 | 5.21 × 10−14 | 1.26 | (1.24 to 1.29) | 1.27 | (1.23 to 1.30) |
| Oxidative Stress Response Genes | 5.30 × 10−10 | 3.71 × 10−9 | 1.11 | (1.09 to 1.13) | 1.08 | (1.05 to 1.12) |
| Tumor suppressor gene | 1.37 × 10−4 | 3.82 × 10−4 | −1.19 | (−1.24 to −1.15) | −1.08 | (−1.14 to −1.02) |
| DNA Repair Mechanisms | 4.96 × 10−2 | 7.71 × 10−2 | 1.04 | (1.03 to 1.05) | 1.05 | (1.03 to 1.07) |
| Probeset ID | Gene | Interaction p-Value | FDR (p-Value) | CRC MSI vs. Normal MSI | CRC MSS vs. Normal MSS) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Fold-Change | (95% CI) | p-Value | Fold-Change | (95% CI) | p-Value | ||||
| ILMN_2386100 | BUB3 | 3.45 × 10−7 | 0.0001 | 1.31 | (1.14–1.50) | 0.000233 | 1.06 | (−1.02–1.16) | 0.13 |
| ILMN_1806040 | TYMS | 1.01 × 10−6 | 0.0001 | 1.65 | (1.27–2.13) | 0.000181 | 1.19 | (1.02–1.39) | 0.02 |
| ILMN_1720282 | NQO1 | 2.22 × 10−6 | 0.0002 | −1.38 | (−1.92–1.01) | 0.05 | 1.68 | (1.38–2.04) | 6.20 × 10−7 |
| ILMN_2385220 | DFFA | 6.92 × 10−6 | 0.0005 | −1.22 | (−1.45–−1.03) | 0.01 | 1.1 | (−1.01–1.21) | 0.07 |
| ILMN_1699265 | TNFRSF10B | 1.67 × 10−5 | 0.001 | 1.69 | (1.42–2.02) | 2.17 × 10−8 | 1.17 | (1.06–1.30) | 0.002 |
| ILMN_2331010 | TNFRSF10B | 5.03 × 10−5 | 0.002 | 1.4 | (1.25–1.55) | 6.09 × 10−9 | 1.16 | (1.09–1.24) | 5.29 × 10−7 |
| ILMN_1741477 | SMAD4 | 8.71 × 10−5 | 0.004 | 1.02 | (−1.15–1.20) | 0.8 | −1.28 | (−1.41–−1.16) | 1.22 × 10−6 |
| ILMN_2358457 | ATF4 | 0.000143 | 0.005 | 1.27 | (1.10–1.45) | 0.00097 | 1.23 | (1.13–1.34) | 4.13 × 10−6 |
| ILMN_1769911 | SLC38A1 | 0.000202 | 0.01 | −1.59 | (−1.91–−1.33) | 1.25 × 10−6 | −1.05 | (−1.17–1.06) | 0.37 |
| ILMN_1757995 | PARP2 | 0.000298 | 0.01 | 1.27 | (1.15–1.39) | 1.68 × 10−6 | 1.13 | (1.07–1.19) | 3.50 × 10−5 |
| ILMN_2402341 | MAPK3 | 0.000375 | 0.01 | −2.02 | (−2.46–−1.67) | 3.22 × 10−11 | −1.52 | (−1.70–−1.35) | 4.32 × 10−11 |
| ILMN_1787212 | CDKN1A | 0.000497 | 0.01 | 1.14 | (1.06–1.22) | 0.000216 | 1.05 | (1.01–1.09) | 0.01 |
| ILMN_1667260 | MAPK3 | 0.000665 | 0.01 | −2.05 | (−2.51–−1.67) | 1.17 × 10−10 | −1.55 | (−1.75–−1.37) | 4.89 × 10−11 |
| ILMN_1743784 | SHMT1 | 0.000782 | 0.01 | 1.02 | (−1.07–1.12) | 0.64 | 1.001 | (−1.05–1.06) | 0.97 |
| ILMN_1662438 | SOD1 | 0.00086 | 0.01 | 1.2 | (1.04–1.40) | 0.01 | 1.04 | (−1.05–1.14) | 0.36 |
| ILMN_1701134 | PTEN | 0.001077 | 0.02 | −1.2 | (−1.33–−1.07) | 0.001 | −1.22 | (−1.31–−1.15) | 7.77 × 10−9 |
| ILMN_2319077 | FAS | 0.001165 | 0.02 | −1.32 | (−1.56–−1.13) | 0.0007 | −1.58 | (−1.74–−1.44) | 1.64 × 10−16 |
| ILMN_1811933 | SHMT1 | 0.001383 | 0.02 | −1.07 | (−1.33–1.15) | 0.51 | −1.13 | (−1.28–1.01) | 0.06 |
| ILMN_1779376 | GSK3B | 0.001934 | 0.03 | 1.03 | (−1.10–1.18) | 0.61 | 1.19 | (1.10–1.28) | 2.04 × 10−5 |
| ILMN_1727855 | PEMT | 0.002 | 0.042 | 1.23 | (1.11–1.38) | 0.0002 | 1.08 | (1.01–1.16) | 0.015 |
| ILMN_1678669 | RRM2 | 0.002 | 0.042 | 1.32 | (1.13–1.55) | 0.0007 | 1.25 | (1.13–1.37) | 1.03 × 10−5 |
| Gene Symbol | Interaction p-Value | MSS in CRC vs. Normal | MSI in CRC vs. Normal | ||
|---|---|---|---|---|---|
| KRAS Wild | KRAS Mutant | KRAS Wild | KRAS Mutant | ||
| FC (95% CI) | FC (95% CI) | FC (95% CI) | FC (95% CI) | ||
| BUB3 | 1.10 × 10−5 | 1.05 (−1.06 to 1.16) | 1.11 (−1.05 to 1.30) | 1.27 (1.08 to 1.49) | 1.49 (1.12 to 1.99) |
| NQO1 | 2.24 × 10−5 | 1.51 (1.20 to 1.91) | 2.16 (1.51 to 3.09) | −1.53 (−2.24 to −1.05) | −1.08 (−2.10 to 1.81) |
| TYMS | 2.94 × 10−5 | 1.17 (−1.03 to 1.40) | 1.27 (−1.04 to 1.68) | 1.58 (1.17 to 2.12) | 2.02 (1.19 to 3.42) |
| TNFRSF10B | 2.00 × 10−4 | 1.11 (−1.02 to 1.26) | 1.35 (1.11 to 1.63) | 1.65 (1.35 to 2.02) | 1.86 (1.30 to 2.66) |
| DFFA | 3.00 × 10−4 | 1.10 (−1.03 to 1.24) | 1.10 (−1.10 to 1.32) | −1.20 (−1.46 to 1.02) | −1.36 (−1.93 to 1.04) |
| TNFRSF10B | 5.00 × 10−4 | 1.14 (1.06 to 1.23) | 1.23 (1.09 to 1.38) | 1.40 (1.24 to 1.59) | 1.39 (1.12 to 1.73) |
| GSK3B | 7.00 × 10−4 | 1.12 (1.02 to 1.23) | 1.38 (1.20 to 1.59) | 1.11 (−1.05 to 1.28) | −1.22 (−1.58 to 1.07) |
| SMAD4 | 1.10 × 10−3 | −1.25 (−1.40 to −1.11) | −1.36 (−1.62 to −1.14) | −1.05 (−1.27 to 1.15) | 1.29 (−1.08 to 1.80) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.R.; Jasmine, F.; Vasiljevs, D.; Raza, M.; Almazan, A.; Ahsan, H.; Kibriya, M.G. Mutation and Microsatellite Instability (MSI) Affect the Differential Gene Expression of Folic Acid and 5-Flourouracil Metabolism-Related Genes in Colorectal Carcinoma. Curr. Oncol. 2025, 32, 661. https://doi.org/10.3390/curroncol32120661
Islam MR, Jasmine F, Vasiljevs D, Raza M, Almazan A, Ahsan H, Kibriya MG. Mutation and Microsatellite Instability (MSI) Affect the Differential Gene Expression of Folic Acid and 5-Flourouracil Metabolism-Related Genes in Colorectal Carcinoma. Current Oncology. 2025; 32(12):661. https://doi.org/10.3390/curroncol32120661
Chicago/Turabian StyleIslam, Muhammad Rafiqul, Farzana Jasmine, Daniil Vasiljevs, Maruf Raza, Armando Almazan, Habibul Ahsan, and Muhammad G. Kibriya. 2025. "Mutation and Microsatellite Instability (MSI) Affect the Differential Gene Expression of Folic Acid and 5-Flourouracil Metabolism-Related Genes in Colorectal Carcinoma" Current Oncology 32, no. 12: 661. https://doi.org/10.3390/curroncol32120661
APA StyleIslam, M. R., Jasmine, F., Vasiljevs, D., Raza, M., Almazan, A., Ahsan, H., & Kibriya, M. G. (2025). Mutation and Microsatellite Instability (MSI) Affect the Differential Gene Expression of Folic Acid and 5-Flourouracil Metabolism-Related Genes in Colorectal Carcinoma. Current Oncology, 32(12), 661. https://doi.org/10.3390/curroncol32120661