
Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation

 ,
, 
 , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Advances, Trends, and Challenges in High-Throughput Screening (HTS)
2.1. Lab-Based HTS
2.2. Structure-Based Virtual HTS, MoA Prediction, New Trends, and Challenges
3. Advances in HT Analytical Techniques for NP Dereplication
4. Dereplication Advances, Databases, Informatic Tools, and Case Studies
4.1. LC-MS/MS Data Visualization and Annotation Methods
4.1.1. LC-MS/MS Data Visualization and Annotation—Case Studies
4.2. Dereplication Using LC-MS/MS, NP Databases, and Informatic Tools
4.2.1. GNPS Database, GNPS-Combined Databases, Integrated Analytical and Informatic Tools, and Other NP Databases to Aid LC-MS/MS Dereplication
4.3. Dereplication Using MS or MS/MS Advanced Computational Prediction Tools
4.4. Dereplication Using Gas Chromatography-Mass Spectrometry (GC-MS), LC-MS Integrated Ion Mobility Spectrometry (IMS), and LC-Matrix Assisted Laser Desorption/lonization Mass Spectrometry MALDI-MS
4.5. Dereplication Using NMR Spectroscopy
4.5.1. NMR and NP Databases for Dereplication
5. Genome Sequencing Methods for Dereplication and Structure Elucidation
5.1. Genome Sequencing Techniques
5.2. High Throughput Next-Generation Sequencing (HT/NGS)
5.3. Dereplication Using Genomics Methods
5.3.1. Retrieving the Microbial/Environmental DNA
5.3.2. Steps and Tools in Genome Mining
5.3.3. Chemoinformatics Approaches for Dereplication Using BGCs Diversity
6. Natural Products Determination of Relative and Absolute Configurations
6.1. X-ray Diffraction
6.2. Chiroptical Spectroscopy
6.3. Low Temperature Atomic Force Microscopy (AFM)
6.4. Relative Configuration by NMR
6.5. Absolute Configuration by NMR
6.5.1. Derivatization with Chiral Anisotropic Reagents
6.5.2. Chiral Solvating Agents (CSA)
6.5.3. Absolute Configuration of Amino Acids by Marfey’s Derivatization Method
6.5.4. Quantum Chemical Calculations of NMR Parameters
6.6. Relative and Absolute Configuration Aided by Genomics
7. Computer Assisted Structure Elucidation and Related NP Databases
8. Chemoinformatics Tools to Facilitate Drug-Lead Discovery
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rotter, A.; Barbier, M.; Bertoni, F.; Bones, A.M.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Ceglowska, M.; Chirivella-Martorell, J.; Dalay, M.C.; et al. The Essentials of Marine Biotechnology. Front. Mar. Sci. 2021, 8, 629629. [Google Scholar] [CrossRef]
- Barreca, M.; Spane, V.; Montalbano, A.; Cueto, M.; Marrero, A.R.D.; Deniz, I.; Erdogan, A.; Bilela, L.L.; Moulin, C.; Taffin-de-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Gaudencio, S.P.; Pereira, F. Dereplication: Racing to speed up the natural products discovery process. Nat. Prod. Rep. 2015, 32, 779–810. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.-L.; Litaudon, M.; Touboul, D.; Queiroz, E.F. Innovative omics-based approaches for prioritisation and targeted isolation of natural products—New strategies for drug discovery. Nat. Prod. Rep. 2019, 36, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.-L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef] [PubMed]
- Moumbock, A.F.A.; Ntie-Kang, F.; Akone, S.H.; Li, L.; Gao, M.; Telukunta, K.K.; Guenther, S. An overview of tools, software, and methods for natural product fragment and mass spectral analysis. Phys. Sci. Rev. 2019, 4, 1368–1374. [Google Scholar] [CrossRef]
- Barbosa, A.J.M.; Roque, A.C.A. Free Marine Natural Products Databases for Biotechnology and Bioengineering. Biotechnol. J. 2019, 14, 1800607. [Google Scholar] [CrossRef]
- Wilson, B.A.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminformatics 2020, 12, 20. [Google Scholar] [CrossRef]
- van Santen, J.A.; Kautsar, S.A.; Medema, M.H.; Linington, R.G. Microbial natural product databases: Moving forward in the multi-omics era. Nat. Prod. Rep. 2021, 38, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Wang, M.; Dorrestein, P.C. The critical role that spectral libraries play in capturing the metabolomics community knowledge. Metabolomics 2022, 18, 94. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminformatics 2021, 13, 2. [Google Scholar] [CrossRef]
- Ramos, A.E.F.; Evanno, L.; Poupon, E.; Champy, P.; Beniddir, M.A. Natural products targeting strategies involving molecular networking: Different manners, one goal. Nat. Prod. Rep. 2019, 36, 960–980. [Google Scholar] [CrossRef]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Covington, B.C.; McLean, J.A.; Bachmann, B.O. Comparative mass spectrometry-based metabolomics strategies for the investigation of microbial secondary metabolites. Nat. Prod. Rep. 2017, 34, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Jarmusch, S.A.; van der Hooft, J.J.J.; Dorrestein, P.C.; Jarmusch, A.K. Advancements in capturing and mining mass spectrometry data are transforming natural products research. Nat. Prod. Rep. 2021, 38, 2066–2082. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Canh Hao, N.; Mamitsuka, H. Current status and prospects of computational resources for natural product dereplication: A review. Brief. Bioinform. 2016, 17, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, E.J.N.; Reiter, S.; Piel, J. Recent advances in genome-based polyketide discovery. Curr. Opin. Biotechnol. 2014, 29, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Albanese, D.; Donati, C. Genome Recovery, Functional Profiling, and Taxonomic Classification from Metagenomes. Methods Mol. Biol. 2021, 2242, 153–172. [Google Scholar] [CrossRef]
- Cruesemann, M. Coupling Mass Spectral and Genomic Information to Improve Bacterial Natural Product Discovery Workflows. Mar. Drugs 2021, 19, 142. [Google Scholar] [CrossRef] [PubMed]
- Krause, J. Applications and Restrictions of Integrated Genomic and Metabolomic Screening: An Accelerator for Drug Discovery from Actinomycetes? Molecules 2021, 26, 5450. [Google Scholar] [CrossRef]
- Chevrette, M.G.; Handelsman, J. Needles in haystacks: Reevaluating old paradigms for the discovery of bacterial secondary metabolites. Nat. Prod. Rep. 2021, 38, 2083–2099. [Google Scholar] [CrossRef] [PubMed]
- Voser, T.M.; Campbell, M.D.; Carroll, A.R. How different are marine microbial natural products compared to their terrestrial counterparts? Nat. Prod. Rep. 2022, 39, 7–19. [Google Scholar] [CrossRef]
- Li, G.; Lin, P.; Wang, K.; Gu, C.-C.; Kusari, S. Artificial intelligence-guided discovery of anticancer lead compounds from plants and associated microorganisms. Trends Cancer 2022, 8, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Sahayasheela, V.J.; Lankadasari, M.B.; Dan, V.M.; Dastager, S.G.; Pandian, G.N.; Sugiyama, H. Artificial intelligence in microbial natural product drug discovery: Current and emerging role. Nat. Prod. Rep. 2022, 39, 2215–2230. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H. The year 2020 in natural product bioinformatics: An overview of the latest tools and databases. Nat. Prod. Rep. 2021, 38, 301–306. [Google Scholar] [CrossRef]
- Ren, H.; Shi, C.; Zhao, H. Computational Tools for Discovering and Engineering Natural Product Biosynthetic Pathways. iScience 2020, 23, 100795. [Google Scholar] [CrossRef]
- Prihoda, D.; Maritz, J.M.; Klempir, O.; Dzamba, D.; Woelk, C.H.; Hazuda, D.J.; Bitton, D.A.; Hannigan, G.D. The application potential of machine learning and genomics for understanding natural product diversity, chemistry, and therapeutic translatability. Nat. Prod. Rep. 2021, 38, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.N.L.; Angrisani, B.R.P.; Lima, M.E.D.; da Silva, S.M.P.; Schettini, V.H.; Chagas, H.A.; dos Santos, F.M., Jr.; Batista, J.M., Jr.; Valverde, A.L. Absolute Configuration Reassignment of Natural Products: An Overview of the Last Decade. J. Braz. Chem. Soc. 2021, 32, 1499–1518. [Google Scholar] [CrossRef]
- Chhetri, B.K.; Lavoie, S.; Sweeney-Jones, A.M.; Kubanek, J. Recent trends in the structural revision of natural products. Nat. Prod. Rep. 2018, 35, 514–531. [Google Scholar] [CrossRef]
- Marcarino, M.O.; Cicetti, S.; Zanardi, M.M.; Sarotti, A.M. A critical review on the use of DP4+ in the structural elucidation of natural products: The good, the bad and the ugly. A practical guide. Nat. Prod. Rep. 2022, 39, 58–76. [Google Scholar] [CrossRef]
- Kim, C.S.; Oh, J.; Lee, T.H. Structure elucidation of small organic molecules by contemporary computational chemistry methods. Arch. Pharmacal Res. 2020, 43, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Lauro, G.; Bifulco, G. Elucidating the Relative and Absolute Configuration of Organic Compounds by Quantum Mechanical Approaches. Eur. J. Org. Chem. 2020, 2020, 3929–3941. [Google Scholar] [CrossRef]
- Nugroho, A.E.; Morita, H. Computationally-assisted discovery and structure elucidation of natural products. J. Nat. Med. 2019, 73, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. [Google Scholar] [CrossRef]
- Superchi, S.; Scafato, P.; Gorecki, M.; Pescitelli, G. Absolute Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef]
- Mandi, A.; Kurtan, T. Applications of OR/ECD/VCD to the Structure Elucidation of Natural Products Dedicated to Professor Dr Sandor Antus on the Occasion of His 75th Anniversary. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Elyashberg, M.; Argyropoulos, D. Computer Assisted Structure Elucidation (CASE): Current and future perspectives. Magn. Reson. Chem. 2021, 59, 669–690. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.C.; Mazzola, E.P.; Reynolds, W.F. The role of computer-assisted structure elucidation (CASE) programs in the structure elucidation of complex natural products. Nat. Prod. Rep. 2019, 36, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Elyashberg, M.; Williams, A. ACD/Structure Elucidator: 20 Years in the History of Development. Molecules 2021, 26, 6623. [Google Scholar] [CrossRef] [PubMed]
- Yirik, M.A.; Steinbeck, C. Chemical graph generators. PLoS Comput. Biol. 2021, 17, e1008504. [Google Scholar] [CrossRef] [PubMed]
- Buevich, A.V.; Elyashberg, M.E. Enhancing computer-assisted structure elucidation with DFT analysis of J-couplings. Magn. Reson. Chem. 2020, 58, 594–606. [Google Scholar] [CrossRef]
- Buevich, A.V.; Elyashberg, M.E. Synergistic Combination of CASE Algorithms and DFT Chemical Shift Predictions: A Powerful Approach for Structure Elucidation, Verification, and Revision. J. Nat. Prod. 2016, 79, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Buevich, A.V.; Elyashberg, M.E. Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem. 2018, 56, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Kountz, D.J.; Balskus, E.P. Leveraging Microbial Genomes and Genomic Context for Chemical Discovery. Acc. Chem. Res. 2021, 54, 2788–2797. [Google Scholar] [CrossRef]
- Sagita, R.; Quax, W.J.; Haslinger, K. Current State and Future Directions of Genetics and Genomics of Endophytic Fungi for Bioprospecting Efforts. Front. Bioeng. Biotechnol. 2021, 9, e1002290. [Google Scholar] [CrossRef]
- Tietz, J.I.; Mitchell, D.A. Using Genomics for Natural Product Structure Elucidation. Curr. Top. Med. Chem. 2016, 16, 1645–1694. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Hemmerling, F.; Piel, J. Strategies to access biosynthetic novelty in bacterial genomes for drug discovery. Nat. Rev. Drug Discov. 2022, 21, 359–378. [Google Scholar] [CrossRef]
- Schneider, X.T.; Stroil, B.K.; Tourapi, C.; Rebours, C.; Gaudencio, S.P.; Novoveska, L.; Vasquez, M.I. Responsible Research and Innovation Framework, the Nagoya Protocol and Other European Blue Biotechnology Strategies and Regulations: Gaps Analysis and Recommendations for Increased Knowledge in the Marine Biotechnology Community. Mar. Drugs 2022, 20, 290. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851. [Google Scholar] [CrossRef]
- Robinson, S.L.; Piel, J.; Sunagawa, S. A roadmap for metagenomic enzyme discovery. Nat. Prod. Rep. 2021, 38, 1994–2023. [Google Scholar] [CrossRef] [PubMed]
- Rotter, A.; Bacu, A.; Barbier, M.; Bertoni, F.; Bones, A.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Ceglowska, M.; Dalay, M.C.; et al. A New Network for the Advancement of Marine Biotechnology in Europe and beyond. Front. Mar. Sci. 2020, 7, 278. [Google Scholar] [CrossRef]
- Rotter, A.; Gaudencio, S.P.; Klun, K.; Macher, J.-N.; Thomas, O.P.; Deniz, I.; Edwards, C.; Grigalionyte-Bembic, E.; Ljubesic, Z.; Robbens, J.; et al. A New Tool for Faster Construction of Marine Biotechnology Collaborative Networks. Front. Mar. Sci. 2021, 8, 685164. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef]
- Rocha-Martin, J.; Harrington, C.; Dobson, A.D.W.; O’Gara, F. Emerging strategies and integrated systems microbiology technologies for biodiscovery of marine bioactive compounds. Mar. Drugs 2014, 12, 3516–3559. [Google Scholar] [CrossRef]
- Navarro, G.; Cheng, A.T.; Peach, K.C.; Bray, W.M.; Bernan, V.S.; Yildiz, F.H.; Linington, R.G. Image-Based 384-Well High-Throughput Screening Method for the Discovery of Skyllamycins A to C as Biofilm Inhibitors and Inducers of Biofilm Detachment in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 1092–1099. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Cooper, S.; Heigwer, F.; Warchal, S.; Qiu, P.; Molnar, C.; Vasilevich, A.S.; Barry, J.D.; Bansal, H.S.; Kraus, O.; et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 2017, 14, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Laubscher, W.E.; Rautenbach, M. Direct Detection of Antibacterial-Producing Soil Isolates Utilizing a Novel High-Throughput Screening Assay. Microorganisms 2022, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Orlov, A.; Semenov, S.; Rukhovich, G.; Sarycheva, A.; Kovaleva, O.; Semenov, A.; Ermakova, E.; Gubareva, E.; Bugrova, A.E.; Kononikhin, A.; et al. Hepatoprotective Activity of Lignin-Derived Polyphenols Dereplicated Using High-Resolution Mass Spectrometry, in vivo Experiments, and Deep Learning. Int. J. Mol. Sci. 2022, 23, 16025. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.E.Z.; Shinn, P.; Itkin, Z.; Eastman, R.T.; Bostwick, R.; Rasmussen, L.; Huang, R.L.; Shen, M.; Hu, X.; Wilson, K.M.; et al. Drug Repurposing Screen for Compounds Inhibiting the Cytopathic Effect of SARS-CoV-2. Front. Pharmacol. 2021, 11, 592737. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Azzollini, A.; Nievergelt, A.; Boccard, J.; Rudaz, S.; Cuendet, M.; Wolfender, J.-L. Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures. Molecules 2016, 21, 259. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef]
- Wang, M.X.; Jarmusch, A.K.; Vargas, F.; Aksenov, A.A.; Gauglitz, J.M.; Weldon, K.; Petras, D.; da Silva, R.; Quinn, R.; Melnik, A.V.; et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 2020, 38, 23–26. [Google Scholar] [CrossRef]
- Bauermeister, A.; Pereira, F.; Grilo, I.R.; Godinho, C.C.; Paulino, M.; Almeida, V.; Gobbo-Neto, L.; Prieto-Davo, A.; Sobral, R.G.; Lopes, N.P.; et al. Intra-clade metabolomic profiling of MAR4 Streptomyces from the Macaronesia Atlantic region reveals a source of anti-biofilm metabolites. Environ. Microbiol. 2019, 21, 1099–1112. [Google Scholar] [CrossRef]
- Pereira, F.; Almeida, J.R.; Paulino, M.; Grilo, I.R.; Macedo, H.; Cunha, I.; Sobral, R.G.; Vasconcelos, V.; Gaudencio, S.P. Antifouling Napyradiomycins from Marine-Derived Actinomycetes Streptomyces aculeolatus. Mar. Drugs 2020, 18, 63. [Google Scholar] [CrossRef]
- Blanco, C.; Verbanic, S.; Seelig, B.; Chen, I.A. EasyDIVER: A Pipeline for Assembling and Counting High-Throughput Sequencing Data from in vitro Evolution of Nucleic Acids or Peptides. J. Mol. Evol. 2020, 88, 477–481. [Google Scholar] [CrossRef]
- Shafranskaya, D.; Chori, A.; Korobeynikov, A. Graph-Based Approaches Significantly Improve the Recovery of Antibiotic Resistance Genes from Complex Metagenomic Datasets. Front. Microbiol. 2021, 12, 714836. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K.L.; Glassey, E.; Linington, R.G. Integration of high-content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc. Natl. Acad. Sci. USA 2015, 112, 11999–12004. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; van Santen, J.A.; Farzaneh, N.; Liu, D.Y.; Pye, C.R.; Baumeister, T.U.H.; Wong, W.R.; Linington, R.G. NP Analyst: An Open Online Platform for Compound Activity Mapping. ACS Cent. Sci. 2022, 8, 223–234. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, A.; Beyhan, S.; Choi, Y.; Morales, P.; Chan, A.P.; Espinoza, J.L.; Dupont, C.L.; Meyer, K.J.; Spoering, A.; Lewis, K.; et al. Mechanism-Of-Action Classification of Antibiotics by Global Transcriptome Profiling. Antimicrob. Agents Chemother. 2020, 64, e01207–e01219. [Google Scholar] [CrossRef]
- Shady, N.H.; Abdelmohsen, U.R.; AboulMagd, A.M.; Amin, M.N.; Ahmed, S.; Fouad, M.A.; Kamel, M.S. Cytotoxic potential of the Red Sea sponge Amphimedon sp. supported by in silico modelling and dereplication analysis. Nat. Prod. Res. 2021, 35, 6093–6098. [Google Scholar] [CrossRef]
- Gallardo, V.E.; Varshney, G.K.; Lee, M.; Bupp, S.; Xu, L.S.; Shinn, P.; Crawford, N.P.; Inglese, J.; Burgess, S.M. Phenotype-driven chemical screening in zebrafish for compounds that inhibit collective cell migration identifies multiple pathways potentially involved in metastatic invasion. Dis. Model. Mech. 2015, 8, 565–576. [Google Scholar] [CrossRef]
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for Natural Product Discovery: A Publicly-Accessible Library of Natural Product Fractions for High-Throughput Screening. ACS Chem. Biol. 2018, 13, 2484–2497. [Google Scholar] [CrossRef]
- Judson, R.; Houck, K.; Martin, M.; Richard, A.M.; Knudsen, T.B.; Shah, I.; Little, S.; Wambaugh, J.; Setzer, R.W.; Kothya, P.; et al. Analysis of the Effects of Cell Stress and Cytotoxicity on In Vitro Assay Activity Across a Diverse Chemical and Assay Space. Toxicol. Sci. 2016, 152, 323–339. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Bisson, J.; McAlpine, J.B.; Friesen, J.B.; Chen, S.N.; Graham, J.; Pauli, G.F. Can Invalid Bioactives Undermine Natural Product-Based Drug Discovery? J. Med. Chem. 2016, 59, 1671–1690. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.R.; Fraga, C.A.M.; Dantas, R.F.; Silva, F.P. Filtering promiscuous compounds in early drug discovery: Is it a good idea? Drug Discov. Today 2016, 21, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Carcache, P.J.B.; Addo, E.M.; Kinghorn, A.D. Current status and contemporary approaches to the discovery of antitumor agents from higher plants. Biotechnol. Adv. 2020, 38, 107337. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef]
- Ernst, M.; Kang, K.B.; Caraballo-Rodriguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef]
- Feng, G.F.; Zheng, Y.; Sun, Y.; Liu, S.; Pi, Z.F.; Song, F.R.; Liu, Z.Q. A targeted strategy for analyzing untargeted mass spectral data to identify lanostane-type triterpene acids in Poria cocos by integrating a scientific information system and liquid chromatography-tandem mass spectrometry combined with ion mobility spectrometry. Anal. Chim. Acta 2018, 1033, 87–99. [Google Scholar]
- Quinn, R.A.; Nothias, L.-F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular Networking As a Drug Discovery, Drug Metabolism, and Precision Medicine Strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef]
- van Der Hooft, J.J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar] [CrossRef]
- Dias, D.A.; Jones, O.A.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.-L.; Wishart, D.S. Current and future perspectives on the structural identification of small molecules in biological systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef]
- Alvarez-Rivera, G.; Ballesteros-Vivas, D.; Parada-Alfonso, F.; Ibañez, E.; Cifuentes, A. Recent applications of high resolution mass spectrometry for the characterization of plant natural products. TrAC Trends Anal. Chem. 2019, 112, 87–101. [Google Scholar] [CrossRef]
- Lianza, M.; Leroy, R.; Machado Rodrigues, C.; Borie, N.; Sayagh, C.; Remy, S.; Kuhn, S.; Renault, J.-H.; Nuzillard, J.-M. The Three Pillars of Natural Product Dereplication. Alkaloids from the Bulbs of Urceolina peruviana (C. Presl) J.F. Macbr. as a Preliminary Test Case. Molecules 2021, 26, 637. [Google Scholar] [CrossRef] [PubMed]
- van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castano-Espriu, L.; Chang, C.; Clark, T.N.; et al. The Natural Products Atlas: An Open Access Knowledge Base for Microbial Natural Products Discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Pereira, D.M.; Valentao, P.; Andrade, P.B. Hybrid MS/NMR methods on the prioritization of natural products: Applications in drug discovery. J. Pharm. Biomed. Anal. 2018, 147, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.N.; Houriet, J.; Vidar, W.S.; Kellogg, J.J.; Todd, D.A.; Cech, N.B.; Linington, R.G. Interlaboratory Comparison of Untargeted Mass Spectrometry Data Uncovers Underlying Causes for Variability. J. Nat. Prod. 2021, 84, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Chanana, S.; Thomas, C.S.; Braun, D.R.; Hou, Y.; Wyche, T.P.; Bugni, T.S. Natural Product Discovery Using Planes of Principal Component Analysis in R (PoPCAR). Metabolites 2017, 7, 34. [Google Scholar] [CrossRef]
- van der Hooft, J.J.J.; Padmanabhan, S.; Burgess, K.E.V.; Barrett, M.P. Urinary antihypertensive drug metabolite screening using molecular networking coupled to high-resolution mass spectrometry fragmentation. Metabolomics 2016, 12, 125. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Schmid, R.; Petras, D.; Nothias, L.-F.; Wang, M.; Aron, A.T.; Jagels, A.; Tsugawa, H.; Rainer, J.; Garcia-Aloy, M.; Duhrkop, K.; et al. Ion identity molecular networking for mass spectrometry-based metabolomics in the GNPS environment. Nat. Commun. 2021, 12, 3832. [Google Scholar] [CrossRef]
- da Silva, R.R.; Wang, M.X.; Nothias, L.F.; van der Hooft, J.J.J.; Caraballo-Rodriguez, A.M.; Fox, E.; Balunas, M.J.; Klassen, J.L.; Lopes, N.P.; Dorrestein, P.C. Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 2018, 14, e1006089. [Google Scholar] [CrossRef]
- Liu, F.J.; Jiang, Y.; Li, P.; Liu, Y.D.; Xin, G.Z.; Yao, Z.P.; Li, H.J. Diagnostic fragmentation-assisted mass spectral networking coupled with in silico dereplication for deep annotation of steroidal alkaloids in medicinal Fritillariae Bulbus. J. Mass Spectrom. 2020, 55, e4528. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Duehrkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Afoullouss, S.; Balsam, A.; Allcock, A.L.; Thomas, O.P. Optimization of LC-MS2 Data Acquisition Parameters for Molecular Networking Applied to Marine Natural Products. Metabolites 2022, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.-F.; Zhang, X.; Zhu, F.; Huo, Z.-Q.; Yao, Q.-Q.; Feng, Q.; Liu, Z.; Zhang, G.-M.; Yao, J.-C.; Liang, H.-B. MS/MS-Based Molecular Networking: An Efficient Approach for Natural Products Dereplication. Molecules 2023, 28, 157. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.M.; Peresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.L. Integration of molecular networking & in-silico MS/MS fragmentation: A novel dereplication strategy in natural products chemistry. Planta Med. 2016, 82, 3317–3323. [Google Scholar] [CrossRef]
- McAvoy, A.C.; Garg, N. Molecular networking-based strategies in mass spectrometry coupled with in silico dereplication of peptidic natural products and gene cluster analysis. Methods Enzymol. 2022, 663, 273–302. [Google Scholar] [CrossRef]
- Moura, M.d.S.; Bellete, B.S.; Vieira, L.C.C.; Sampaio, O.M. Use of Molecular Networking for Compound Annotation in Metabolomics. Rev. Virtual De Quim. 2021, 14, 214–223. [Google Scholar] [CrossRef]
- Treen, D.G.C.; Wang, M.; Xing, S.; Louie, K.B.; Huan, T.; Dorrestein, P.C.; Northen, T.R.; Bowen, B.P. SIMILE enables alignment of tandem mass spectra with statistical significance. Nat. Commun. 2022, 13, 5210. [Google Scholar] [CrossRef]
- Wang, E.; Sorolla, M.A.; Krishnan, P.D.G.; Sorolla, A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules 2020, 10, 248. [Google Scholar] [CrossRef]
- Aron, A.T.; Petras, D.; Schmid, R.; Gauglitz, J.M.; Buttel, I.; Antelo, L.; Zhi, H.; Nuccio, S.-P.; Saak, C.C.; Malarney, K.P.; et al. Native mass spectrometry-based metabolomics identifies metal-binding compounds. Nat. Chem. 2022, 14, 100–109. [Google Scholar] [CrossRef]
- Tripathi, A.; Vázquez-Baeza, Y.; Gauglitz, J.M.; Wang, M.; Dührkop, K.; Nothias-Esposito, M.; Acharya, D.D.; Ernst, M.; van der Hooft, J.J.J.; Zhu, Q.; et al. Chemically informed analyses of metabolomics mass spectrometry data with Qemistree. Nat. Chem. Biol. 2021, 17, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Maansson, M.; Vynne, N.G.; Klitgaard, A.; Nybo, J.L.; Melchiorsen, J.; Nguyen, D.D.; Sanchez, L.M.; Ziemert, N.; Dorrestein, P.C.; Andersen, M.R.; et al. An Integrated Metabolomic and Genomic Mining Workflow to Uncover the Biosynthetic Potential of Bacteria. Msystems 2016, 1, e00028-15. [Google Scholar] [CrossRef]
- Sigrist, R.; Paulo, B.S.; Angolini, C.F.F.; De Oliveira, L.G. Mass Spectrometry-Guided Genome Mining as a Tool to Uncover Novel Natural Products. JoVE 2020, e60825. [Google Scholar] [CrossRef]
- Li, Y.; Ma, B.; Hua, K.; Gong, H.; He, R.; Luo, R.; Bi, D.; Zhou, R.; Langford, P.R.; Jin, H. PPNet: Identifying Functional Association Networks by Phylogenetic Profiling of Prokaryotic Genomes. Microbiol. Spectr. 2023, 11, e0387122. [Google Scholar] [CrossRef] [PubMed]
- Petras, D.; Caraballo-Rodriguez, A.M.; Jarmusch, A.K.; Molina-Santiago, C.; Gauglitz, J.M.; Gentry, E.C.; Belda-Ferre, P.; Romero, D.; Tsunoda, S.M.; Dorrestein, P.C.; et al. Chemical Proportionality within Molecular Networks. Anal. Chem. 2021, 93, 12833–12839. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, K.; Fedarko, M.W.; Rahman, G.; McDonald, D.; Yang, Y.; Zaw, T.; Gonzalez, A.; Janssen, S.; Estaki, M.; Haiminen, N.; et al. EMPress Enables Tree-Guided, Interactive, and Exploratory Analyses of Multi-Omic Data Sets. mSystems 2021, 6, e01216–e01220. [Google Scholar] [CrossRef]
- Protsyuk, I.; Melnik, A.V.; Nothias, L.F.; Rappez, L.; Phapale, P.; Aksenov, A.A.; Bouslimani, A.; Ryazanov, S.; Dorrestein, P.C.; Alexandrov, T. 3D molecular cartography using LC-MS facilitated by Optimus and ‘ili software. Nat. Protoc. 2018, 13, 134–154. [Google Scholar] [CrossRef]
- Floros, D.J.; Jensen, P.R.; Dorrestein, P.C.; Koyama, N. A metabolomics guided exploration of marine natural product chemical space. Metabolomics 2016, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Crusemann, M.; O’Neill, E.C.; Larson, C.B.; Melnik, A.V.; Floros, D.J.; da Silva, R.R.; Jensen, P.R.; Dorrestein, P.C.; Moore, B.S. Prioritizing Natural Product Diversity in a Collection of 146 Bacterial Strains Based on Growth and Extraction Protocols. J. Nat. Prod. 2017, 80, 588–597. [Google Scholar] [CrossRef]
- Fan, B.; Parrot, D.; Bluemel, M.; Labes, A.; Tasdemir, D. Influence of OSMAC-Based Cultivation in Metabolome and Anticancer Activity of Fungi Associated with the Brown Alga Fucus vesiculosus. Mar. Drugs 2019, 17, 67. [Google Scholar] [CrossRef]
- Bracegirdle, J.; Stevenson, L.J.; Page, M.J.; Owen, J.G.; Keyzers, R.A. Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui. Mar. Drugs 2020, 18, 337. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Gaquerel, E. Next-Generation Mass Spectrometry Metabolomics Revives the Functional Analysis of Plant Metabolic Diversity. Annu. Rev. Plant Biol. 2021, 72, 867–891. [Google Scholar] [CrossRef] [PubMed]
- Buedenbender, L.; Astone, F.A.; Tasdemir, D. Bioactive molecular networking for mapping the antimicrobial constituents of the baltic brown alga Fucus vesiculosus. Mar. Drugs 2020, 18, 311. [Google Scholar] [CrossRef]
- Buedenbender, L.; Kumar, A.; Bluemel, M.; Kempken, F.; Tasdemir, D. Genomics- and Metabolomics-Based Investigation of the Deep-Sea Sediment-Derived Yeast, Rhodotorula mucilaginosa 50-3-19/20B. Mar. Drugs 2021, 19, 14. [Google Scholar] [CrossRef]
- Bauermeister, A.; Velasco-Alzate, K.; Dias, T.; Macedo, H.; Ferreira, E.G.; Jimenez, P.C.; Lotufo, T.M.C.; Lopes, N.P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Metabolomic Fingerprinting of Salinispora from Atlantic Oceanic Islands. Front. Microbiol. 2018, 9, 3021. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.R.; Cruesemann, M.; Lechner, A.; Sarkar, A.; Li, J.; Ziemert, N.; Wang, M.; Bandeira, N.; Moore, B.S.; Dorrestein, P.C.; et al. Molecular Networking and Pattern-Based Genome Mining Improves Discovery of Biosynthetic Gene Clusters and Their Products from Salinispora Species. Chem. Biol. 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Almeida, A.; Bauermeister, A.; Luppino, L.; Grilo, I.R.; Oliveira, J.; Sousa, J.R.; Petras, D.; Rodrigues, C.F.; Prieto-Davo, A.; Tasdemir, D.; et al. The Diversity, Metabolomics Profiling, and the Pharmacological Potential of Actinomycetes Isolated from the Estremadura Spur Pockmarks (Portugal). Mar. Drugs 2022, 20, 21. [Google Scholar] [CrossRef]
- Petras, D.; Phelan, V.V.; Acharya, D.; Allen, A.E.; Aron, A.T.; Bandeira, N.; Bowen, B.P.; Belle-Oudry, D.; Boecker, S.; Cummings, D.A., Jr.; et al. GNPS Dashboard: Collaborative exploration of mass spectrometry data in the web browser. Nat. Methods 2022, 19, 134–136. [Google Scholar] [CrossRef]
- Wohlgemuth, G.; Mehta, S.S.; Mejia, R.F.; Neumann, S.; Pedrosa, D.; Pluskal, T.; Schymanski, E.L.; Willighagen, E.L.; Wilson, M.; Wishart, D.S.; et al. SPLASH, a hashed identifier for mass spectra. Nat. Biotechnol. 2016, 34, 1099–1101. [Google Scholar] [CrossRef]
- Ramos, A.E.F.; Le Pogam, P.; Alcover, C.F.; N’Nang, E.O.; Cauchie, G.; Hazni, H.; Awang, K.; Breard, D.; Echavarren, A.M.; Frederich, M.; et al. Collected mass spectrometry data on monoterpene indole alkaloids from natural product chemistry research. Sci. Data 2019, 6, 15. [Google Scholar] [CrossRef]
- Le Pogam, P.; Poupon, E.; Champy, P.; Beniddir, M.A. Implementation of an MS/MS Spectral Library for Monoterpene Indole Alkaloids. Methods Mol. Biol. 2022, 2505, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Soares, V.; Taujale, R.; Garrett, R.; da Silva, A.J.R.; Borges, R.M. Extending compound identification for molecular network using the LipidXplorer database independent method: A proof of concept using glycoalkaloids from Solanum pseudoquina A. St.-Hil. Phytochem. Anal. 2019, 30, 132–138. [Google Scholar] [CrossRef]
- Scotti, M.T.; Herrera-Acevedo, C.; Oliveira, T.B.; Oliveira Costa, R.P.; Konno de Oliveira Santos, S.Y.; Rodrigues, R.P.; Scotti, L.; Da-Costa, F.B. SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites. Molecules 2018, 23, 103. [Google Scholar] [CrossRef]
- Vargas, F.; Weldon, K.C.; Sikora, N.; Wang, M.; Zhang, Z.; Gentry, E.C.; Panitchpakdi, M.W.; Caraballo-Rodriguez, A.M.; Dorrestein, P.C.; Jarmusch, A.K. Protocol for Community-Created Public MS/MS Reference Spectra within the Global Natural Products Social Molecular Networking Infrastructure. Rapid Commun. Mass Spectrom. 2020, 34, e8725. [Google Scholar] [CrossRef]
- Leao, T.F.; Clark, C.M.; Bauermeister, A.; Elijah, E.O.; Gentry, E.C.; Husband, M.; Oliveira, M.F.; Bandeira, N.; Wang, M.; Dorrestein, P.C. Quick-start infrastructure for untargeted metabolomics analysis in GNPS. Nat. Metab. 2021, 3, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Leao, T.; Wang, M.; Moss, N.; da Silva, R.; Sanders, J.; Nurk, S.; Gurevich, A.; Humphrey, G.; Reher, R.; Zhu, Q.; et al. A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria. Mar. Drugs 2021, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Aron, A.T.; Gentry, E.C.; McPhail, K.L.; Nothias, L.-F.; Nothias-Esposito, M.; Bouslimani, A.; Petras, D.; Gauglitz, J.M.; Sikora, N.; Vargas, F.; et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat. Protoc. 2020, 15, 1954–1991. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vazquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, web-enabled microbiome meta-analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef]
- Ma, H.; Liang, H.; Cai, S.; O’Keefe, B.R.; Mooberry, S.L.; Cichewicz, R.H. An Integrated Strategy for the Detection, Dereplication, and Identification of DNA-Binding Biomolecules from Complex Natural Product Mixtures. J. Nat. Prod. 2021, 84, 750–761. [Google Scholar] [CrossRef]
- Quinlan, Z.A.A.; Koester, I.; Aron, A.T.T.; Petras, D.; Aluwihare, L.I.I.; Dorrestein, P.C.C.; Nelson, C.E.E.; Kelly, L.W. ConCISE: Consensus Annotation Propagation of Ion Features in Untargeted Tandem Mass Spectrometry Combining Molecular Networking and in Silico Metabolite Structure Prediction. Metabolites 2022, 12, 1275. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Levitsky, L.; Pilz, M.; Sachsenberg, T.; Huber, F.; Wang, M.; Dorrestein, P.C. Unified and Standardized Mass Spectrometry Data Processing in Python Using Spectrum_utils. J. Proteome Res. 2023, 22, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Covington, B.C.; Seyedsayamdost, M.R. MetEx, a Metabolomics Explorer Application for Natural Product Discovery. ACS Chem. Biol. 2021, 16, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Naake, T.; Gaquerel, E. MetCirc: Navigating mass spectral similarity in high-resolution MS/MS metabolomics data. Bioinformatics 2017, 33, 2419–2420. [Google Scholar] [CrossRef]
- Duhrkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Bocker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Duehrkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Boecker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.-F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30–37. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Shlemov, A.; Mikheenko, A.; Korobeynikov, A.; Cao, L.; Shcherbin, E.; Nothias, L.-F.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 4035. [Google Scholar] [CrossRef]
- Ricart, E.; Pupin, M.; Muller, M.; Lisacek, F. Automatic Annotation and Dereplication of Tandem Mass Spectra of Peptidic Natural Products. Anal. Chem. 2020, 92, 15862–15871. [Google Scholar] [CrossRef]
- Gurevich, A.; Mikheenko, A.; Shlemov, A.; Korobeynikov, A.; Mohimani, H.; Pevzner, P.A. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat. Microbiol. 2018, 3, 319–327. [Google Scholar] [CrossRef]
- Olivon, F.; Roussi, F.; Litaudon, M.; Touboul, D. Optimized experimental workflow for tandem mass spectrometry molecular networking in metabolomics. Anal. Bioanal. Chem. 2017, 409, 5767–5778. [Google Scholar] [CrossRef]
- Wolf, S.; Schmidt, S.; Mueller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, M.; Neumann, S. MetFusion: Integration of compound identification strategies. J. Mass Spectrom. 2013, 48, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S.; de Vos, R.C.H.; Bino, R.J.; Vervoort, J. Automatic Chemical Structure Annotation of an LC-MSn Based Metabolic Profile from Green Tea. Anal. Chem. 2013, 85, 6033–6040. [Google Scholar] [CrossRef]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A Database-Searching Algorithm for Metabolite Identification in Metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef]
- Rasche, F.; Scheubert, K.; Hufsky, F.; Zichner, T.; Kai, M.; Svatos, A.; Boecker, S. Identifying the Unknowns by Aligning Fragmentation Trees. Anal. Chem. 2012, 84, 3417–3426. [Google Scholar] [CrossRef]
- Kangas, L.J.; Metz, T.O.; Isaac, G.; Schrom, B.T.; Ginovska-Pangovska, B.; Wang, L.; Tan, L.; Lewis, R.R.; Miller, J.H. In silico identification software (ISIS): A machine learning approach to tandem mass spectral identification of lipids. Bioinformatics 2012, 28, 1705–1713. [Google Scholar] [CrossRef]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Duehrkop, K.; Nothias, L.-F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef]
- Hoffmann, M.A.; Nothias, L.-F.; Ludwig, M.; Fleischauer, M.; Gentry, E.C.; Witting, M.; Dorrestein, P.C.; Duhrkop, K.; Bocker, S. High-confidence structural annotation of metabolites absent from spectral libraries. Nat. Biotechnol. 2021, 40, 411–421. [Google Scholar] [CrossRef]
- Ludwig, M.; Nothias, L.-F.; Dührkop, K.; Koester, I.; Fleischauer, M.; Hoffmann, M.A.; Petras, D.; Vargas, F.; Morsy, M.; Aluwihare, L.; et al. ZODIAC: Database-independent molecular formula annotation using Gibbs sampling reveals unknown small molecules. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.-F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A Deep Neural Network-Based Structural Classification Tool for Natural Products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Ruttkies, C.; Krauss, M.; Brouard, C.; Kind, T.; Duhrkop, K.; Allen, F.; Vaniya, A.; Verdegem, D.; Bocker, S.; et al. Critical Assessment of Small Molecule Identification 2016: Automated methods. J. Cheminformatics 2017, 9, 22. [Google Scholar] [CrossRef]
- Nikolic, D. CASMI 2016: A manual approach for dereplication of natural products using tandem mass spectrometry. Phytochem. Lett. 2017, 21, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Vaniya, A.; Samra, S.N.; Palazoglu, M.; Tsugawa, H.; Fiehn, O. Using MS-FINDER for identifying 19 natural products in the CASMI 2016 contest. Phytochem. Lett. 2017, 21, 306–312. [Google Scholar] [CrossRef]
- Roullier, C.; Guitton, Y.; Valery, M.; Amand, S.; Prado, S.; du Pont, T.R.; Grovel, O.; Pouchus, Y.F. Automated Detection of Natural Halogenated Compounds from LC-MS Profiles-Application to the Isolation of Bioactive Chlorinated Compounds from Marine-Derived Fungi. Anal. Chem. 2016, 88, 9143–9150. [Google Scholar] [CrossRef]
- Neto, F.C.; Pilon, A.C.; Selegato, D.M.; Freire, R.T.; Gu, H.; Raftery, D.; Lopes, N.P.; Castro-Gamboa, I. Dereplication of Natural Products Using GC-TOF Mass Spectrometry: Improved Metabolite Identification by Spectral Deconvolution Ratio Analysis. Front. Mol. Biosci. 2016, 3, 59. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Cote, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The Proteomics Identifications (PRIDE) Database and Associated Tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef]
- Ternent, T.; Csordas, A.; Qi, D.; Gomez-Baena, G.; Beynon, R.J.; Jones, A.R.; Hermjakob, H.; Vizcaino, J.A. How to submit MS proteomics data to ProteomeXchange via the PRIDE database. Proteomics 2014, 14, 2233–2241. [Google Scholar] [CrossRef]
- Aksenov, A.A.; Laponogov, I.; Zhang, Z.; Doran, S.L.F.; Belluomo, I.; Veselkov, D.; Bittremieux, W.; Nothias, L.F.; Nothias-Esposito, M.; Maloney, K.N.; et al. Auto-deconvolution and molecular networking of gas chromatography-mass spectrometry data. Nat. Biotechnol. 2021, 39, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.P.; Johnson, A.R.; Vega, M.M.; Thomson, R.J.; Carlson, E.E. Ion Mobility Mass Spectrometry as an Efficient Tool for Identification of Streptorubin B in Streptomyces coelicolor M145. J. Nat. Prod. 2020, 83, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Neto, F.C.; Clark, T.N.; Lopes, N.P.; Linington, R.G. Evaluation of Ion Mobility Spectrometry for Improving Constitutional Assignment in Natural Product Mixtures. J. Nat. Prod. 2022, 85, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-Cell MALDI-TOF MS versus 16S rRNA Gene Analysis for Identification and Dereplication of Recurrent Bacterial Isolates. Front. Microbiol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H. The Face of a Molecule. J. Nat. Prod. 2017, 80, 2583–2588. [Google Scholar] [CrossRef]
- Dumolin, C.; Aerts, M.; Verheyde, B.; Schellaert, S.; Vandamme, T.; Van der Jeugt, F.; De Canck, E.; Cnockaert, M.; Wieme, A.D.; Cleenwerck, I.; et al. Introducing SPeDE: High-Throughput Dereplication and Accurate Determination of Microbial Diversity from Matrix-Assisted Laser Desorption-Ionization Time of Flight Mass Spectrometry Data. Msystems 2019, 4, e00437-19. [Google Scholar] [CrossRef]
- Oetjen, J.; Veselkov, K.; Watrous, J.; McKenzie, J.S.; Becker, M.; Hauberg-Lotte, L.; Kobarg, J.H.; Strittmatter, N.; Mroz, A.K.; Hoffmann, F.; et al. Benchmark datasets for 3D MALDI- and DESI-imaging mass spectrometry. Gigascience 2015, 4, 20. [Google Scholar] [CrossRef]
- Petras, D.; Jarmusch, A.K.; Dorrestein, P.C. From single cells to our planet-recent advances in using mass spectrometry for spatially resolved metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 24–31. [Google Scholar] [CrossRef]
- Carneiro, K.; de Brito, J.M.; Rossi, M.I.D. Development by Three-Dimensional Approaches and Four-Dimensional Imaging: To the Knowledge Frontier and beyond. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 1–8. [Google Scholar] [CrossRef]
- Li, L.F.; Zhou, Q.; Voss, T.C.; Quick, K.L.; LaBarbera, D.V. High-throughput imaging: Focusing in on drug discovery in 3D. Methods 2016, 96, 97–102. [Google Scholar] [CrossRef]
- Corcoran, O. Hit Discovery from Natural Products in Pharmaceutical R&D. Emagres 2015, 4, 455–461. [Google Scholar] [CrossRef]
- Pauli, G.F.; Niemitz, M.; Bisson, J.; Lodewyk, M.W.; Soldi, C.; Shaw, J.T.; Tantillo, D.J.; Saya, J.M.; Vos, K.; Kleinnijenhuis, R.A.; et al. Toward Structural Correctness: Aquatolide and the Importance of 1D Proton NMR FID Archiving. J. Org. Chem. 2016, 81, 878–889. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Simmler, C.; McAlpine, J.B.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Digital NMR Profiles as Building Blocks: Assembling H-1 Fingerprints of Steviol Glycosides. J. Nat. Prod. 2015, 78, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Chen, S.-N.; Lankin, D.C.; Bisson, J.; Case, R.J.; Chadwick, L.R.; Goedecke, T.; Inui, T.; Krunic, A.; Jaki, B.U.; et al. Essential Parameters for Structural Analysis and Dereplication by H-1 NMR Spectroscopy. J. Nat. Prod. 2014, 77, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Bruguiere, A.; Derbre, S.; Dietsch, J.; Leguy, J.; Rahier, V.; Pottier, Q.; Breard, D.; Suor-Cherer, S.; Viault, G.; Le Ray, A.-M.; et al. MixONat, a Software for the Dereplication of Mixtures Based on C-13 NMR Spectroscopy. Anal. Chem. 2020, 92, 8793–8801. [Google Scholar] [CrossRef]
- Bruguiere, A.; Derbre, S.; Breard, D.; Tomi, F.; Nuzillard, J.-M.; Richomme, P. 13C NMR Dereplication Using MixONat Software: A Practical Guide to Decipher Natural Products Mixtures. Planta Med. 2021, 87, 1061–1068. [Google Scholar] [CrossRef]
- Bakiri, A.; Hubert, J.; Reynaud, R.; Lanthony, S.; Harakat, D.; Renault, J.-H.; Nuzillard, J.-M. Computer-Aided C-13 NMR Chemical Profiling of Crude Natural Extracts without Fractionation. J. Nat. Prod. 2017, 80, 1387–1396. [Google Scholar] [CrossRef]
- Martinez-Trevino, S.H.; Uc-Cetina, V.; Fernandez-Herrera, M.A.; Merino, G. Prediction of Natural Product Classes Using Machine Learning and C-13 NMR Spectroscopic Data. J. Chem. Inf. Model. 2020, 60, 3376–3386. [Google Scholar] [CrossRef]
- Qiu, F.; McAlpine, J.B.; Lankin, D.C.; Burton, I.; Karakach, T.; Chen, S.-N.; Pauli, G.F. 2D NMR Barcoding and Differential Analysis of Complex Mixtures for Chemical Identification: The Actaea Triterpenes. Anal. Chem. 2014, 86, 3964–3972. [Google Scholar] [CrossRef]
- Bakiri, A.; Hubert, J.; Reynaud, R.; Lambert, C.; Martinez, A.; Renault, J.-H.; Nuzillard, J.-M. Reconstruction of HMBC Correlation Networks: A Novel NMR-Based Contribution to Metabolite Mixture Analysis. J. Chem. Inf. Model. 2018, 58, 262–270. [Google Scholar] [CrossRef]
- Kuhn, S.; Colreavy-Donnelly, S.; de Souza, J.S.; Borges, R.M. An integrated approach for mixture analysis using MS and NMR techniques. Faraday Discuss. 2019, 218, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Idelbayev, Y.; Roberts, N.; Tao, Y.; Nannapaneni, Y.; Duggan, B.M.; Min, J.; Lin, E.C.; Gerwick, E.C.; Cottrell, G.W.; et al. Small Molecule Accurate Recognition Technology (SMART) to Enhance Natural Products Research. Sci. Rep. 2017, 7, 14243. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Zhang, C.; Cottrell, G.W.; Gerwick, W.H. SMART-Miner: A convolutional neural network-based metabolite identification from H-1-C-13 HSQC spectra. Magn. Reson. Chem. 2021, 60, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Reher, R.; Kim, H.W.; Zhang, C.; Mao, H.H.; Wang, M.; Nothias, L.-F.; Caraballo-Rodriguez, A.M.; Glukhov, E.; Teke, B.; Leao, T.; et al. A Convolutional Neural Network-Based Approach for the Rapid Annotation of Molecularly Diverse Natural Products. J. Am. Chem. Soc. 2020, 142, 4114–4120. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.-P.; Yu, Y.; Liu, Q.-H.; Zhou, M.-Y.; Zhu, G.-Y.; Bai, L.-P.; Zhang, W.; Jiang, Z.-H. 2D NMR-Based MatchNat Dereplication Strategy Enables Explosive Discovery of Novel Diterpenoid Alkaloids. Chin. J. Chem. 2022, 40, 2169–2178. [Google Scholar] [CrossRef]
- Zani, C.L.; Carroll, A.R. Database for Rapid Dereplication of Known Natural Products Using Data from MS and Fast NMR Experiments. J. Nat. Prod. 2017, 80, 1758–1766. [Google Scholar] [CrossRef]
- Kleks, G.; Holland, D.C.; Porter, J.; Carroll, A.R. Natural products dereplication by diffusion ordered NMR spectroscopy (DOSY). Chem. Sci. 2021, 12, 10930–10943. [Google Scholar] [CrossRef]
- Diaz-Allen, C.; Spjut, R.W.; Kinghorn, A.D.; Rakotondraibe, H.L. Prioritizing natural product compounds using 1D-TOCSY NMR spectroscopy. Trends Org. Chem. 2021, 22, 99–114. [Google Scholar]
- Borges, R.M.; Mendes Resende, J.V.; Pinto, A.P.; Garrido, B.C. Exploring correlations between MS and NMR for compound identification using essential oils: A pilot study. Phytochem. Anal. 2022, 33, 533–542. [Google Scholar] [CrossRef]
- Egan, J.M.; van Santen, J.A.; Liu, D.Y.; Linington, R.G. Development of an NMR-Based Platform for the Direct Structural Annotation of Complex Natural Products Mixtures. J. Nat. Prod. 2021, 84, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Flores-Bocanegra, L.; Al Subeh, Z.Y.; Egan, J.M.; El-Elimat, T.; Raja, H.A.; Burdette, J.E.; Pearce, C.J.; Linington, R.G.; Oberlies, N.H. Dereplication of Fungal Metabolites by NMR-Based Compound Networking Using MADByTE. J. Nat. Prod. 2022, 85, 614–624. [Google Scholar] [CrossRef]
- van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2022, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Pinto, E.; Torres, M.A.; Dorr, F.; Mazur-Marzec, H.; Szubert, K.; Tartaglione, L.; Dell’Aversano, C.; Miles, C.O.; Beach, D.G.; et al. CyanoMetDB, a comprehensive public database of secondary metabolites from cyanobacteria. Water Res. 2021, 196, 117017. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sayeeda, Z.; Budinski, Z.; Guo, A.; Lee, B.L.; Berjanskii, M.; Rout, M.; Peters, H.; Dizon, R.; Mah, R.; et al. NP-MRD: The Natural Products Magnetic Resonance Database. Nucleic Acids Res. 2022, 50, D665–D677. [Google Scholar] [CrossRef]
- Moumbock, A.F.A.; Gao, M.; Qaseem, A.; Li, J.; Kirchner, P.A.; Ndingkokhar, B.; Bekono, B.D.; Simoben, C.V.; Babiaka, S.B.; Malange, Y.I.; et al. StreptomeDB 3.0: An updated compendium of streptomycetes natural products. Nucleic Acids Res. 2021, 49, D600–D604. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: A comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2021, 49, D509–D515. [Google Scholar] [CrossRef]
- Scott, T.A.; Piel, J. The hidden enzymology of bacterial natural product biosynthesis. Nat. Rev. Chem. 2019, 3, 404–425. [Google Scholar] [CrossRef]
- Reen, F.J.; Romano, S.; Dobson, A.D.W.; O’Gara, F. The Sound of Silence: Activating Silent Biosynthetic Gene Clusters in Marine Microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef]
- Paoli, L.; Ruscheweyh, H.J.; Forneris, C.C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A.; et al. Biosynthetic potential of the global ocean microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef]
- Kurita, K.L.; Linington, R.G. Connecting Phenotype and Chemotype: High-Content Discovery Strategies for Natural Products Research. J. Nat. Prod. 2015, 78, 587–596. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Scholz, M.B.; Lo, C.C.; Chain, P.S.G. Next generation sequencing and bioinformatic bottlenecks: The current state of metagenomic data analysis. Curr. Opin. Biotechnol. 2012, 23, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef]
- Fakruddin, M.; Chowdhury, A.; Hossain, M.; Mannan, K.S.B.; Mazumdar, R.M. Pyrosequencing-principles and applications. Life 2012, 2, 65–76. [Google Scholar]
- Kawashima, E.H.; Farinelli, L.; Mayer, P. Method of Nuclec Acid Amplification. WO1998GB00961. 1 April 1997. [Google Scholar]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genom. 2017, 18, 602. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Hwang, S.; Kim, J.; Cho, S.; Palsson, B.; Cho, B.-K. Mini review: Genome mining approaches for the identification of secondary metabolite biosynthetic gene clusters in Streptomyces. Comput. Struct. Biotechnol. J. 2020, 18, 1548–1556. [Google Scholar] [CrossRef]
- Medema, M.H.; Trefzer, A.; Kovalchuk, A.; van den Berg, M.; Muller, U.; Heijne, W.; Wu, L.; Alam, M.T.; Ronning, C.M.; Nierman, W.C.; et al. The Sequence of a 1.8-Mb Bacterial Linear Plasmid Reveals a Rich Evolutionary Reservoir of Secondary Metabolic Pathways. Genome Biol. Evol. 2010, 2, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Jeong, H.; Yu, D.S.; Fischbach, M.A.; Park, H.-S.; Kim, J.J.; Seo, J.-S.; Jensen, S.E.; Oh, T.K.; Lee, K.J.; et al. Draft Genome Sequence of Streptomyces clavuligerus NRRL 3585, a Producer of Diverse Secondary Metabolites. J. Bacteriol. 2010, 192, 6317–6318. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, N.; Jeong, Y.; Lee, Y.; Kim, W.; Cho, S.; Palsson, B.O.; Cho, B.-K. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 2019, 47, 6114–6129. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, W.; Hwang, S.; Lee, Y.; Cho, S.; Palsson, B.; Cho, B.-K. Thirty complete Streptomyces genome sequences for mining novel secondary metabolite biosynthetic gene clusters. Sci. Data 2020, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef]
- Corre, C.; Challis, G.L. New natural product biosynthetic chemistry discovered by genome mining. Nat. Prod. Rep. 2009, 26, 977–986. [Google Scholar] [CrossRef]
- Chevrette, M.G.; Aicheler, F.; Kohlbacher, O.; Currie, C.R.; Medema, M.H. SANDPUMA: Ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 2017, 33, 3202–3210. [Google Scholar] [CrossRef]
- Behsaz, B.; Bode, E.; Gurevich, A.; Shi, Y.-N.; Grundmann, F.; Acharya, D.; Caraballo-Rodriguez, A.M.; Bouslimani, A.; Panitchpakdi, M.; Linck, A.; et al. Integrating genomics and metabolomics for scalable non-ribosomal peptide discovery. Nat. Commun. 2021, 12, 3225. [Google Scholar] [CrossRef]
- Kunyavskaya, O.; Tagirdzhanov, A.M.; Caraballo-Rodriguez, A.M.; Nothias, L.-F.; Dorrestein, P.C.; Korobeynikov, A.; Mohimani, H.; Gurevich, A. Nerpa: A Tool for Discovering Biosynthetic Gene Clusters of Bacterial Nonribosomal Peptides. Metabolites 2021, 11, 693. [Google Scholar] [CrossRef]
- Novak, J.; Lemr, K.; Schug, K.A.; Havlicek, V. CycloBranch: De Novo Sequencing of Nonribosomal Peptides from Accurate Product Ion Mass Spectra. J. Am. Soc. Mass Spectrom. 2015, 26, 1780–1786. [Google Scholar] [CrossRef]
- Privratsky, J.; Novak, J. MassSpecBlocks: A web-based tool to create building blocks and sequences of nonribosomal peptides and polyketides for tandem mass spectra analysis. J. Cheminformatics 2021, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ibrahim, A.; Johnston, C.W.; Skinnider, M.A.; Ma, B.; Magarvey, N.A. Exploration of Nonribosomal Peptide Families with an Automated Informatic Search Algorithm. Chem. Biol. 2015, 22, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Sundaramurthi, J.C.; Lee, J.; Kandimalla, M.; Chen, I.A.; Kyrpides, N.C.; Reddy, T.B.K. Genomes OnLine Database (GOLD) v.8: Overview and updates. Nucleic Acids Res. 2021, 49, D723–D733. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Kleigrewe, K.; Almaliti, J.; Tian, I.Y.; Kinnel, R.B.; Korobeynikov, A.; Monroe, E.A.; Duggan, B.M.; Di Marzo, V.; Sherman, D.H.; Dorrestein, P.C.; et al. Combining Mass Spectrometric Metabolic Profiling with Genomic Analysis: A Powerful Approach for Discovering Natural Products from Cyanobacteria. J. Nat. Prod. 2015, 78, 1671–1682. [Google Scholar] [CrossRef]
- Moss, N.A.; Bertin, M.J.; Kleigrewe, K.; Leao, T.F.; Gerwick, L.; Gerwick, W.H. Integrating mass spectrometry and genomics for cyanobacterial metabolite discovery. J. Ind. Microbiol. Biotechnol. 2016, 43, 313–324. [Google Scholar] [CrossRef]
- Ishaque, N.M.; Burgsdorf, I.; Malit, J.J.L.; Saha, S.; Teta, R.; Ewe, D.; Kannabiran, K.; Hrouzek, P.; Steindler, L.; Costantino, V.; et al. Isolation, Genomic and Metabolomic Characterization of Streptomyces tendae VITAKN with Quorum Sensing Inhibitory Activity from Southern India. Microorganisms 2020, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Welzel, M.; Lange, A.; Heider, D.; Schwarz, M.; Freisleben, B.; Jensen, M.; Boenigk, J.; Beisser, D. Natrix: A Snakemake-based workflow for processing, clustering, and taxonomically assigning amplicon sequencing reads. BMC Bioinform. 2020, 21, 526. [Google Scholar] [CrossRef]
- Schorn, M.A.; Verhoeven, S.; Ridder, L.; Huber, F.; Acharya, D.D.; Aksenov, A.A.; Aleti, G.; Moghaddam, J.A.; Aron, A.T.; Aziz, S.; et al. A community resource for paired genomic and metabolomic data mining. Nat. Chem. Biol. 2021, 17, 363–368. [Google Scholar] [CrossRef]
- Walker, A.S.; Clardy, J. A Machine Learning Bioinformatics Method to Predict Biological Activity from Biosynthetic Gene Clusters. J. Chem. Inf. Model. 2021, 61, 2560–2571. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, H.-R.; Jeong, D.-E.; Choi, S.-K. Cytosine Base Editor-Mediated Multiplex Genome Editing to Accelerate Discovery of Novel Antibiotics in Bacillus subtilis and Paenibacillus polymyxa. Front. Microbiol. 2021, 12, 691839. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar] [CrossRef]
- Wilson, M.C.; Mori, T.; Rückert, C.; Uria, A.R.; Helf, M.J.; Takada, K.; Gernert, C.; Steffens, U.A.; Heycke, N.; Schmitt, S.; et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 2014, 506, 58–62. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Chen, C.; Wang, L.; Ning, K. Integrating pan-genome with metagenome for microbial community profiling. Comput. Struct. Biotechnol. J. 2021, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Vernikos, G.S. A Review of Pangenome Tools and Recent Studies. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 89–112. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]
- Mohite, O.S.; Lloyd, C.J.; Monk, J.M.; Weber, T.; Palsson, B.O. Pangenome Analysis of Enterobacteria Reveals Richness of Secondary Metabolite Gene Clusters and their Associated Gene Sets. bioRxiv 2019. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Aires-de-Sousa, J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Albanese, D.; Donati, C. Large-scale quality assessment of prokaryotic genomes with metashot/prok-quality. F1000Research 2021, 10, 822. [Google Scholar] [CrossRef]
- Meleshko, D.; Mohimani, H.; Tracanna, V.; Hajirasouliha, I.; Medema, M.H.; Korobeynikov, A.; Pevzner, P.A. BiosyntheticSPAdes: Reconstructing biosynthetic gene clusters from assembly graphs. Genome Res. 2019, 29, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef]
- van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]
- Santos-Aberturas, J.; Chandra, G.; Frattaruolo, L.; Lacret, R.; Pham, T.H.; Vior, N.M.; Eyles, T.H.; Truman, A.W. Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res. 2019, 47, 4624–4637. [Google Scholar] [CrossRef]
- Mungan, M.D.; Alanjary, M.; Blin, K.; Weber, T.; Medema, M.H.; Ziemert, N. ARTS 2.0: Feature Updates and Expansion of the Antibiotic Resistant Target Seeker for Comparative Genome Mining. Nucleic Acids Res. 2020, 48, W546–W552. [Google Scholar] [CrossRef]
- Almeida, H.; Palys, S.; Tsang, A.; Diallo, A.B. TOUCAN: A framework for fungal biosynthetic gene cluster discovery. NAR Genom. Bioinform. 2020, 2, lqaa098. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kautsar, S.A.; Medema, M.H.; Weber, T. The antiSMASH database version 3: Increased taxonomic coverage and new query features for modular enzymes. Nucleic Acids Res. 2021, 49, D639–D643. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Mueller, R.; Wohlleben, W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef]
- Palaniappan, K.; Chen, I.M.A.; Chu, K.; Ratner, A.; Seshadri, R.; Kyrpides, N.C.; Ivanova, N.N.; Mouncey, N.J. IMG-ABC v.5.0: An Update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res. 2020, 48, D422–D430. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; van der Hooft, J.J.J.; de Ridder, D.; Medema, M.H. BiG-SLiCE: A Highly Scalable Tool Maps the Diversity of 1.2 Million Biosynthetic Gene Clusters. GigaScience 2021, 10, giaa154. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Weber, T.; Medema, M.H. BiG-FAM: The biosynthetic gene cluster families database. Nucleic Acids Res. 2021, 49, D490–D497. [Google Scholar] [CrossRef]
- Merwin, N.J.; Mousa, W.K.; Dejong, C.A.; Skinnider, M.A.; Cannon, M.J.; Li, H.; Dial, K.; Gunabalasingam, M.; Johnston, C.; Magarvey, N.A. DeepRiPP integrates multiomics data to automate discovery of novel ribosomally synthesized natural products. Proc. Natl. Acad. Sci. USA 2020, 117, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Khater, S.; Gupta, M.; Sain, N.; Mohanty, D. RiPPMiner: A bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res. 2017, 45, W80–W88. [Google Scholar] [CrossRef] [PubMed]
- Tietz, J.I.; Schwalen, C.J.; Patel, P.S.; Maxson, T.; Blair, P.M.; Tai, H.C.; Zakai, U.I.; Mitchell, D.A. A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 2017, 13, 470–478. [Google Scholar] [CrossRef]
- Mohimani, H.; Liu, W.-T.; Kersten, R.D.; Moore, B.S.; Dorrestein, P.C.; Pevzner, P.A. NRPquest: Coupling Mass Spectrometry and Genome Mining for Nonribosomal Peptide Discovery. J. Nat. Prod. 2014, 77, 1902–1909. [Google Scholar] [CrossRef]
- Cao, L.; Gurevich, A.; Alexander, K.L.; Naman, C.B.; Leao, T.; Glukhov, E.; Luzzatto-Knaan, T.; Vargas, F.; Quinn, R.; Bouslimani, A.; et al. MetaMiner: A Scalable Peptidogenomics Approach for Discovery of Ribosomal Peptide Natural Products with Blind Modifications from Microbial Communities. Cell Syst. 2019, 9, 600.e4–608.e4. [Google Scholar] [CrossRef]
- Behsaz, B.; Mohimani, H.; Gurevich, A.; Prjibelski, A.; Fisher, M.; Vargas, F.; Smarr, L.; Dorrestein, P.C.; Mylne, J.S.; Pevzner, P.A. De Novo Peptide Sequencing Reveals Many Cyclopeptides in the Human Gut and Other Environments. Cell Syst. 2020, 10, 99.e105–108.e105. [Google Scholar] [CrossRef]
- Hjoerleifsson Eldjarn, G.; Ramsay, A.; van der Hooft, J.J.J.; Duncan, K.R.; Soldatou, S.; Rousu, J.; Daly, R.; Wandy, J.; Rogers, S. Ranking microbial metabolomic and genomic links in the NPLinker framework using complementary scoring functions. PLoS Comput. Biol. 2021, 17, 1008920. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Paalvast, Y.; Nguyen, D.D.; Melnik, A.; Dorrestein, P.C.; Takano, E.; Breitling, R. Pep2Path: Automated Mass Spectrometry-Guided Genome Mining of Peptidic Natural Products. PLoS Comput. Biol. 2014, 10, 1003822. [Google Scholar] [CrossRef]
- Williams, A.N.; Sorout, N.; Cameron, A.J.; Stavrinides, J. The Integration of Genome Mining, Comparative Genomics, and Functional Genetics for Biosynthetic Gene Cluster Identification. Front. Genet. 2020, 11, 1543. [Google Scholar] [CrossRef] [PubMed]
- Leonard, R.R.; Leleu, M.; Van Vlierberghe, M.; Cornet, L.; Kerff, F.; Baurain, D. ToRQuEMaDA: Tool for retrieving queried Eubacteria, metadata and dereplicating assemblies. Peerj 2021, 9, e11348. [Google Scholar] [CrossRef] [PubMed]
- Vandova, G.A.; Nivina, A.; Khosla, C.; Davis, R.W.; Fisher, C.R.; Hillenmeyer, M.E. Identification of polyketide biosynthetic gene clusters that harbor self-resistance target genes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Bhattacharya, N.; Olm, M.R.; Song, Y.S.; Banfield, J.F. Transporter genes in biosynthetic gene clusters predict metabolite characteristics and siderophore activity. Genome Res. 2021, 31, 239–250. [Google Scholar] [CrossRef]
- Iglesias, A.; Latorre-Perez, A.; Stach, J.E.M.; Porcar, M.; Pascual, J. Out of the Abyss: Genome and Metagenome Mining Reveals Unexpected Environmental Distribution of Abyssomicins. Front. Microbiol. 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Johns, N.I.; Gomes, A.L.C.; Yim, S.S.; Yang, A.; Blazejewski, T.; Smillie, C.S.; Smith, M.B.; Alm, E.J.; Kosuri, S.; Wang, H.H. Metagenomic mining of regulatory elements enables programmable species-selective gene expression. Nat. Methods 2018, 15, 323–329. [Google Scholar] [CrossRef]
- Sheth, R.U.; Cabral, V.; Chen, S.P.; Wang, H.H. Manipulating Bacterial Communities by in situ Microbiome Engineering. Trends Genet. 2016, 32, 189–200. [Google Scholar] [CrossRef]
- Adnani, N.; Rajski, S.R.; Bugni, T.S. Symbiosis-inspired approaches to antibiotic discovery. Nat. Prod. Rep. 2017, 34, 784–814. [Google Scholar] [CrossRef] [PubMed]
- Atencio, L.A.; Boya, P.C.A.; Martin, H.C.; Mejía, L.C.; Dorrestein, P.C.; Gutiérrez, M. Genome Mining, Microbial Interactions, and Molecular Networking Reveals New Dibromoalterochromides from Strains of Pseudoalteromonas of Coiba National Park-Panama. Mar. Drugs 2020, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.M.; Hirschmann, M.; Shi, Y.N.; Ahmed, S.; Abebew, D.; Tobias, N.J.; Grun, P.; Crames, J.J.; Poschel, L.; Kuttenlochner, W.; et al. Global analysis of biosynthetic gene clusters reveals conserved and unique natural products in entomopathogenic nematode-symbiotic bacteria. Nat. Chem. 2022, 14, 701–712. [Google Scholar] [CrossRef]
- Wilkins, L.G.E.; Ettinger, C.L.; Jospin, G.; Eisen, J.A. Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal pools in Kamchatka, Russia. Sci. Rep. 2019, 9, 3059. [Google Scholar] [CrossRef]
- Sysoev, M.; Grötzinger, S.W.; Renn, D.; Eppinger, J.; Rueping, M.; Karan, R. Bioprospecting of Novel Extremozymes from Prokaryotes—The Advent of Culture-Independent Methods. Front. Microbiol. 2021, 12, 196. [Google Scholar] [CrossRef]
- Borer, B.; Or, D. Spatiotemporal metabolic modeling of bacterial life in complex habitats. Curr. Opin. Biotechnol. 2021, 67, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Trivella, D.B.B.; de Felicio, R. The Tripod for Bacterial Natural Product Discovery: Genome Mining, Silent Pathway Induction, and Mass Spectrometry-Based Molecular Networking. mSystems 2018, 3, e00160-17. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Baichoo, S.; Blencowe, B.J.; Bork, P.; Borodovsky, M.; Brooksbank, C.; Chain, P.S.G.; Colwell, R.R.; Daffonchio, D.G.; Danchin, A.; et al. Toward unrestricted use of public genomic data. Science 2019, 363, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Imperatore, C.; Mangoni, A.; Della Sala, G.; Taglialatela-Scafati, O. Challenges in the configuration assignment of natural products. A case-selective perspective. Nat. Prod. Rep. 2019, 36, 476–489. [Google Scholar] [CrossRef]
- Cen-Pacheco, F.; Rodriguez, J.; Norte, M.; Fernandez, J.J.; Daranas, A.H. Connecting Discrete Stereoclusters by Using DFT and NMR Spectroscopy: The Case of Nivariol. Chem. A Eur. J. 2013, 19, 8525–8532. [Google Scholar] [CrossRef]
- Huo, Z.Q.; Zhu, F.; Zhang, X.W.; Zhang, X.; Liang, H.B.; Yao, J.C.; Liu, Z.; Zhang, G.M.; Yao, Q.Q.; Qin, G.F. Approaches to Configuration Determinations of Flexible Marine Natural Products: Advances and Prospects. Mar. Drugs 2022, 20, 333. [Google Scholar] [CrossRef]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466. [Google Scholar] [CrossRef]
- Sairenji, S.; Kikuchi, T.; Abozeid, M.A.; Takizawa, S.; Sasai, H.; Ando, Y.; Ohmatsu, K.; Ooi, T.; Fujita, M. Determination of the absolute configuration of compounds bearing chiral quaternary carbon centers using the crystalline sponge method. Chem. Sci. 2017, 8, 5132–5136. [Google Scholar] [CrossRef]
- Urban, S.; Brkljaca, R.; Hoshino, M.; Lee, S.; Fujita, M. Determination of the Absolute Configuration of the Pseudo-Symmetric Natural Product Elatenyne by the Crystalline Sponge Method. Angew. Chem. Int. Ed. 2016, 55, 2678–2682. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Awakawa, T.; Mori, T.; Abe, I. Unusual chemistries in fungal meroterpenoid biosynthesis. Curr. Opin. Chem. Biol. 2016, 31, 1–7. [Google Scholar] [CrossRef]
- Cardenal, A.D.; Ramadhar, T.R. The crystalline sponge method: Quantum chemical in silico derivation and analysis of guest binding energies. Crystengcomm 2021, 23, 7570–7575. [Google Scholar] [CrossRef]
- Gee, W.J. The growing importance of crystalline molecular flasks and the crystalline sponge method. Dalton Trans. 2017, 46, 15979–15986. [Google Scholar] [CrossRef]
- de Poel, W.; Tinnemans, P.T.; Duchateau, A.L.L.; Honing, M.; Rutjes, F.; Vlieg, E.; de Gelder, R. Racemic and Enantiopure Camphene and Pinene Studied by the Crystalline Sponge Method. Cryst. Growth Des. 2018, 18, 126–132. [Google Scholar] [CrossRef]
- Schlesinger, C.; Tapmeyer, L.; Gumbert, S.D.; Prill, D.; Bolte, M.; Schmidt, M.U.; Saal, C. Absolute Configuration of Pharmaceutical Research Compounds Determined by X-ray Powder Diffraction. Angew. Chem. Int. Ed. Engl. 2018, 57, 9150–9153. [Google Scholar] [CrossRef]
- Santoro, E.; Vergura, S.; Scafato, P.; Belviso, S.; Masi, M.; Evidente, A.; Superchi, S. Absolute Configuration Assignment to Chiral Natural Products by Biphenyl Chiroptical Probes: The Case of the Phytotoxins Colletochlorin A and Agropyrenol. J. Nat. Prod. 2020, 83, 1061–1068. [Google Scholar] [CrossRef]
- Masi, M.; Cimmino, A.; Boari, A.; Tuzi, A.; Zonno, M.C.; Baroncelli, R.; Vurro, M.; Evidente, A. Colletochlorins E and F, New Phytotoxic Tetrasubstituted Pyran-2-One and Dihydrobenzofuran, Isolated from Colletotrichum higginsianum with Potential Herbicidal Activity. J. Agric. Food Chem. 2017, 65, 7903–7905. [Google Scholar] [CrossRef]
- Andolfi, A.; Cimmino, A.; Vurro, M.; Berestetskiy, A.; Troise, C.; Zonno, M.C.; Motta, A.; Evidente, A.; Arya, N.; Mishra, S.K.; et al. Application of crystalline matrices for the structural determination of organic molecules. Phytochemistry 2012, 79, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, D.J. Walking in the woods with quantum chemistry—Applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 2013, 30, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- McCann, D.M.; Stephens, P.J. Determination of absolute configuration using density functional theory calculations of optical rotation and electronic circular dichroism: Chiral alkenes. J. Org. Chem. 2006, 71, 6074–6098. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, D.; Šekutor, M.; Stiefermann, M.; Tschakert, J.; Dahl, J.E.P.; Carlson, R.M.K.; Schirmeisen, A.; Schreiner, P.R. Assigning the absolute configuration of single aliphatic molecules by visual inspection. Nat. Commun. 2018, 9, 2420. [Google Scholar] [CrossRef] [PubMed]
- Saito, F.; Gerbig, D.; Becker, J.; Schreiner, P.R. Absolute Configuration of Trans-Perhydroazulene. Org. Lett. 2020, 22, 3895–3899. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Morales-Amador, A.; de Vera, C.R.; Marquez-Fernandez, O.; Daranas, A.H.; Padron, J.M.; Fernandez, J.J.; Souto, M.L.; Norte, M. Pinnatifidenyne-Derived Ethynyl Oxirane Acetogenins from Laurencia viridis. Mar. Drugs 2018, 16, 5. [Google Scholar] [CrossRef]
- Domínguez, H.J.; Napolitano, J.G.; Fernández-Sánchez, M.T.; Cabrera-García, D.; Novelli, A.; Norte, M.; Fernández, J.J.; Daranas, A.H.; Dorta, E.; Díaz-Marrero, A.R.; et al. Belizentrin, a Highly Bioactive Macrocycle from the Dinoflagellate Prorocentrum belizeanum. Org. Lett. 2014, 16, 4546–4549. [Google Scholar] [CrossRef]
- Parella, T.; Espinosa, J.F.; Porto, S.; Seco, J.M.; Quiñoá, E.; Riguera, R. Long-range proton-carbon coupling constants: NMR methods and applications. Resin-bound chiral derivatizing agents for assignment of configuration by NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 17–55. [Google Scholar] [CrossRef]
- Saurí, J.; Nolis, P.; Parella, T. How to measure long-range proton-carbon coupling constants from 1H-selective HSQMBC experiments. Magn. Reson. Chem. MRC 2020, 58, 363–375. [Google Scholar] [CrossRef]
- Motiram-Corral, K.; Nolis, P.; Saurí, J.; Parella, T. LR-HSQMBC versus LR-selHSQMBC: Enhancing the Observation of Tiny Long-Range Heteronuclear NMR Correlations. J. Nat. Prod. 2020, 83, 1275–1282. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, K.-Z.; Yan, H.-W.; Feng, Z.-M.; Yang, Y.-N.; Jiang, J.-S.; Zhang, P.-C. An ingenious method for the determination of the relative and absolute configurations of compounds containing aryl-glycerol fragments by 1H NMR spectroscopy. RSC Adv. 2021, 11, 686–689. [Google Scholar] [CrossRef]
- Xu, G.; Elkin, M.; Tantillo, D.J.; Newhouse, T.R.; Maimone, T.J. Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem. Int. Ed. 2017, 56, 12498–12502. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, Y.; Lehr, K.; Colas, L.; Breit, B. Assignment of relative configuration of desoxypropionates by 1H NMR spectroscopy: Method development, proof of principle by asymmetric total synthesis of xylarinic acid A and applications. Chemistry 2012, 18, 7071–7081. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.; Brito, I.; Cueto, M.; D’Croz, L.; Darias, J. C-13 NMR-based empirical rules to determine the configuration of fatty acid butanolides. Novel gamma-dilactones from Pterogorgia spp. Org. Lett. 2006, 8, 5001–5004. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Marrero, A.R.; Cueto, M.; Dorta, E.; Rovirosa, J.; San-Martin, A.; Darias, J. Geometry and halogen regiochemistry determination of vicinal vinyl dihalides by H-1 and C-13 NMR. Application to the structure elucidation of prefuroplocamioid, an unusual marine monoterpene. Org. Lett. 2002, 4, 2949–2952. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.A.; Mosher, H.S.; de Poel, W.; Tinnemans, P.; Duchateau, A.L.L.; Honing, M.; Rutjes, F.P.J.T.; Vlieg, E.; de Gelder, R. The crystalline sponge method in water. J. Am. Chem. Soc. 1973, 95, 8311–8317. [Google Scholar]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of configuration and f-19 chemical-shifts of alpha-methoxy-alpha-trifluoromethylphenylacetate derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher method—The absolute-configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Kusumi, T.; Ohtani, I.I. Determination of the absolute configuration of biologically active compounds by the modified Mosher’s method. In The Biology—Chemistry Interface: A Tribute To Koji Nakanishi; CRC Press: Boca Raton, FL, USA, 1999; pp. 103–137. [Google Scholar]
- Nagai, Y.; Kusumi, T.; Ohtani, I.; Kashman, Y.; Kakisawa, H. The absolute configurations of marine terpenoids. Tetrahedron Lett. 1995, 36, 1275–1282. [Google Scholar]
- Ferreiro, M.J.; Latypov, S.K.; Quinoa, E.; Riguera, R. Assignment of the absolute configuration of alpha-chiral carboxylic acids by 1H NMR spectroscopy. J. Org. Chem. 2000, 65, 2658–2666. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R.; Stephens, P.J.; McCann, D.M.; Devlin, F.J.; Smith, A.B., 3rd; Sullivan, G.R.; Dale, J.A.; Mosher, H.S.; et al. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron Asymmetry 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of anthrax tetrasaccharide and related tetrasaccharide. J. Org. Chem. 2008, 73, 5211–5220. [Google Scholar] [CrossRef]
- Porto, S.; Seco, J.M.; Espinosa, J.F.; Quinoa, E.; Riguera, R. Resin-bound chiral derivatizing agents for assignment of configuration by NMR spectroscopy. J. Org. Chem. 2008, 73, 5714–5722. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. Assignment of the absolute configuration of polyfunctional compounds by NMR using chiral derivatizing agents. Chem. Rev. 2012, 112, 4603–4641. [Google Scholar] [CrossRef]
- Louzao, I.; Seco, J.M.; Quiñoá, E.; Riguera, R. 13C NMR as a general tool for the assignment of absolute configuration. Chem. Commun. 2010, 46, 5001–5004. [Google Scholar] [CrossRef] [PubMed]
- Latypov, S.; Franck, X.; Jullian, J.-C.; Hocquemiller, R.; Figadère, B.; Lorenzo, M.; Brito, I.; Cueto, M.; D’Croz, L.; Darias, J. NMR determination of absolute configuration of butenolides of annonaceous type. Chem. Eur. J. 2002, 8, 5662–5666. [Google Scholar] [CrossRef]
- Diaz-Marrero, A.R.; Brito, I.; Cueto, M.; San-Martin, A.; Darias, J. Conformational analysis and absolute stereochemistry of ‘spongian’-related metabolites. Tetrahedron 2004, 60, 1073–1078. [Google Scholar] [CrossRef]
- Dorta, E.; Diaz-Marrero, A.R.; Brito, I.; Cueto, M.; D’Croz, L.; Darias, J. The oxidation profile at C-18 of furanocembranolides may provide a taxonomical marker for several genera of octocorals. Tetrahedron 2007, 63, 9057–9062. [Google Scholar] [CrossRef]
- Díaz-Marrero, A.R.; Brito, I.; de la Rosa, J.M.; D’Croz, L.; Fabelo, O.; Ruiz-Pérez, C.; Darias, J.; Cueto, M. Novel lactone Chamigrene-derived metabolites from Laurencia majuscula. Eur. J Org. Chem. 2009, 2009, 1407–1411. [Google Scholar] [CrossRef]
- Arya, N.; Mishra, S.K.; Suryaprakash, N. A simple ternary ion-pair complexation protocol for testing the enantiopurity and the absolute configurational analysis of acid and ester derivatives. New J. Chem. 2018, 42, 9920–9929. [Google Scholar] [CrossRef]
- Chen, S.; Ding, M.; Liu, W.; Huang, X.; Liu, Z.; Lu, Y.; Liu, H.; She, Z. Chiral sensors for determining the absolute configurations of α-amino acid derivatives. Org. Biomol. Chem. 2018, 16, 8311–8317. [Google Scholar] [CrossRef]
- Mishra, S.K.; Suryaprakash, N. Some new protocols for the assignment of absolute configuration by NMR spectroscopy using chiral solvating agents and CDAs. Tetrahedron Asymmetry 2017, 28, 1220–1232. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bourne, C.E.; Clark, R.L.; Xu, K.; Yang, P.-F.; Yang, Y.-N.; Feng, Z.-M.; Jiang, J.-S.; Zhang, P.-C. (18-Crown-6)-2,3,11,12-tetracarboxylic acid as a chiral NMR solvating agent for determining the enantiomeric purity and absolute configuration of β-amino acids. Tetrahedron Asymmetry 2009, 20, 2678–2682. [Google Scholar] [CrossRef]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591. [Google Scholar] [CrossRef]
- Cueto, M.; Jensen, P.R.; Fenical, W. N-Methylsansalvamide, a cytotoxic cyclic depsipeptide from a marine fungus of the genus Fusarium. Phytochemistry 2000, 55, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Austin, P.; Diaz-Marrero, A.R.; Soest, R.V.; Matainaho, T.; Roskelley, C.D.; Roberge, M.; Andersen, R.J. Neopetrosiamides, Peptides from the Marine Sponge Neopetrosia sp. That Inhibit Amoeboid Invasion by Human Tumor Cells. Org. Lett. 2005, 7, 4173–4176. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Cueto, M.; Jensen, P.R.; Fenical, W.; Silverman, R.B. Microsporins A and B: New histone deacetylase inhibitors from the marine-derived fungus Microsporum cf. gypseum and the solid-phase synthesis of microsporin A. Tetrahedron 2007, 63, 6535–6541. [Google Scholar] [CrossRef]
- Bhushan, R.; Brückner, H. Marfey’s reagent for chiral amino acid analysis: A review. Amino Acids 2004, 27, 231–247. [Google Scholar] [CrossRef]
- Harada, K.-I.; Fujii, K.; Mayumi, T.; Hibino, Y.; Suzuki, M.; Ikai, Y.; Oka, H. A method using LC/MS for determination of absolute configuration of constituent amino acids in peptide—Advanced Marfey’s method. Tetrahedron Lett. 1995, 36, 1515–1518. [Google Scholar] [CrossRef]
- Takiguchi, S.; Hirota-Takahata, Y.; Nishi, T. Application of the advanced Marfey’s method for the determination of the absolute configuration of ogipeptins. Tetrahedron Lett. 2022, 96, 153760. [Google Scholar] [CrossRef]
- Vijayasarathy, S.; Prasad, P.; Fremlin, L.J.; Ratnayake, R.; Salim, A.A.; Khalil, Z.; Capon, R.J. C3 and 2D C3 Marfey’s Methods for Amino Acid Analysis in Natural Products. J. Nat. Prod. 2016, 79, 421–427. [Google Scholar] [CrossRef]
- Pérez-Victoria, I.; Crespo, G.; Reyes, F. Expanding the utility of Marfey’s analysis by using HPLC-SPE-NMR to determine the Cβ configuration of threonine and isoleucine residues in natural peptides. Anal. Bioanal. Chem. 2022, 414, 8063–8070. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning the Stereochemistry of Pairs of Diastereoisomers Using GIAO NMR Shift Calculation. J. Org. Chem. 2009, 74, 4597–4607. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds Using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Grimblat, N.; Gavin, J.A.; Daranas, A.H.; Sarotti, A.M. Combining the Power of J Coupling and DP4 Analysis on Stereochemical Assignments: The J-DP4 Methods. Org. Lett. 2019, 21, 4003–4007. [Google Scholar] [CrossRef]
- Cairns, E.; Hashimi, M.A.; Singh, A.J.; Eakins, G.; Lein, M.; Keyzers, R. Structure of Echivulgarine, a Pyrrolizidine Alkaloid Isolated from the Pollen of Echium vulgare. J. Agric. Food Chem. 2015, 63, 7421–7427. [Google Scholar] [CrossRef]
- Cooper, J.K.; Li, K.L.; Aube, J.; Coppage, D.A.; Konopelski, J.P. Application of the DP4 Probability Method to Flexible Cyclic Peptides with Multiple Independent Stereocenters: The True Structure of Cyclocinamide A. Org. Lett. 2018, 20, 4314–4317. [Google Scholar] [CrossRef]
- Gutierrez-Cepeda, A.; Daranas, A.H.; Fernandez, J.J.; Norte, M.; Souto, M.L. Stereochemical Determination of Five-Membered Cyclic Ether Acetogenins Using a Spin-Spin Coupling Constant Approach and DFT Calculations. Mar. Drugs 2014, 12, 4031–4044. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, H.J.; Crespin, G.D.; Santiago-Benitez, A.J.; Gavin, J.A.; Norte, M.; Fernandez, J.J.; Daranas, A.H. Stereochemistry of Complex Marine Natural Products by Quantum Mechanical Calculations of NMR Chemical Shifts: Solvent and Conformational Effects on Okadaic Acid. Mar. Drugs 2014, 12, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Nguyen, Q.N.; Na, M.W.; Kim, K.H.; Guo, Y.; Yim, J.H.; Shim, S.H.; Kim, J.-J.; Kang, K.S.; Lee, D. A new alpha-pyrone from Arthrinium pseudosinense culture medium and its estrogenic activity in MCF-7 cells. J. Antibiot. 2021, 74, 893–897. [Google Scholar] [CrossRef]
- Ermanis, K.; Parkes, K.E.B.; Agback, T.; Goodman, J.M. The optimal DFT approach in DP4 NMR structure analysis—Pushing the limits of relative configuration elucidation. Org. Biomol. Chem. 2019, 17, 5886–5890. [Google Scholar] [CrossRef]
- Xin, D.Y.; Jones, P.J.; Gonnella, N.C. DiCE: Diastereomeric in Silico Chiral Elucidation, Expanded DP4 Probability Theory Method for Diastereomer and Structural Assignment. J. Org. Chem. 2018, 83, 5035–5043. [Google Scholar] [CrossRef]
- Daranas, A.H.; Sarotti, A.M. Are Computational Methods Useful for Structure Elucidation of Large and Flexible Molecules? Belizentrin as a Case Study. Org. Lett. 2021, 23, 503–507. [Google Scholar] [CrossRef]
- Hu, Y.Y.; Wang, M.; Wu, C.Y.; Tan, Y.; Li, J.; Hao, X.M.; Duan, Y.B.; Guan, Y.; Shang, X.Y.; Wang, Y.G.; et al. Identification and Proposed Relative and Absolute Configurations of Niphimycins C-E from the Marine-Derived Streptomyces sp IMB7-145 by Genomic Analysis. J. Nat. Prod. 2018, 81, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Machado, H.; Jang, K.H.; Trzoss, L.; Jensen, P.R.; Fenical, W. Integration of Genomic Data with NMR Analysis Enables Assignment of the Full Stereostructure of Neaumycin B, a Potent Inhibitor of Glioblastoma from a Marine-Derived Micromonospora. J. Am. Chem. Soc. 2018, 140, 10775–10784. [Google Scholar] [CrossRef] [PubMed]
- An, J.S.; Lee, J.Y.; Kim, E.; Ahn, H.; Jang, Y.J.; Shin, B.; Hwang, S.; Shin, J.; Yoon, Y.J.; Lee, S.K.; et al. Formicolides A and B, Antioxidative and Antiangiogenic 20-Membered Macrolides from a Wood Ant Gut Bacterium. J. Nat. Prod. 2020, 83, 2776–2784. [Google Scholar] [CrossRef]
- Sasaki, S.I.; Abe, H.; Hirota, Y.; Ishida, Y.; Kudo, Y.; Ochiai, S.; Saito, K.; Yamasaki, T. Chemics-F—Computer-program system for structure elucidation of organic-compounds. J. Chem. Inf. Comput. Sci. 1978, 18, 211–222. [Google Scholar] [CrossRef]
- Funatsu, K.; Sasaki, S. Recent advances in the automated structure elucidation system, CHEMICS. Utilization of two-dimensional NMR spectral information and development of peripheral functions for examination of candidates. J. Chem. Inf. Comput. Sci. 1996, 36, 190–204. [Google Scholar] [CrossRef]
- Zlatina, L.A.; Elyashberg, M.E. Generation and representation of stereoisomers of a molecular-structure. J. Struct. Chem. 1991, 32, 528–533. [Google Scholar] [CrossRef]
- Pesek, M.; Juvan, A.; Jakos, J.; Kosmrlj, J.; Marolt, M.; Gazvoda, M. Database Independent Automated Structure Elucidation of Organic Molecules Based on IR, H-1 NMR, C-13 NMR, and MS Data. J. Chem. Inf. Model. 2021, 61, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Christie, B.D.; Munk, M.E. Structure generation by reduction—A new strategy for computer-assisted structure elucidation. J. Chem. Inf. Comput. Sci. 1988, 28, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Faulon, J.L. Stochastic generator of chemical-structure.1. Application to the structure elucidation of large molecules. J. Chem. Inf. Comput. Sci. 1994, 34, 1204–1218. [Google Scholar] [CrossRef]
- Lindel, T.; Junker, J.; Kock, M. COCON: From NMR correlation data to molecular constitutions. J. Mol. Model. 1997, 3, 364–368. [Google Scholar] [CrossRef]
- Badertscher, M.; Korytko, A.; Schulz, K.P.; Madison, M.; Munk, M.E.; Portmann, P.; Junghans, M.; Fontana, P.; Pretsch, E. Assemble 2.0: A structure generator. Chemom. Intell. Lab. Syst. 2000, 51, 73–79. [Google Scholar] [CrossRef]
- Korytko, A.; Schulz, K.P.; Madison, M.S.; Munk, M.E. HOUDINI: A new approach to computer-based structure generation. J. Chem. Inf. Comput. Sci. 2003, 43, 1434–1446. [Google Scholar] [CrossRef]
- Schulz, K.P.; Korytko, A.; Munk, M.E. Applications of a HOUDINI-based structure elucidation system. J. Chem. Inf. Comput. Sci. 2003, 43, 1447–1456. [Google Scholar] [CrossRef]
- Elyashberg, M.E.; Blinov, K.A.; Molodtsov, S.G.; Williams, A.J.; Martin, G.E. Fuzzy structure generation: A new efficient tool for computer-aided structure elucidation (CASE). J. Chem. Inf. Model. 2007, 47, 1053–1066. [Google Scholar] [CrossRef]
- Nuzillard, J.M.; Massiot, G. Logic for structure determination. Tetrahedron 1991, 47, 3655–3664. [Google Scholar] [CrossRef]
- Benecke, C.; Grund, R.; Hohberger, R.; Kerber, A.; Laue, R.; Wieland, T. MOLGEN(+), a generator of connectivity isomers and stereoisomers for molecular-structure elucidation. Anal. Chim. Acta 1995, 314, 141–147. [Google Scholar] [CrossRef]
- Benecke, C.; Gruner, T.; Kerber, A.; Laue, R.; Wieland, T. MOLecular structure GENeration with MOLGEN, new features and future developments. Fresenius J. Anal. Chem. 1997, 359, 23–32. [Google Scholar] [CrossRef]
- Meringer, M.; Schymanski, E.L. Small Molecule Identification with MOLGEN and Mass Spectrometry. Metabolites 2013, 3, 440–462. [Google Scholar] [CrossRef]
- Kerber, A. MOLGEN, a Generator for Structural Formulas. Match Commun. Math. Comput. Chem. 2018, 80, 733–744. [Google Scholar]
- Will, M.; Fachinger, W.; Richert, J.R. Fully automated structure elucidation—A spectroscopis’s dream comes true. J. Chem. Inf. Comput. Sci. 1996, 36, 221–227. [Google Scholar] [CrossRef]
- Neudert, R.; Penk, M. Enhanced structure elucidation. J. Chem. Inf. Comput. Sci. 1996, 36, 244–248. [Google Scholar] [CrossRef]
- Meiler, J.; Will, M. Automated structure elucidation of organic molecules from C-13 NMR spectra using genetic algorithms and neural networks. J. Chem. Inf. Comput. Sci. 2001, 41, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Meiler, J.; Will, M. Genius: A genetic algorithm for automated structure elucidation from C-13 NMR spectra. J. Am. Chem. Soc. 2002, 124, 1868–1870. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C. SENECA: A platform-independent, distributed, and parallel system for computer-assisted structure elucidation in organic chemistry. J. Chem. Inf. Comput. Sci. 2001, 41, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.Q.; Steinbeck, C. Evolutionary-algorithm-based strategy for computer-assisted structure elucidation. J. Chem. Inf. Comput. Sci. 2004, 44, 489–498. [Google Scholar] [CrossRef]
- Peng, C.; Yuan, S.G.; Zheng, C.Z.; Hui, Y.Z.; Wu, H.M.; Ma, K.; Han, X.W. Application of expert-system CISOC-SES to the structure elucidation of complex natural-products. J. Chem. Inf. Comput. Sci. 1994, 34, 814–819. [Google Scholar] [CrossRef]
- Koeck, M.; Lindel, T.; Junker, J. Incorporation of (4)J-HMBC and NOE Data into Computer-Assisted Structure Elucidation with WEBCOCON. Molecules 2021, 26, 4846. [Google Scholar] [CrossRef]
- Nuzillard, J.M.; Plainchont, B. Tutorial for the structure elucidation of small molecules by means of the LSD software. Magn. Reson. Chem. 2018, 56, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The Correct Structure of Aquatolide-Experimental Validation of a Theoretically-Predicted Structural Revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.; Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminformatics 2019, 11, 50. [Google Scholar] [CrossRef]
- Kwon, Y.; Lee, D.; Choi, Y.S.; Kang, M.; Kang, S. Neural Message Passing for NMR Chemical Shift Prediction. J. Chem. Inf. Model. 2020, 60, 2024–2030. [Google Scholar] [CrossRef]
- Elyashberg, M.E.; Blinov, K.A.; Williams, A.J.; Molodtsov, S.G.; Martin, G.E.; Martirosian, E.R. Structure elucidator: A versatile expert system for molecular structure elucidation from 1D and 2D NMR data and molecular fragments. J. Chem. Inf. Comput. Sci. 2004, 44, 771–792. [Google Scholar] [CrossRef]
- Elyashberg, M.; Blinov, K.; Williams, A. A systematic approach for the generation and verification of structural hypotheses. Magn. Reson. Chem. 2009, 47, 371–389. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2020. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688. [Google Scholar] [CrossRef]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-Oriented Synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. A Chemoinformatics Approach to the Discovery of Lead-Like Molecules from Marine and Microbial Sources En Route to Antitumor and Antibiotic Drugs. Mar. Drugs 2014, 12, 757–778. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef]
- Jayaseelan, K.V.; Steinbeck, C. Building blocks for automated elucidation of metabolites: Natural product-likeness for candidate ranking. BMC Bioinform. 2014, 15, 234. [Google Scholar] [CrossRef]
- Shang, J.; Hu, B.; Wang, J.; Zhu, F.; Kang, Y.; Li, D.; Sun, H.; Kong, D.-X.; Hou, T. A cheminformatic insight into the differences between terrestrial and marine originated natural products. J. Chem. Inf. Model. 2018, 56, 1180–1193. [Google Scholar] [CrossRef]
- Pereira, F. Machine Learning Methods to Predict the Terrestrial and Marine Origin of Natural Products. Mol. Inform. 2021, 40, e2060034. [Google Scholar] [CrossRef]
- Klementz, D.; Doering, K.; Lucas, X.; Telukunta, K.K.; Erxleben, A.; Deubel, D.; Erber, A.; Santillana, I.; Thomas, O.S.; Bechthold, A.; et al. StreptomeDB 2.0—An extended resource of natural products produced by streptomycetes. Nucleic Acids Res. 2016, 44, D509–D514. [Google Scholar] [CrossRef]
- Christoforow, A.; Wilke, J.; Binici, A.; Pahl, A.; Ostermann, C.; Sievers, S.; Waldmann, H. Design, Synthesis, and Phenotypic Profiling of Pyrano-Furo-Pyridone Pseudo Natural Products. Angew. Chem. Int. Ed. 2019, 58, 14715–14723. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Hu, J.; Wang, Y.; Zhou, X.; Li, Y.; Zhang, L.; Liu, Z. Privileged Scaffold Analysis of Natural Products with Deep Learning-Based Indication Prediction Model. Mol. Inform. 2020, 39, e2000057. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Hernandez, A.L.; Sanchez-Cruz, N.; Medina-Franco, J.L. A Fragment Library of Natural Products and Its Comparative Chemoinformatic Characterization. Mol. Inform. 2020, 39, e2000050. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittala, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef]
- Liang, J.-W.; Wang, M.-Y.; Wang, S.; Li, X.-Y.; Meng, F.-H. Fragment-Based Structural Optimization of a Natural Product Itampolin A as a p38 Inhibitor for Lung Cancer. Mar. Drugs 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.R.; Palmeira, A.; Campos, A.; Cunha, I.; Freitas, M.; Felpeto, A.B.; Turkina, M.V.; Vasconcelos, V.; Pinto, M.; Correia-da-Silva, M.; et al. Structure-Antifouling Activity Relationship and Molecular Targets of Bio-Inspired(thio)xanthones. Biomolecules 2020, 10, 1126. [Google Scholar] [CrossRef]
- Almeida, J.R.; Moreira, J.; Pereira, D.; Pereira, S.; Antunes, J.; Palmeira, A.; Vasconcelos, V.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Potential of synthetic chalcone derivatives to prevent marine biofouling. Sci. Total Environ. 2018, 643, 98–106. [Google Scholar] [CrossRef]
- Wang, Q.; Sciabola, S.; Barreiro, G.; Hou, X.; Bai, G.; Shapiro, M.J.; Koehn, F.; Villalobos, A.; Jacobson, M.P. Dihedral Angle-Based Sampling of Natural Product Polyketide Conformations: Application to Permeability Prediction. J. Chem. Inf. Model. 2016, 56, 2194–2206. [Google Scholar] [CrossRef]
- Davis, G.D.J.; Vasanthi, A.H.R. QSAR based docking studies of marine algal anticancer compounds as inhibitors of protein kinase B (PKB beta). Eur. J. Pharm. Sci. 2015, 76, 110–118. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A Structure- and Ligand-Based Virtual Screening of a Database of “Small” Marine Natural Products for the Identification of “Blue” Sigma-2 Receptor Ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef] [PubMed]
- Gaudencio, S.P.; Pereira, F. Predicting Antifouling Activity and Acetylcholinesterase Inhibition of Marine-Derived Compounds Using a Computer-Aided Drug Design Approach. Mar. Drugs 2022, 20, 129. [Google Scholar] [CrossRef]
- Dias, T.; Gaudencio, S.P.; Pereira, F. A Computer-Driven Approach to Discover Natural Product Leads for Methicillin-Resistant Staphylococcus aureus Infection Therapy. Mar. Drugs 2019, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Zanni, R.; Galvez-Llompart, M.; Machuca, J.; Garcia-Domenech, R.; Recacha, E.; Pascual, A.; Rodriguez-Martinez, J.M.; Galvez, J. Molecular topology: A new strategy for antimicrobial resistance control. Eur. J. Med. Chem. 2017, 137, 233–246. [Google Scholar] [CrossRef]
- Bueso-Bordils, J.I.; Perez-Gracia, M.T.; Suay-Garcia, B.; Duart, M.J.; Algarra, R.V.M.; Zamora, L.L.; Anton-Fos, G.M.; Lopez, P.A.A. Topological pattern for the search of new active drugs against methicillin resistant Staphylococcus aureus. Eur. J. Med. Chem. 2017, 138, 807–815. [Google Scholar] [CrossRef]
- Wang, L.; Le, X.; Li, L.; Ju, Y.C.; Lin, Z.X.; Gu, Q.; Xu, J. Discovering New Agents Active against Methicillin-Resistant Staphylococcus aureus with Ligand-Based Approaches. J. Chem. Inf. Model. 2014, 54, 3186–3197. [Google Scholar] [CrossRef]
- Aswathy, L.; Jisha, R.S.; Masand, V.H.; Gajbhiye, J.M.; Shibi, I.G. Computational strategies to explore antimalarial thiazine alkaloid lead compounds based on an Australian marine sponge Plakortis Lita. J. Biomol. Struct. Dyn. 2017, 35, 2407–2429. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.C.; Marquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. QSAR-Assisted Virtual Screening of Lead-Like Molecules from Marine and Microbial Natural Sources for Antitumor and Antibiotic Drug Discovery. Molecules 2015, 20, 4848–4873. [Google Scholar] [CrossRef]
- Ghosh, K.; Amin, S.A.; Gayen, S.; Jha, T. Chemical-informatics approach to COVID-19 drug discovery: Exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J. Mol. Struct. 2021, 1224, 129026. [Google Scholar] [CrossRef]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR Modeling of SARS-CoV M(pro)Inhibitors Identifies Sufugolix, Cenicriviroc, Proglumetacin, and Other Drugs as Candidates for Repurposing against SARS-CoV-2. Mol. Inform. 2020, 40, e2000113. [Google Scholar] [CrossRef] [PubMed]
- Gaudencio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633. [Google Scholar] [CrossRef]
- vonRanke, N.L.; Ribeiro, M.M.J.; Miceli, L.A.; de Souza, N.P.; Abrahim-Vieira, B.A.; Castro, H.C.; Teixeira, V.L.; Rodrigues, C.R.; Souza, A.M.T. Structure-activity relationship, molecular docking, and molecular dynamic studies of diterpenes from marine natural products with anti-HIV activity. J. Biomol. Struct. Dyn. 2020, 40, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P.; Gaudencio, S.P.; Pereira, F. In Silico HCT116 Human Colon Cancer Cell-Based Models En Route to the Discovery of Lead-Like Anticancer Drugs. Biomolecules 2018, 8, 56. [Google Scholar] [CrossRef]
- Kumar, V.; Roy, K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ. Res. 2020, 31, 511–526. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef]
- Khan, M.T.; Ali, A.; Wang, Q.; Irfan, M.; Khan, A.; Zeb, M.T.; Zhang, Y.-J.; Chinnasamy, S.; Wei, D.-Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2-a molecular dynamic study. J. Biomol. Struct. Dyn. 2021, 39, 3627–3637. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, W.; Xu, Y.; Ren, S.; Zhang, W.; Li, Y. Design, synthesis and biological evaluation of tasiamide B derivatives as BACE1 inhibitors. Bioorganic Med. Chem. 2015, 23, 1963–1974. [Google Scholar] [CrossRef]
- Abdelhameed, R.F.A.; Eltamany, E.E.; Hal, D.M.; Ibrahim, A.K.; AboulMagd, A.M.; Al-Warhi, T.; Youssif, K.A.; Abd El-kader, A.M.; Hassanean, H.A.; Fayez, S.; et al. New Cytotoxic Cerebrosides from the Red Sea Cucumber Holothuria spinifera Supported by In-Silico Studies. Mar. Drugs 2020, 18, 405. [Google Scholar] [CrossRef] [PubMed]





 ) and for absolute configuration determination in green check sign (
) and for absolute configuration determination in green check sign ( ).
) and for absolute configuration determination in green check sign ().
).
) and for absolute configuration determination in green check sign ().










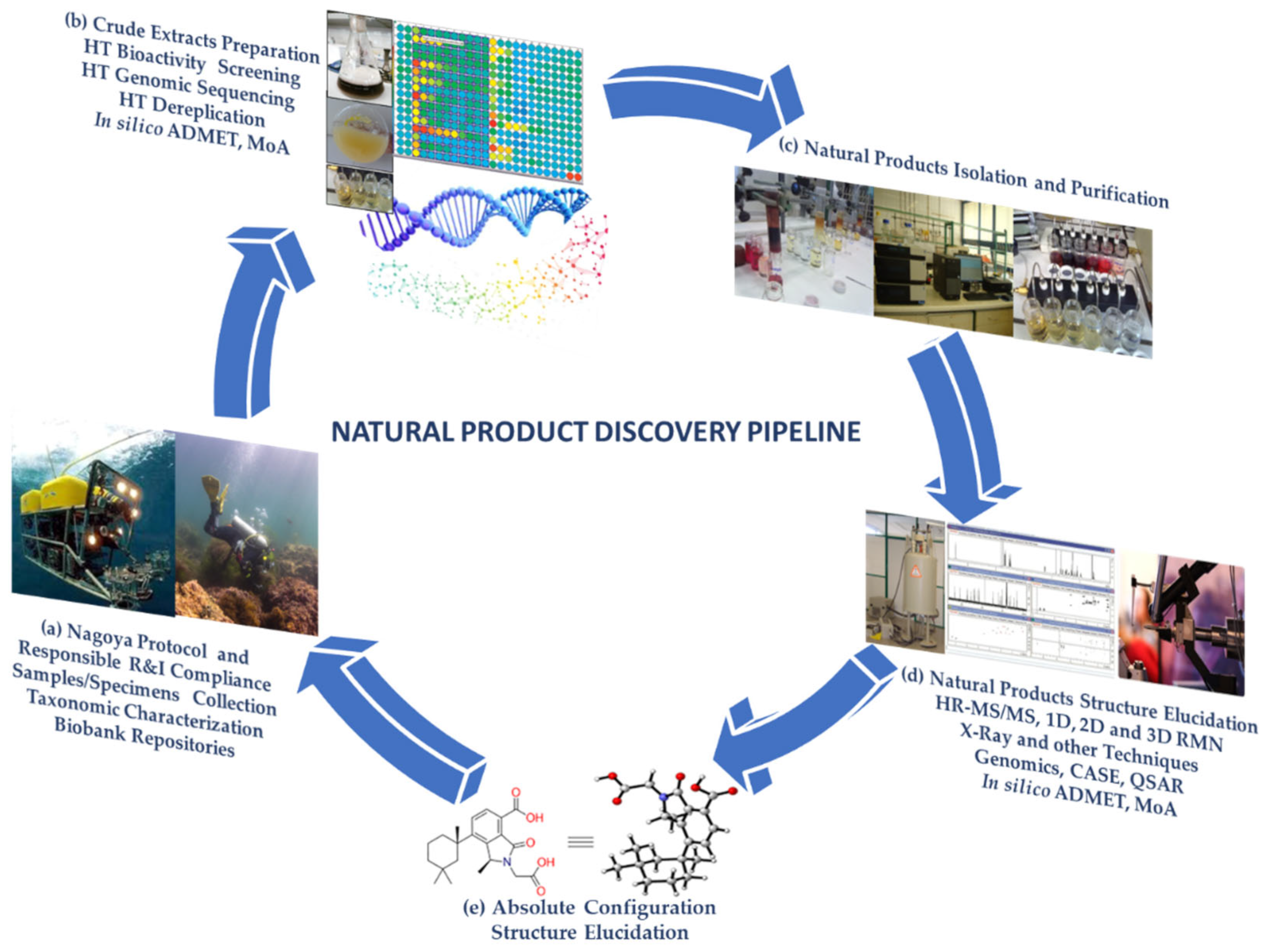{kind=link}
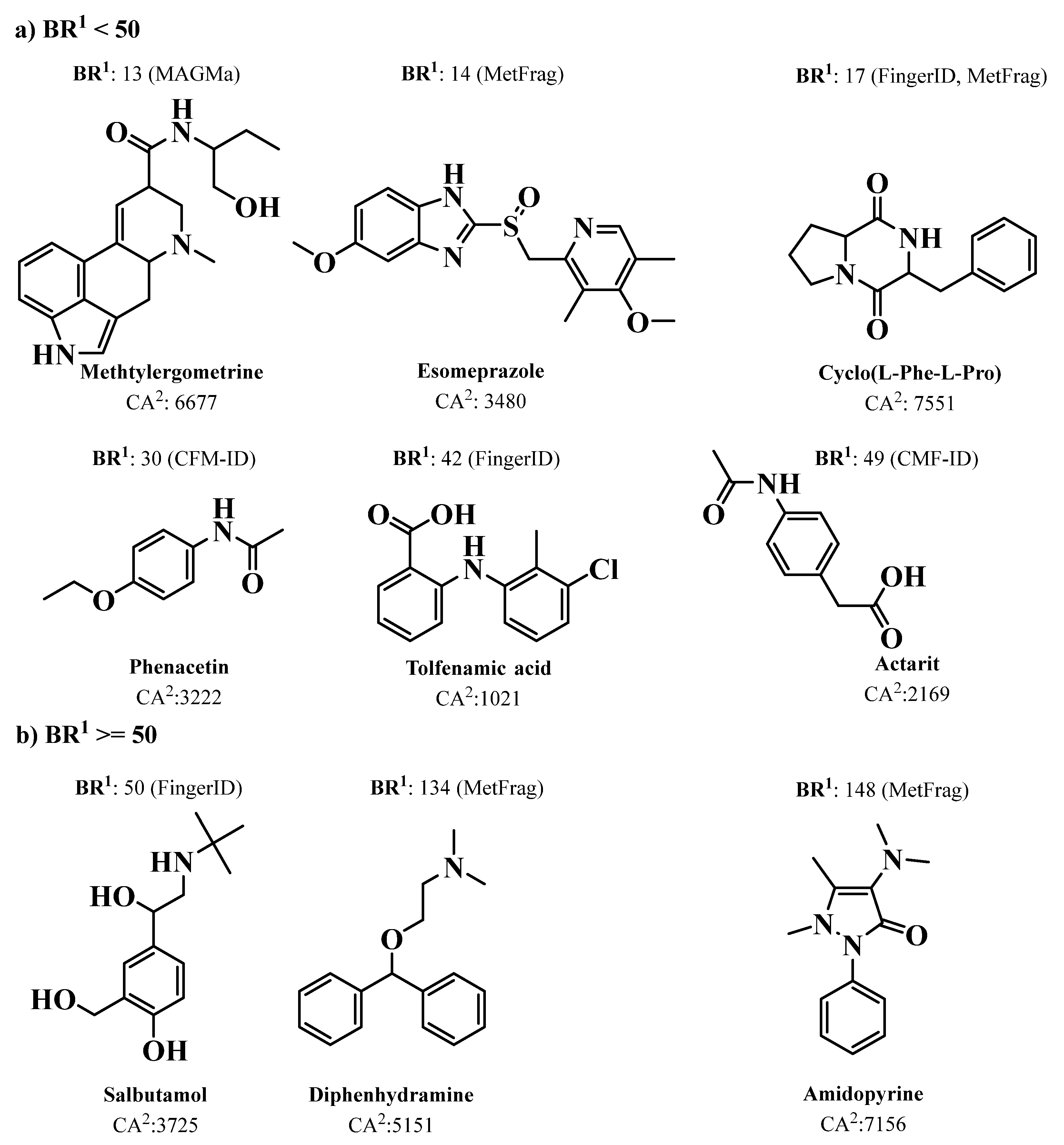{kind=link}
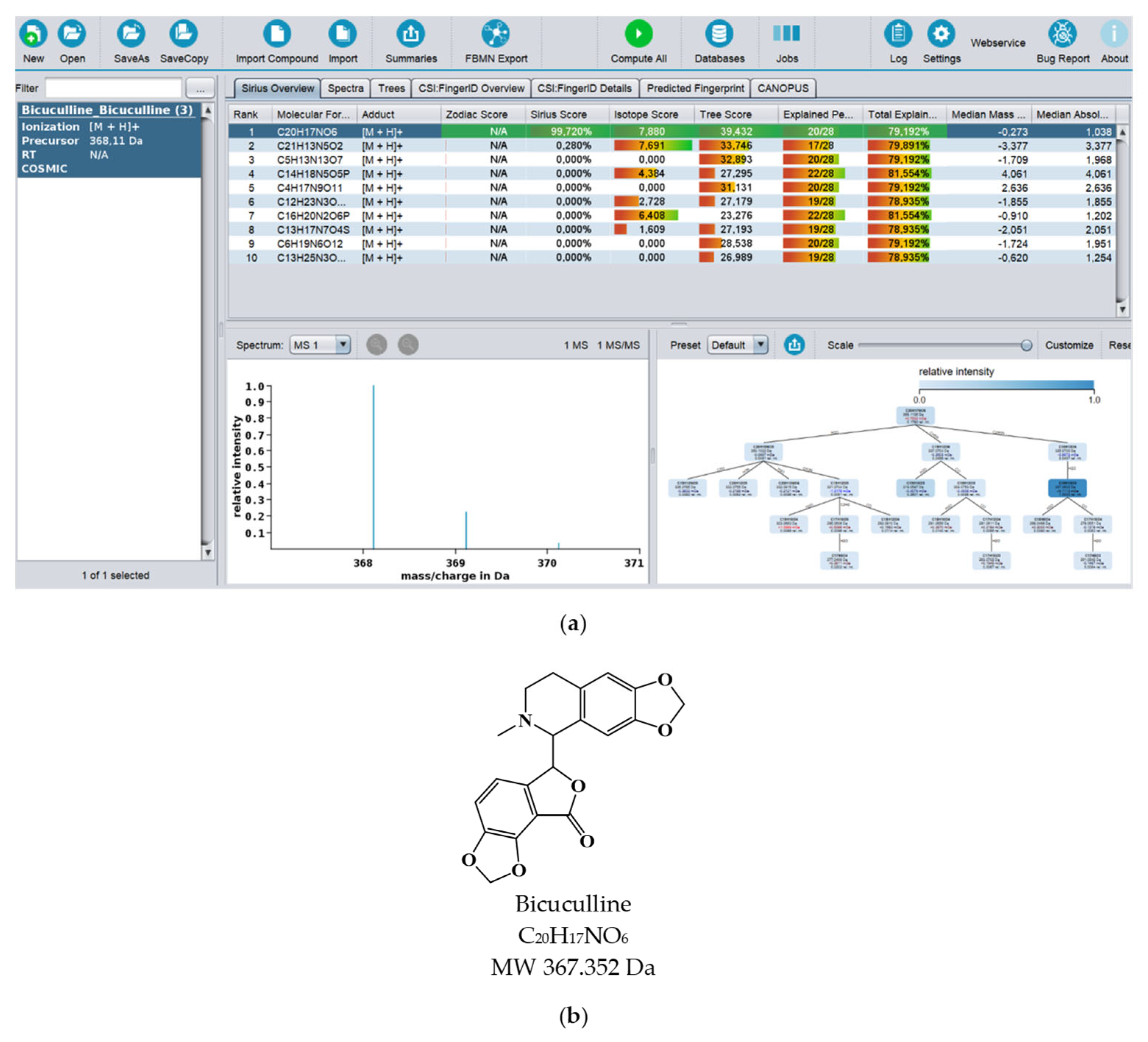{kind=link}
{kind=link}
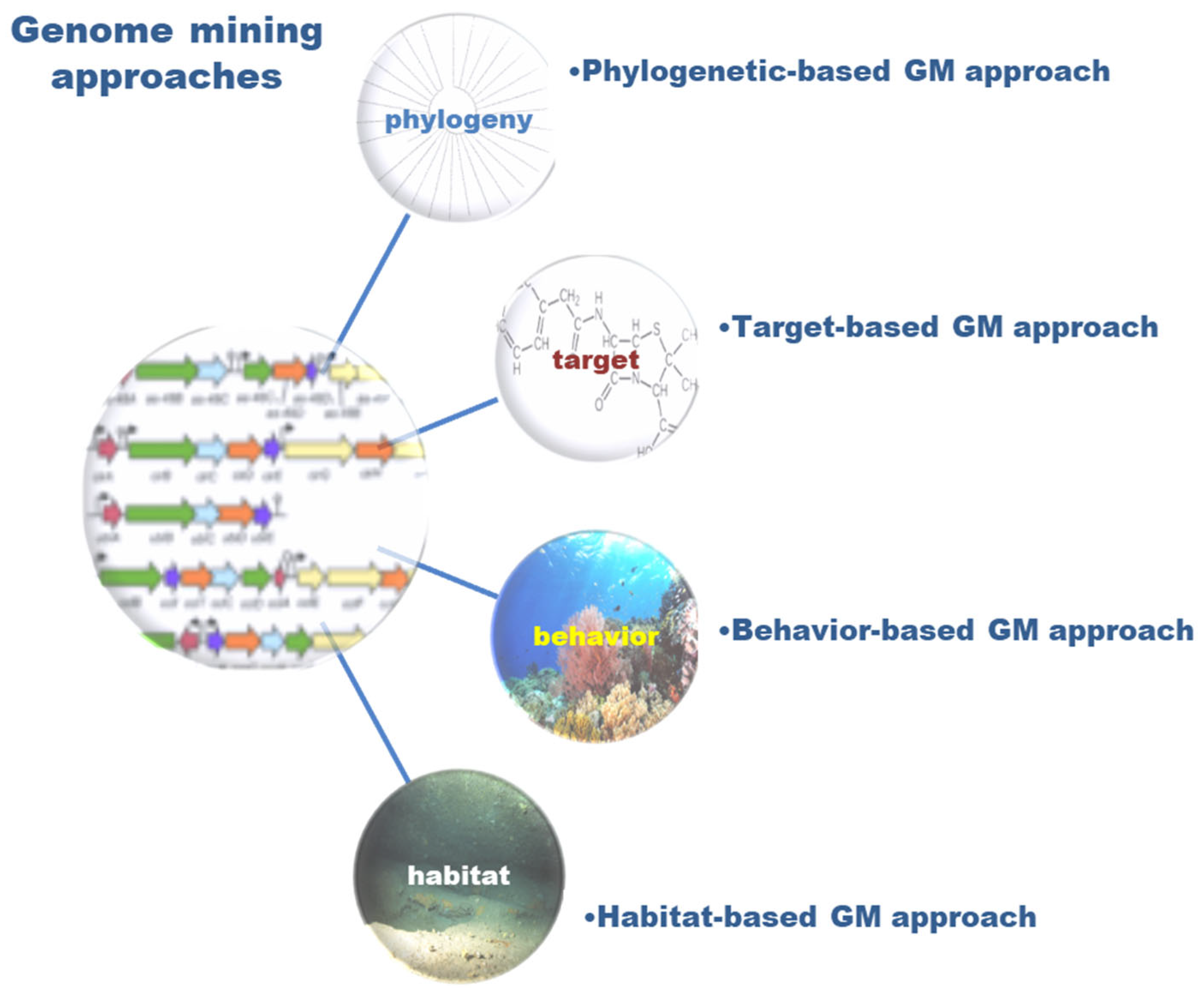{kind=link}
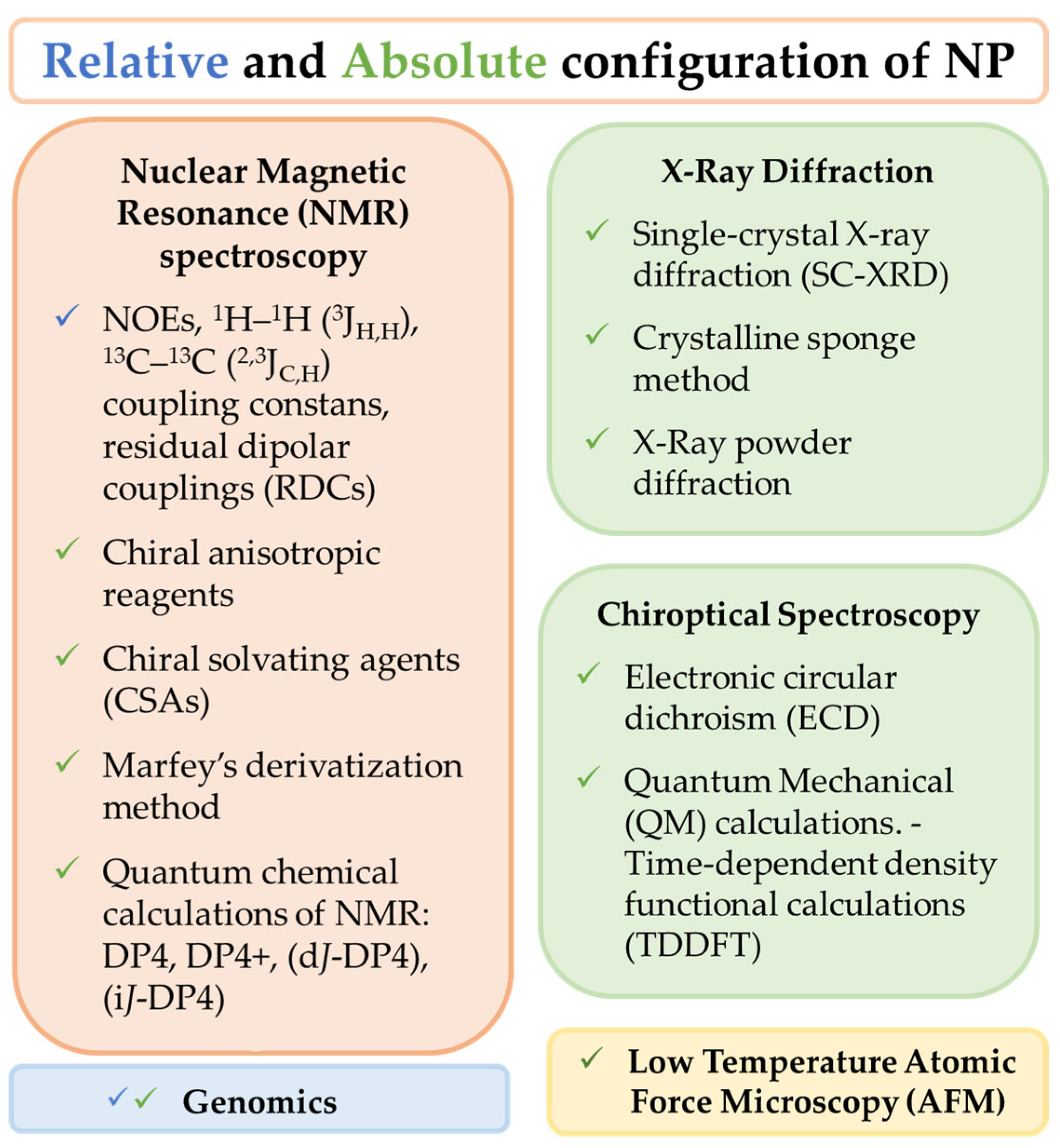{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Databases for MS/MS Dereplication | Databases for GC-MS, IMS, and MALDI Dereplication | ||||
|---|---|---|---|---|---|
| GNPS | GNPS/ MassIVE | Metabolitghts | MarinLit | GNPS/ MassIVE | MetaboLights |
| Metabolomics workbench | MassBank | ReSpect | NIST | MSHub/ GNPS | ProteomeXchange |
| MoNA | mzCloud | SPLASH | LipidXplorer | Metabolomics Workbench | PRIDE |
| Sumner/ Bruker | CASMI | PNNL Lipids | Sirenas/ Gates | GC-MS, IMS, and MALDI-MS Processing Informatic Analysis Tools | |
| EMBL | MCF | SistematX | NPBS | MSHub/ GNPS | |
| CMNPD | MIADB/ Beniddir | NPNPD | Antibase | SpeDE | |
| HMDB | MIADB | SistematX | DNP | AMDIS | |
| UNP | ChemSpider | Reaxys | SciFinder | RAMSY | |
| PubMed | Community-curated data | Users Libraries | - | ||
| MS/MS Visualization and Annotation Tools | |||||
| PCA | PoPCAR | PLS-DA | MN | ||
| MS2LDA | IIMN | NAP | DFMN-ISD | ||
| FBMN | CLMN | BBMN | BMN | ||
| MolNetEnhancer | MS2LDA-MOTIF | DEREPLICATOR | SIMILE | ||
| MetaboAnalyst | MSDIAL | XCMS Online | HMDB | ||
| Fragmentation Trees | GNPS Dashboard | Optimus and ‘ili | EMPress | ||
| Qemistree | ChemProp | PPNet | - | ||
| MS/MS Processing Informatics Analysis Tools | |||||
| GNPS Dashboard | MASST | GNPS | Mzmine.FBmn | ||
| CAM | XCMS Online | HMDB | MSDIAL | ||
| SPLASH | RMassBank | BinBase | MZmine | ||
| Bioclipse | MSDK | SIRIUS 1 to 4 | CSI:FingerID | ||
| DEREPLICATOR | DEREPLICATOR+ | NRPro | ReDU | ||
| QIIME and QIIME 2 | Qiita | CytoScape | Optimus and ‘ili | ||
| MetaboAnalyst | MS/MS-Chooser | ChemProp | PPNet | ||
| MeHaloCoA | SpeDE | ConCise | ClassyFire | ||
| Bioclips | MSDK | XCMS | Qemistree | ||
| EMPress | LLAMAS | NP Analyst | MetFrag | ||
| MetFusion | MAGMa | MIDAS | FT-BLAST | ||
| ISIS | FinderID | CFM-ID | MS-FINDER | ||
| MetEX | MeTCirc | Spectrum_utils | COSMIC | ||
| ZODIAC | CANOPUS | NPClassifier | IPO | ||
| CAMERA | - | - | - | ||
| Databases for NMR Dereplication | |
|---|---|
| Antibase | MarinLit |
| NP-MRD | NP Atlas |
| MIBiG | StreptomeDB 3.0 |
| PNMRNP | COCONUT |
| UNP | KnapsackSearch |
| NPBS | CMNPD |
| NMR Processing Informatic Analysis Tools | |
| HiFSA | MixONat |
| XGBoost classifier | 2D barcodes |
| NMRfilter | COLMAR |
| SMART | SMART 2.0 |
| SMART- Miner | MatchNat |
| DEREP-NP | MADByTE |
| RESTful | NP Classifier |
| ClassyFire | CyanoMetDB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaudêncio, S.P.; Bayram, E.; Lukić Bilela, L.; Cueto, M.; Díaz-Marrero, A.R.; Haznedaroglu, B.Z.; Jimenez, C.; Mandalakis, M.; Pereira, F.; Reyes, F.; et al. Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation. Mar. Drugs 2023, 21, 308. https://doi.org/10.3390/md21050308
Gaudêncio SP, Bayram E, Lukić Bilela L, Cueto M, Díaz-Marrero AR, Haznedaroglu BZ, Jimenez C, Mandalakis M, Pereira F, Reyes F, et al. Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation. Marine Drugs. 2023; 21(5):308. https://doi.org/10.3390/md21050308
Chicago/Turabian StyleGaudêncio, Susana P., Engin Bayram, Lada Lukić Bilela, Mercedes Cueto, Ana R. Díaz-Marrero, Berat Z. Haznedaroglu, Carlos Jimenez, Manolis Mandalakis, Florbela Pereira, Fernando Reyes, and et al. 2023. "Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation" Marine Drugs 21, no. 5: 308. https://doi.org/10.3390/md21050308
APA StyleGaudêncio, S. P., Bayram, E., Lukić Bilela, L., Cueto, M., Díaz-Marrero, A. R., Haznedaroglu, B. Z., Jimenez, C., Mandalakis, M., Pereira, F., Reyes, F., & Tasdemir, D. (2023). Advanced Methods for Natural Products Discovery: Bioactivity Screening, Dereplication, Metabolomics Profiling, Genomic Sequencing, Databases and Informatic Tools, and Structure Elucidation. Marine Drugs, 21(5), 308. https://doi.org/10.3390/md21050308