
Multi-Omics Analysis of Gene and Protein Candidates Possibly Related to Tetrodotoxin Accumulation in the Skin of Takifugu flavidus

 , ,
, ,
Abstract
:1. Introduction
2. Results
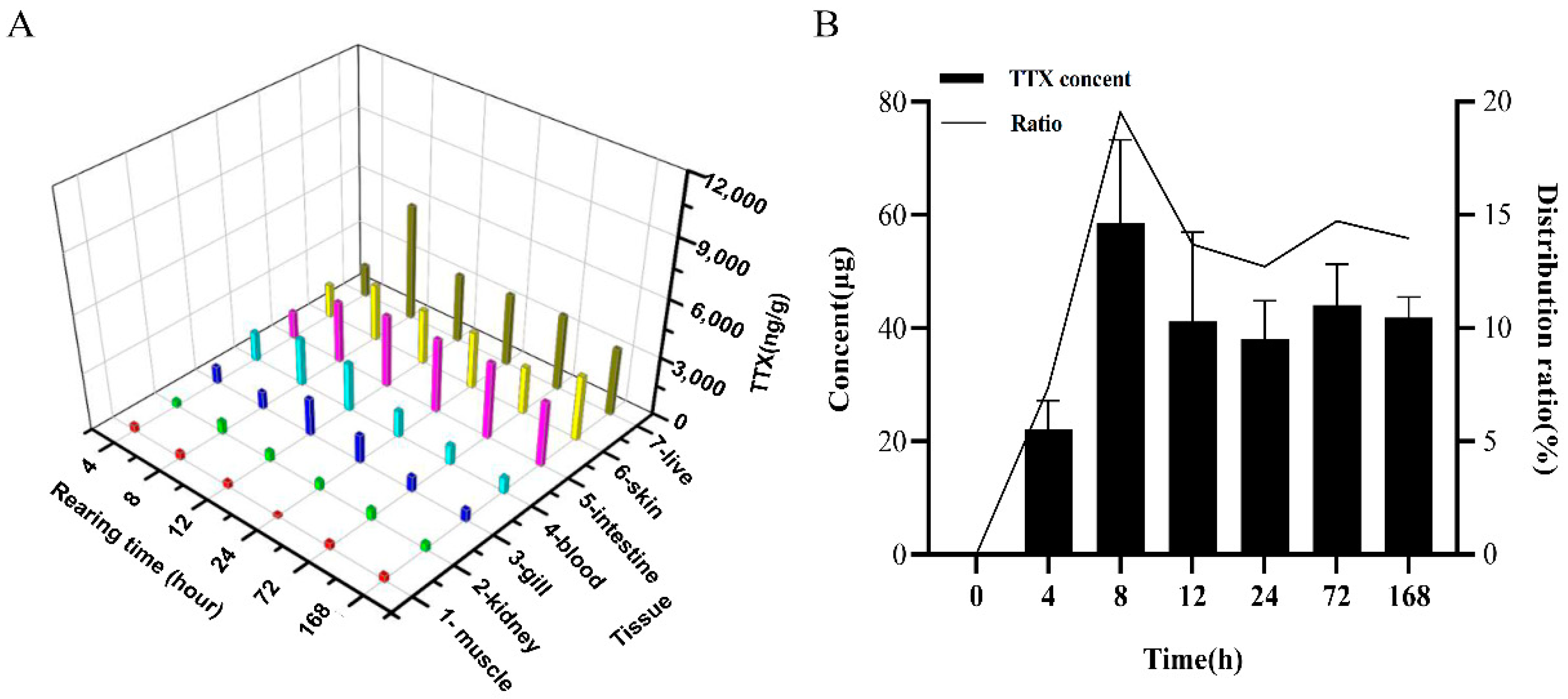
2.1. Accumulation of TTX in the Skin
2.2. Transcriptomic Landscape in the Skin Tissue of T. flavidus and Identification of Differentially Expressed Genes (DEGs)
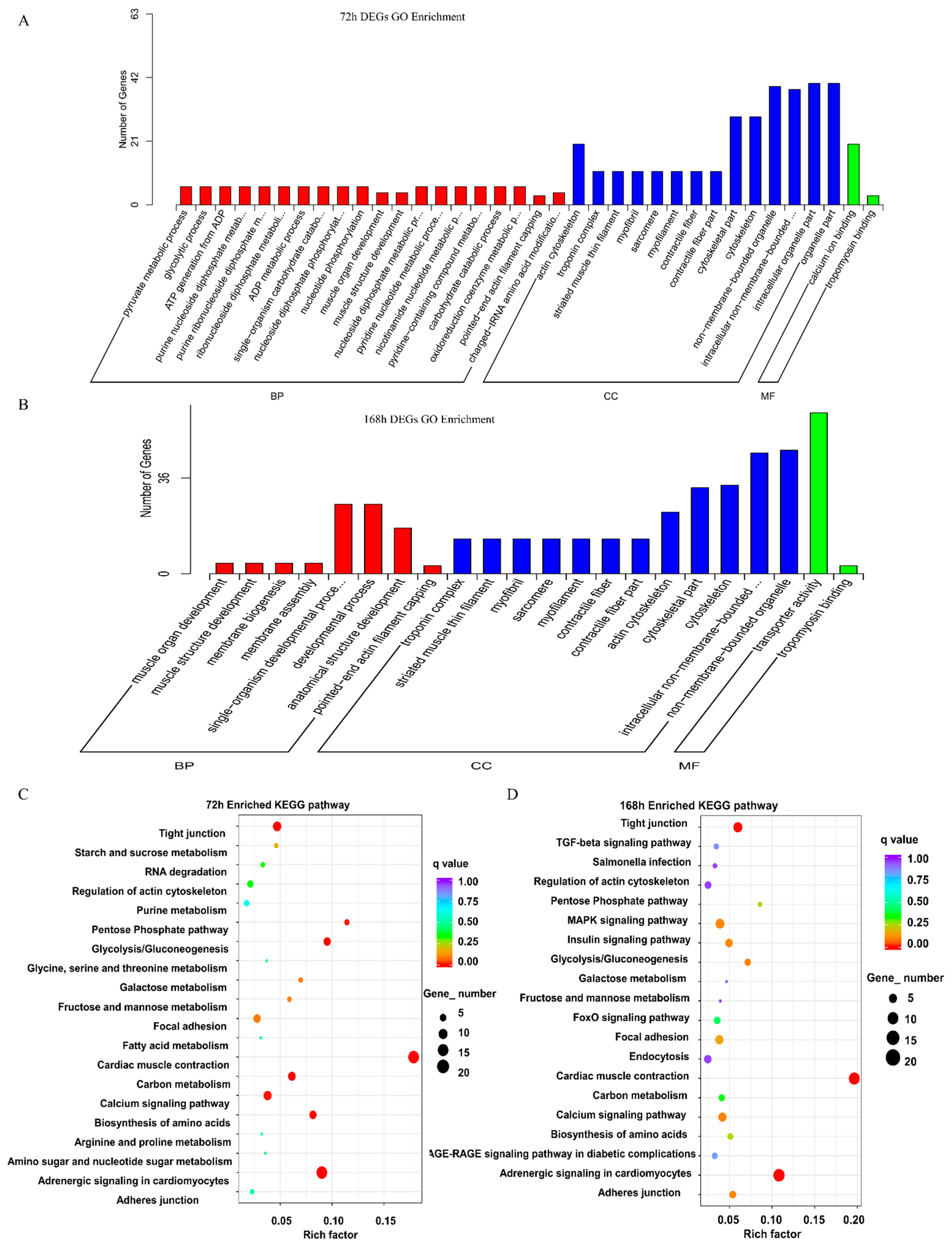
2.3. Functional Enrichment of the DEGs
2.4. Results of Proteomic Study
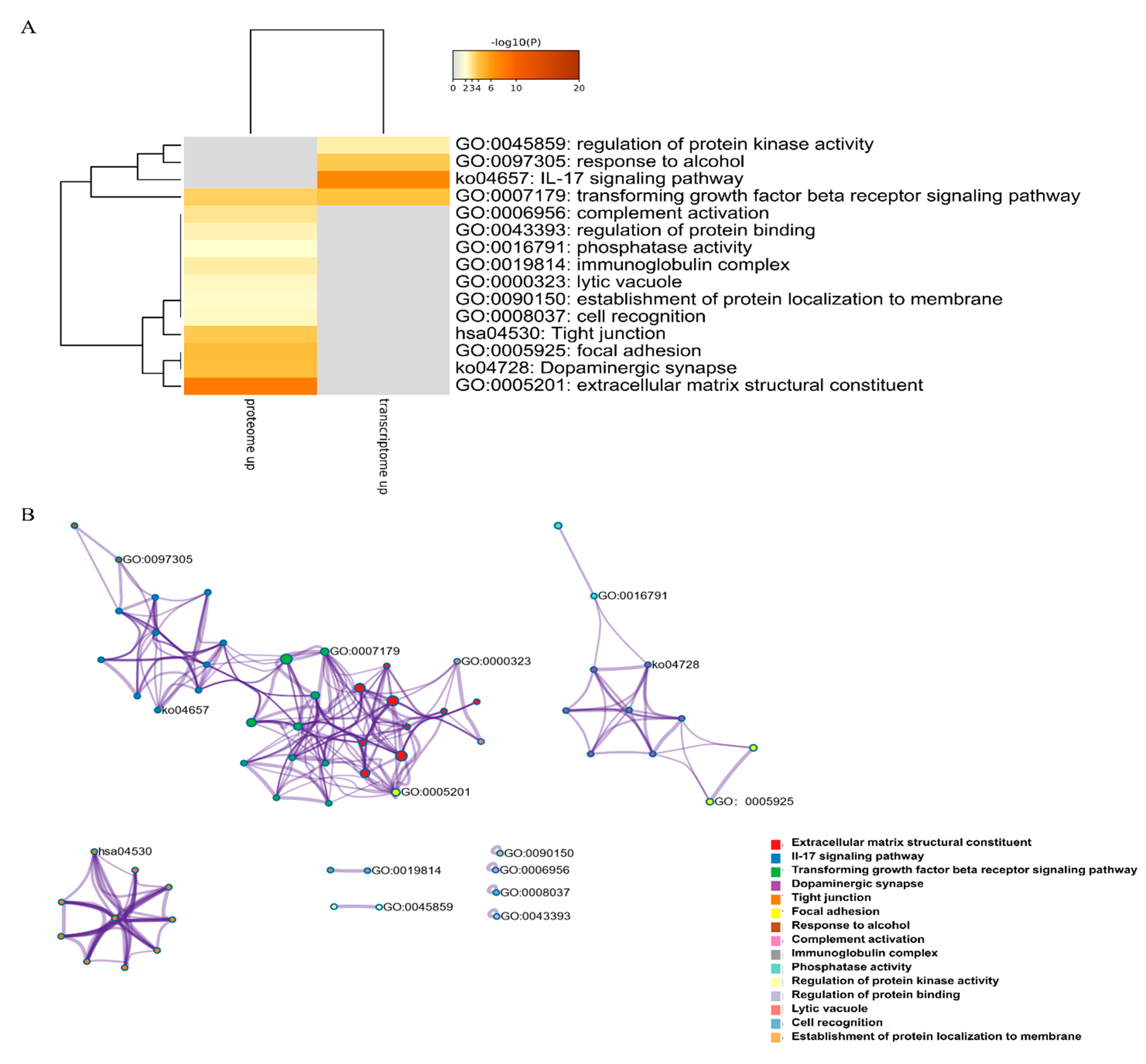
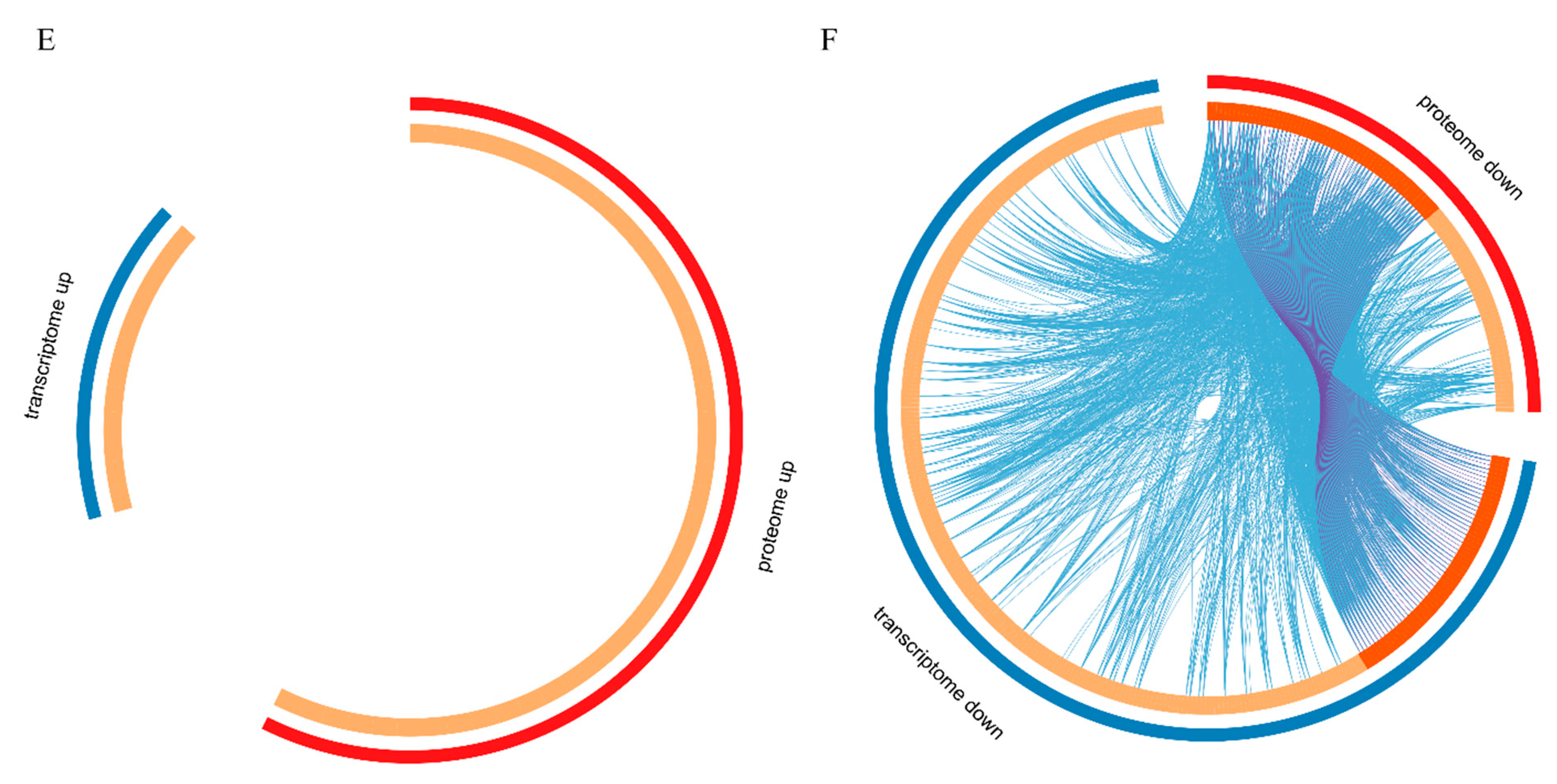
2.5. Summary of Transcriptomic and Proteomic Analyses
3. Discussion
4. Materials and Methods
4.1. Pufferfish Specimens
4.2. Toxin Administration Experiments
4.2.1. Preparation of Samples
4.2.2. LC-MS/MS
4.3. RNA Extraction and Sequencing
4.4. Reads Mapping, Gene Structure Analysis, and Gene Transcript Functional Annotation
4.5. Identification of DEGs
4.6. Validation of Transcriptome Analyses Using Quantitative PCR
4.7. Total Protein Extraction and Tandem Mass Tag (TMT) Labeling of Peptides
4.8. The Identification and Functional Analysis of DEPs
4.9. Enrichment Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- The Editors of Encyclopaedia, T.E.O. Puffer Encyclopedia Britannica. 2018. Available online: https://www.britannica.com/animal/puffer (accessed on 15 November 2021).
- Suehiro, M. Historical review on chemical and medical studies of globefish toxin before World War II. Yakushigaku Zasshi 1994, 29, 428–434. [Google Scholar]
- Goto, T.; Kishi, Y.; Takahashi, S.; Hirata, Y. Tetrodotoxin. Tetrahedron 1965, 21, 2059–2088. [Google Scholar] [CrossRef]
- Tsuda, K.; Ikuma, S.; Kawamura, M.; Tachikawa, R.; Sakai, K. Tetrodotoxin. VII. On the structure of tetrodotoxin and its derivatives. Chem. Pharm. Bull. 1964, 12, 1357–1374. [Google Scholar] [CrossRef] [Green Version]
- Woodward, R.B. The structure of tetrodotoxin. Pure Appl. Chem. 1964, 9, 49–74. [Google Scholar] [CrossRef]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [Green Version]
- Saoudi, M.; Rabeh, F.B.; Jammoussi, K.; Abdelmouleh, A.; Belbahri, L.; El Feki, A. Biochemical and physiological responses in Wistar rat after administration of puffer fish (Lagocephalus lagocephalus) flesh. J. Food Agric. Environ. 2007, 5, 107–111. [Google Scholar]
- Narahashi, T. Tetrodotoxin: A brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008, 84, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Maruta, S.; Yamaoka, K.; Yotsu-Yamashita, M. Two critical residues in p-loop regions of puffer fish Na+ channels on TTX sensitivity. Toxicon 2008, 51, 381–387. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.A.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef]
- Zimmer, T. Effects of tetrodotoxin on the mammalian cardiovascular system. Mar. Drugs 2010, 8, 741–762. [Google Scholar] [CrossRef]
- Zimmer, T.; Haufe, V.; Blechschmidt, S. Voltage-gated sodium channels in the mammalian heart. Glob. Cardiol. Sci. Pract. 2014, 2014, 449–463. [Google Scholar] [CrossRef]
- Zou, S. Comparative transcriptome analysis of toxic and non-toxic nassarius communities and identification of genes involved in TTX-adaptation. Toxins 2020, 12, 761. [Google Scholar] [CrossRef]
- Matsumoto, T.; Feroudj, H.; Kikuchi, R.; Kawana, Y.; Kondo, H.; Hirono, I.; Mochizuki, T.; Nagashima, Y.; Kaneko, G.; Ushio, H.; et al. DNA microarray analysis on the genes differentially expressed in the liver of the pufferfish, Takifugu rubripes, following an intramuscular administration of tetrodotoxin. Microarrays 2014, 3, 226–244. [Google Scholar] [CrossRef]
- Kiriake, A.; Ohta, A.; Suga, E.; Matsumoto, T.; Ishizaki, S.; Nagashima, Y. Comparison of tetrodotoxin uptake and gene expression in the liver between juvenile and adult tiger pufferfish, Takifugu rubripes. Toxicon 2016, 111, 6–12. [Google Scholar] [CrossRef]
- Kamieniarz-Gdula, K.; Proudfoot, N.J. Transcriptional control by premature termination: A forgotten mechanism. Trends Genet. 2019, 35, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, T.; Arakawa, O. Tetrodotoxin–distribution and accumulation in aquatic organisms, and cases of human intoxication. Mar. Drugs 2008, 6, 220–242. [Google Scholar] [CrossRef] [Green Version]
- Itoi, S.; Suzuki, M.; Asahina, K.; Sawayama, E.; Nishikubo, J.; Oyama, H.; Takei, M.; Shiibashi, N.; Takatani, T.; Arakawa, O.; et al. Role of maternal tetrodotoxin in survival of larval pufferfish. Toxicon 2018, 148, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Arakawa, O.; Takatani, T. Toxicity of pufferfish Takifugu rubripes cultured in netcages at sea or aquaria on land. Comp. Biochem. Physiol. Part D Genom. Proteom. 2006, 1, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, R.; Gao, W.; Ibi, K.; Mine, T.; Okita, K.; Nishihara, G.N.; Takatani, T.; Arakawa, O. Profile differences in tetrodotoxin transfer to skin and liver in the pufferfish Takifugu rubripes. Toxicon 2017, 130, 73–78. [Google Scholar] [CrossRef]
- Gao, W.; Yamada, M.; Ohki, R.; Nagashima, Y.; Tatsuno, R.; Ikeda, K.; Kawatsu, K.; Takatani, T.; Arakawa, O. Evaluation of the tetrodotoxin uptake ability of pufferfish Takifugu rubripes tissues according to age using an in vitro tissue slice incubation method. Toxicon 2020, 174, 8–12. [Google Scholar] [CrossRef]
- Ikeda, K.; Murakami, Y.; Emoto, Y.; Ngy, L.; Taniyama, S.; Yagi, M.; Takatani, T.; Arakawa, O. Transfer profile of intramuscularly administered tetrodotoxin to non-toxic cultured specimens of the pufferfish Takifugu rubripes. Toxicon 2009, 53, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, Y.; Okada, K.; Takatani, T.; Kawatsu, K.; Hamano, Y.; Arakawa, O.; Noguchi, T. Intra-tissue distribution of tetrodotoxin in two marine puffers Takifugu vermicularis and Chelonodon patoca. Toxicon 2003, 41, 13–18. [Google Scholar] [CrossRef]
- Mahmud, Y.; Arakawa, O.; Ichinose, A.; Begum Tanu, M.; Takatani, T.; Tsuruda, K.; Kawatsu, K.; Hamano, Y.; Noguchi, T. Intracellular visualization of tetrodotoxin (TTX) in the skin of a puffer Tetraodon nigroviridis by immunoenzymatic technique. Toxicon 2003, 41, 605–611. [Google Scholar] [CrossRef]
- Knauf, F.; Yang, C.L.; Thomson, R.B.; Mentone, S.A.; Giebisch, G.; Aronson, P.S. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9425–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendrick, A.A.; Dickey, A.M.; Redwine, W.B.; Tran, P.T.; Vaites, L.P.; Dzieciatkowska, M.; Harper, J.W.; Reck-Peterson, S.L. Hook3 is a scaffold for the opposite-polarity microtubule-based motors cytoplasmic dynein-1 and KIF1C. J. Cell Biol. 2019, 218, 2982–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Ostriker, A.C.; Jin, Y.; Hu, H.; Sizer, A.J.; Peng, G.; Morris, A.H.; Ryu, C.; Herzog, E.L.; Kyriakides, T.; et al. LMO7 is a negative feedback regulator of transforming growth factor β signaling and fibrosis. Circulation 2019, 139, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Draisci, G.; Iadarola, M.J. Temporal analysis of increases in c-fos, preprodynorphin and preproenkephalin mRNAs in rat spinal cord. Mol. Brain Res. 1989, 6, 31–37. [Google Scholar] [CrossRef]
- Ji, R.R.; Wang, X.M.; Han, J.S. Induction of Fos-like protein in the rat spinal cord following electroacupuncture stimulation. Sheng Li Xue Bao 1992, 44, 394–400. [Google Scholar]
- Marvaldi, L.; Panayotis, N.; Alber, S.; Dagan, S.Y.; Okladnikov, N.; Koppel, I.; Di Pizio, A.; Song, D.A.; Tzur, Y.; Terenzio, M.; et al. Importin α3 regulates chronic pain pathways in peripheral sensory neurons. Science 2020, 369, 842–846. [Google Scholar] [CrossRef]
- Li, Y.; Xu, X.; Liu, L.; Kuang, H.; Xu, L.; Xu, C. A gold nanoparticle-based lateral flow immunosensor for ultrasensitive detection of tetrodotoxin. Analyst 2020, 145, 2143–2151. [Google Scholar] [CrossRef]
- Qiao, K.; Fang, C.; Chen, B.; Liu, Z.; Pan, N.; Peng, H.; Hao, H.; Xu, M.; Wu, J.; Liu, S. Molecular characterization, purification, and antioxidant activity of recombinant superoxide dismutase from the Pacific abalone Haliotis discus hannai Ino. World J. Microbiol. Biotechnol. 2020, 36, 115. [Google Scholar] [CrossRef]
- Li, B.; Song, K.; Meng, J.; Li, L.; Zhang, G. Integrated application of transcriptomics and metabolomics provides insights into glycogen content regulation in the Pacific oyster Crassostrea gigas. BMC Genom. 2017, 18, 713. [Google Scholar] [CrossRef]
- Zhang, H.; Jain, C.; Aluru, S. A comprehensive evaluation of long read error correction methods. BMC Genom. 2020, 21, 889. [Google Scholar] [CrossRef] [PubMed]
- Modi, A.; Vai, S.; Caramelli, D.; Lari, M. The Illuminasequencing protocol and the NovaSeq 6000 system. Methods Mol. Biol. 2021, 2242, 15–42. [Google Scholar]
- Wu, Q.; Zang, F.; Xie, X.; Ma, Y.; Zheng, Y.; Zang, D. Full-length transcriptome sequencing analysis and development of EST-SSR markers for the endangered species Populus wulianensis. Sci. Rep. 2020, 10, 16249. [Google Scholar] [CrossRef] [PubMed]
- Bhoite, R.; Si, P.; Siddique, K.H.M.; Yan, G. Comparative transcriptome analyses for metribuzin tolerance provide insights into key genes and mechanisms restoring photosynthetic efficiency in bread wheat (Triticum aestivum L.). Genomics 2021, 113, 910–918. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, T.; Tatsukami, Y.; Kitahara, N.; Morisaka, H.; Kuroda, K.; Ueda, M. Time-course proteomic profile of Candida albicans during adaptation to a fetal serum. Pathog. Dis. 2013, 67, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Liang, W.; Dai, L.; Zhou, L.; Yao, Y.; Zhong, X.; Chen, H.; Xu, J. Comparative and quantitative proteomic analysis of normal and degenerated human annulus fibrosus cells. Clin. Exp. Pharmacol. Physiol. 2015, 42, 530–536. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Bai, F.; Tang, Z.; Liu, N.; Liu, Q. Integrative transcriptomic, proteomic, and machine learning approach to identifying feature genes of atrial fibrillation using atrial samples from patients with valvular heart disease. BMC Cardiovasc. Disord. 2021, 21, 52. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TTX | anhydro-TTX | 6-epi-TTX | 11-deoxy-TTX |
 | 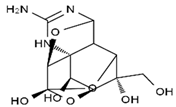 |  | 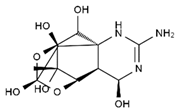 |
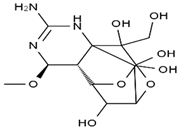
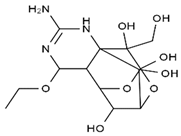
| methoxy-TTX | 4-ethoxy-TTX | 6-dehydroxymentyl-TTX | deoxy-TTX |
 |  | 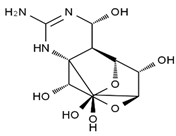 | 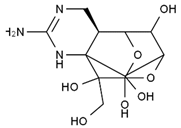 |
| diacetylanhydro-TTX | 11-oxo-TTX | 4-epi-TTX | 4,9-anhydro-TTX |
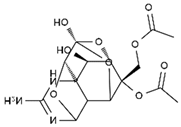 |  |  |  |
| 4,9-anhydro-TTX | 8,11-dideoxy-TTX | 8-deoxy-TTX | 11-nor-TTX-6(R)-ol |
 | 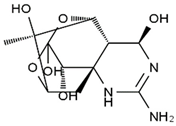 | 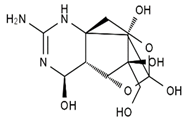 | 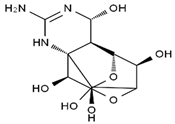 |
| 11-nor-TTX-6(S)-ol | 11-nor-TTX-6,6-diol | 5-deoxy-TTX | 6-deoxy-TTX |
 |  | 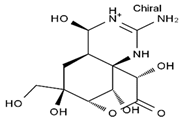 |  |
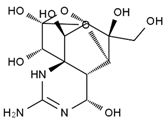
| 6,11-dideoxy-TTX | chiriquitoxin | tetrodaminotoxin | 4-beta-TTX |
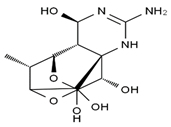 | 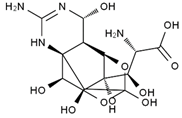 |  |  |
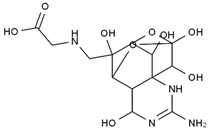
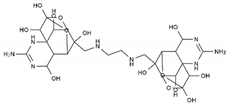
| 11-carboxymethyl amino-11-deoxy-TTX | N,N′-ethylenediaminedi-TTX | ||
 |  | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, H.; Qiao, K.; Wang, C.; Chen, B.; Xu, M.; Hao, H.; Huang, Z.; Liu, Z.; Wang, Q. Multi-Omics Analysis of Gene and Protein Candidates Possibly Related to Tetrodotoxin Accumulation in the Skin of Takifugu flavidus. Mar. Drugs 2021, 19, 639. https://doi.org/10.3390/md19110639
Feng H, Qiao K, Wang C, Chen B, Xu M, Hao H, Huang Z, Liu Z, Wang Q. Multi-Omics Analysis of Gene and Protein Candidates Possibly Related to Tetrodotoxin Accumulation in the Skin of Takifugu flavidus. Marine Drugs. 2021; 19(11):639. https://doi.org/10.3390/md19110639
Chicago/Turabian StyleFeng, Huimin, Kun Qiao, Chunchun Wang, Bei Chen, Min Xu, Hua Hao, Zhen Huang, Zhiyu Liu, and Qin Wang. 2021. "Multi-Omics Analysis of Gene and Protein Candidates Possibly Related to Tetrodotoxin Accumulation in the Skin of Takifugu flavidus" Marine Drugs 19, no. 11: 639. https://doi.org/10.3390/md19110639
APA StyleFeng, H., Qiao, K., Wang, C., Chen, B., Xu, M., Hao, H., Huang, Z., Liu, Z., & Wang, Q. (2021). Multi-Omics Analysis of Gene and Protein Candidates Possibly Related to Tetrodotoxin Accumulation in the Skin of Takifugu flavidus. Marine Drugs, 19(11), 639. https://doi.org/10.3390/md19110639