Computational Methodologies in the Exploration of Marine Natural Product Leads



Abstract
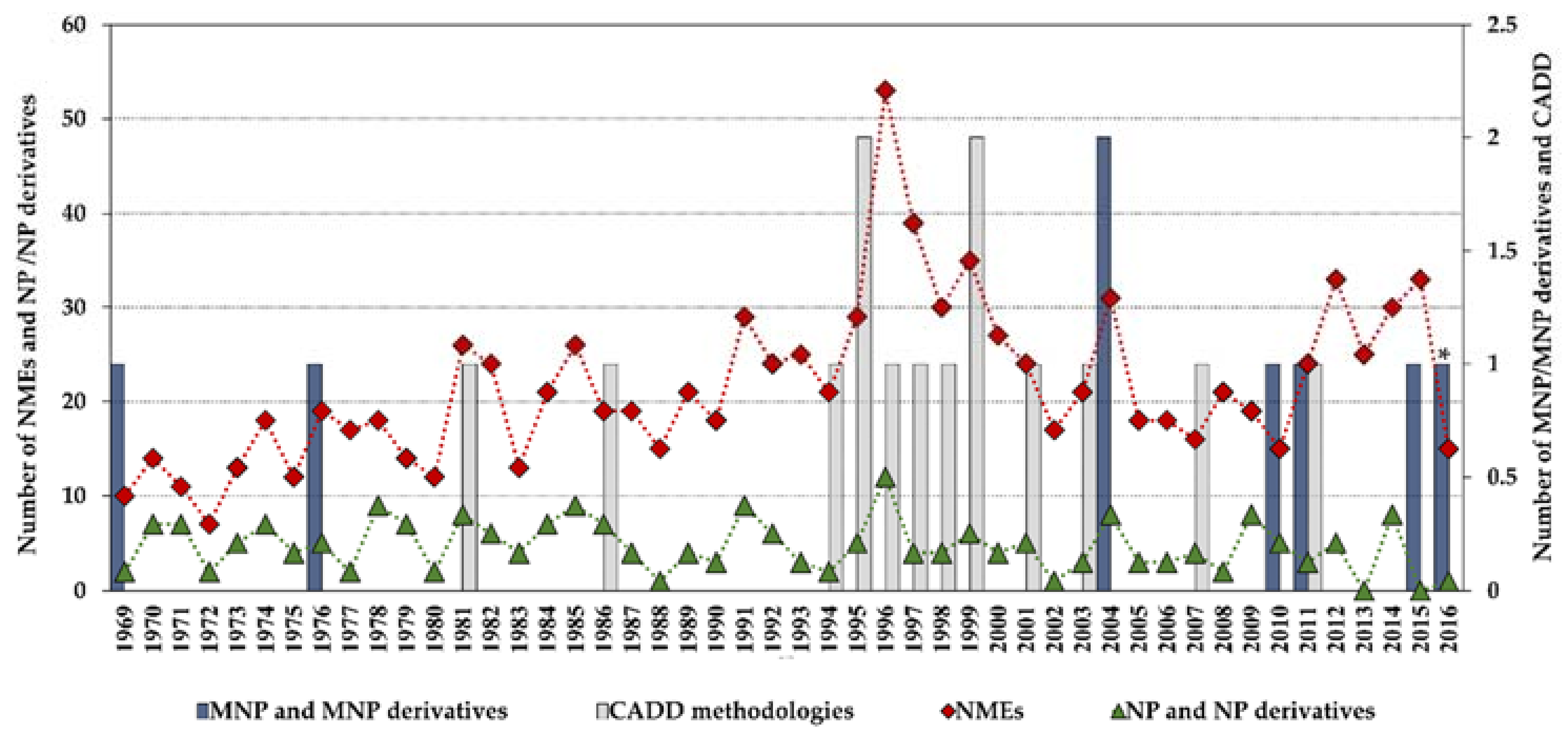
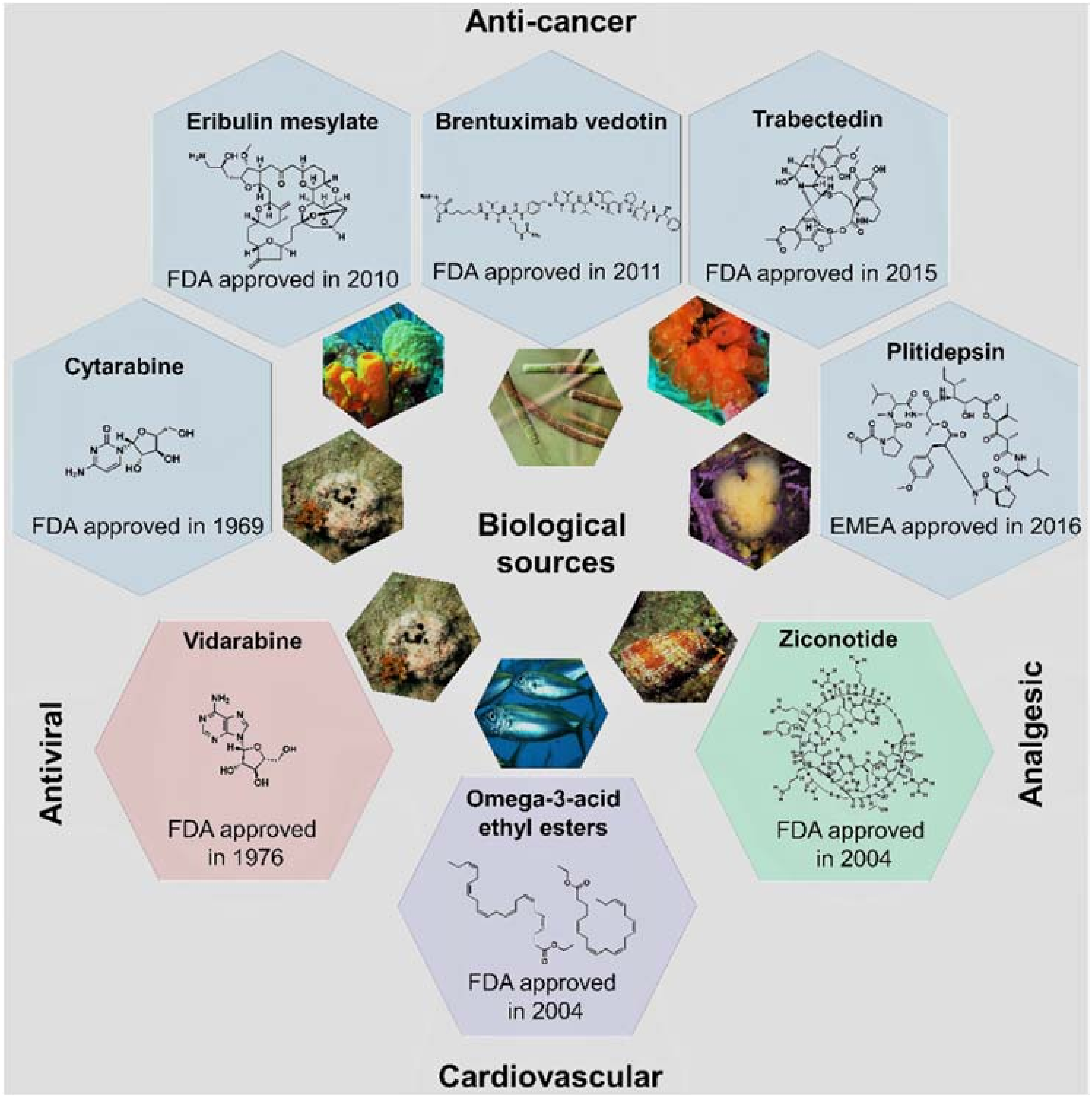
1. Introduction
2. Databases
3. Dereplication
3.1. Computer-Assisted Identification of Compounds
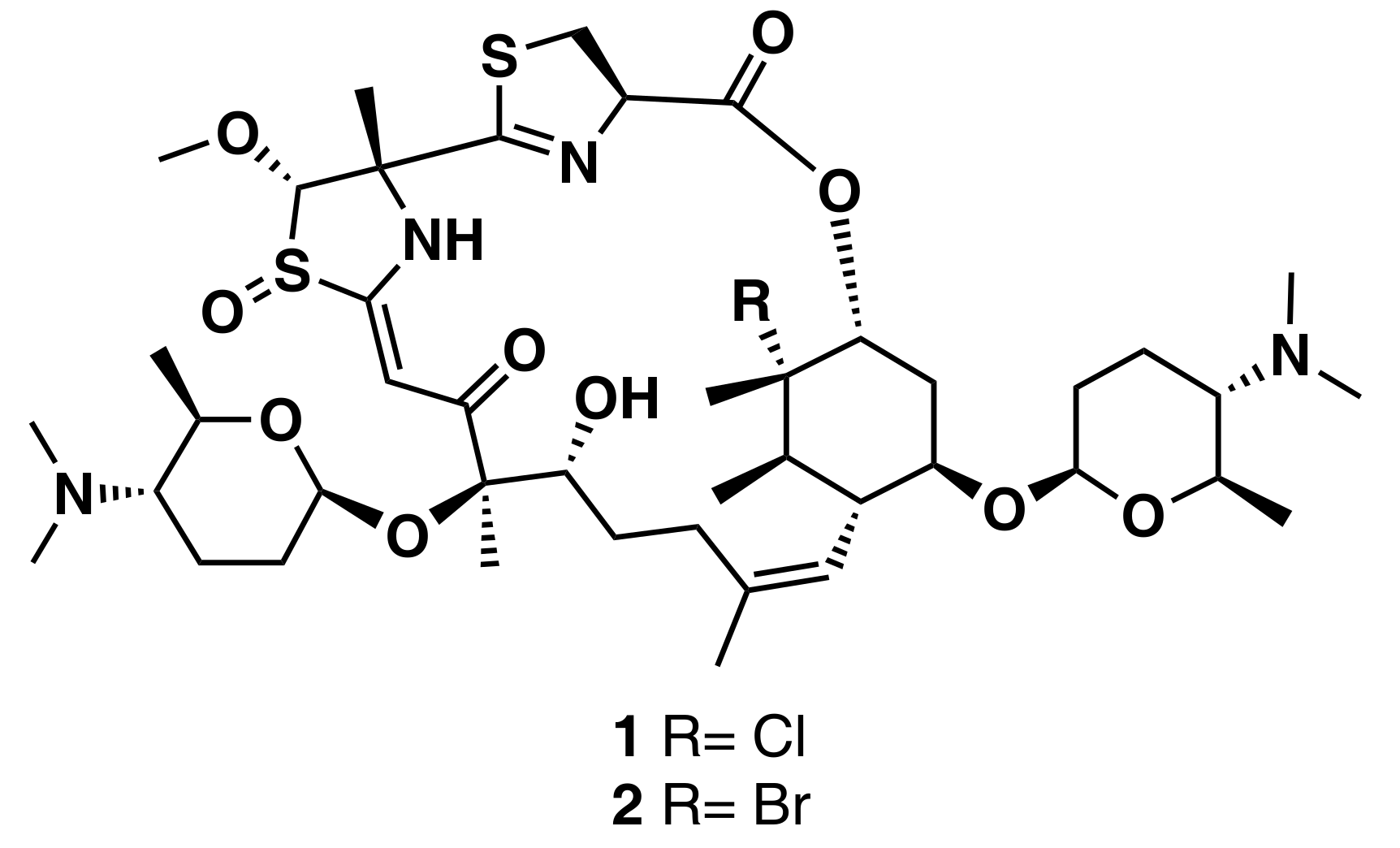
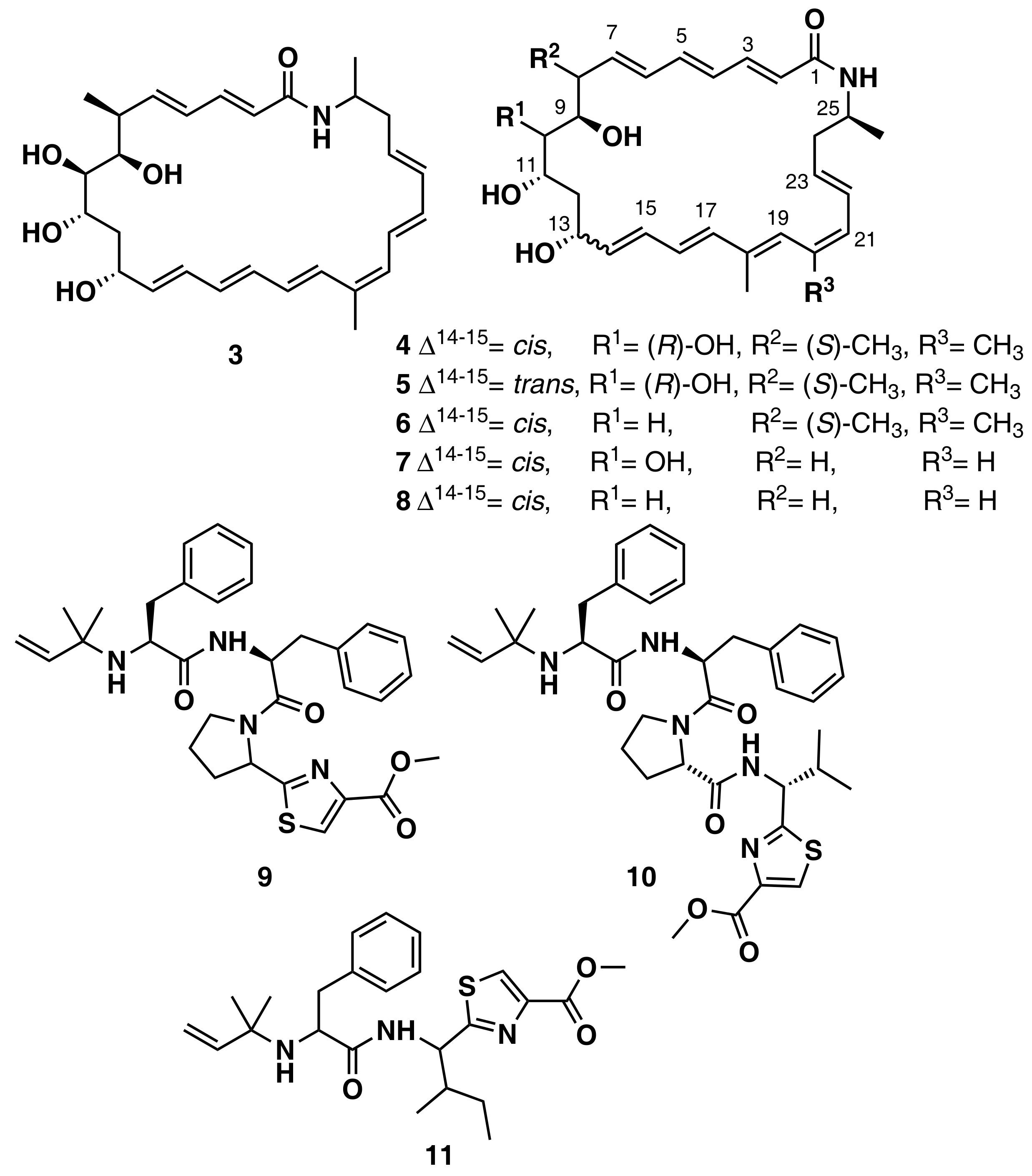
3.1.1. Secondary Metabolite-Guided
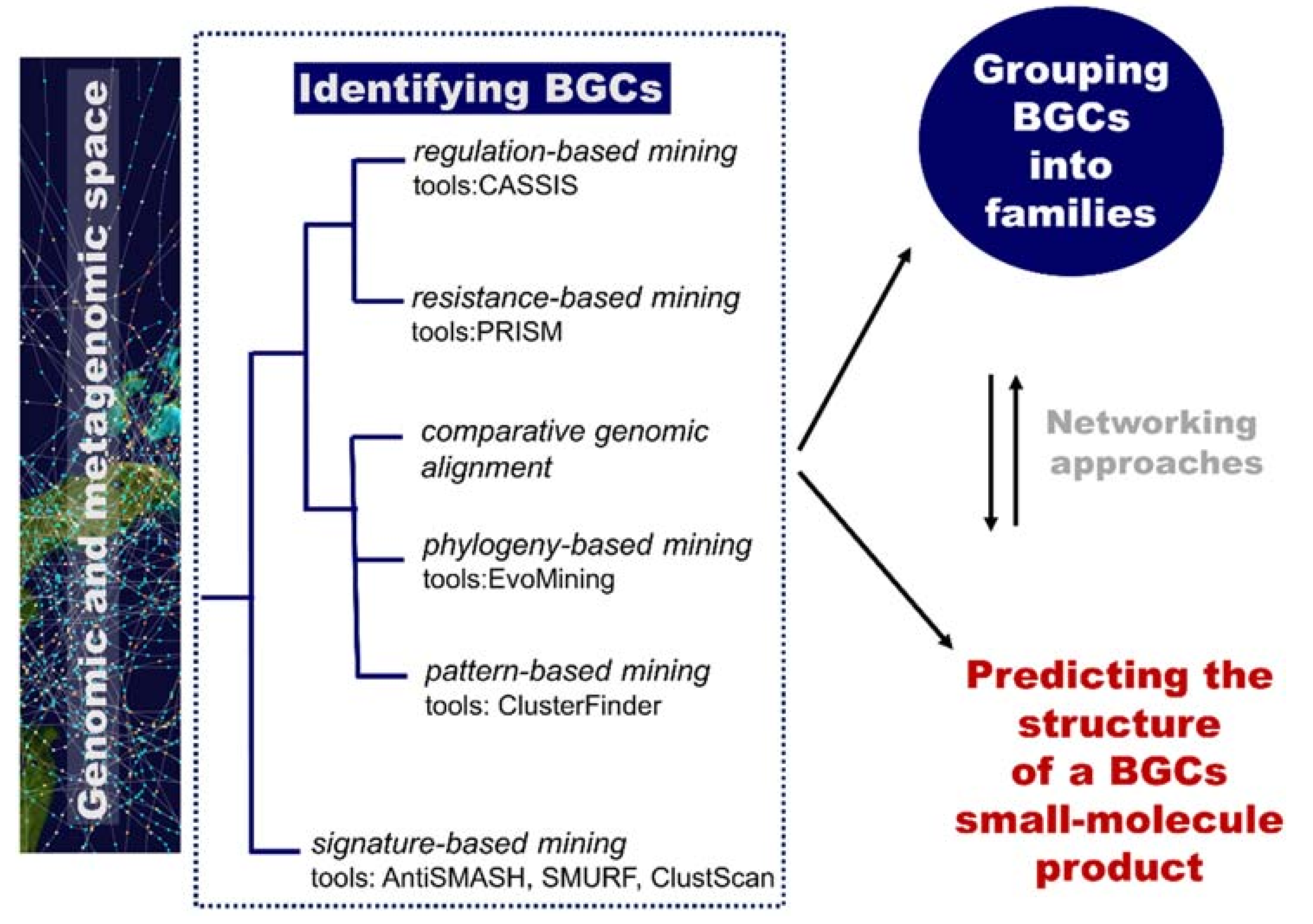
3.1.2. Genome-Guided
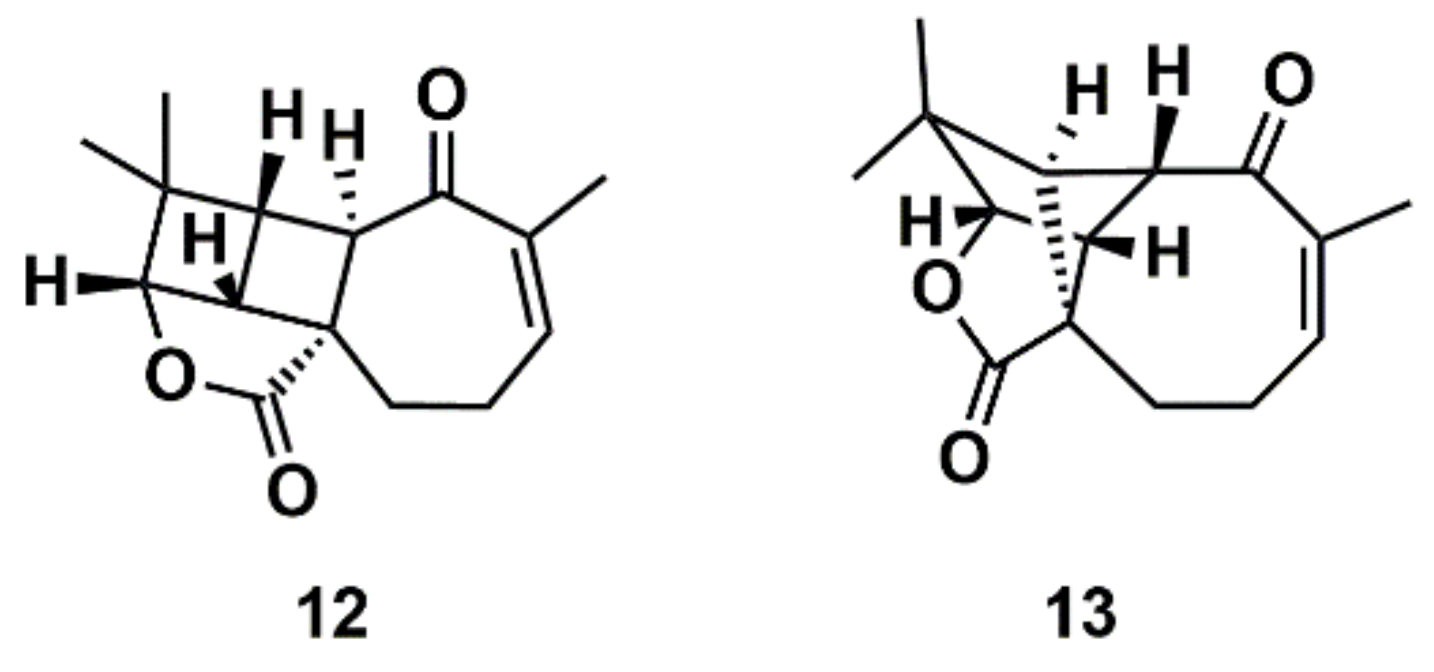
3.2. Computer-Assisted Structure Elucidation (CASE)
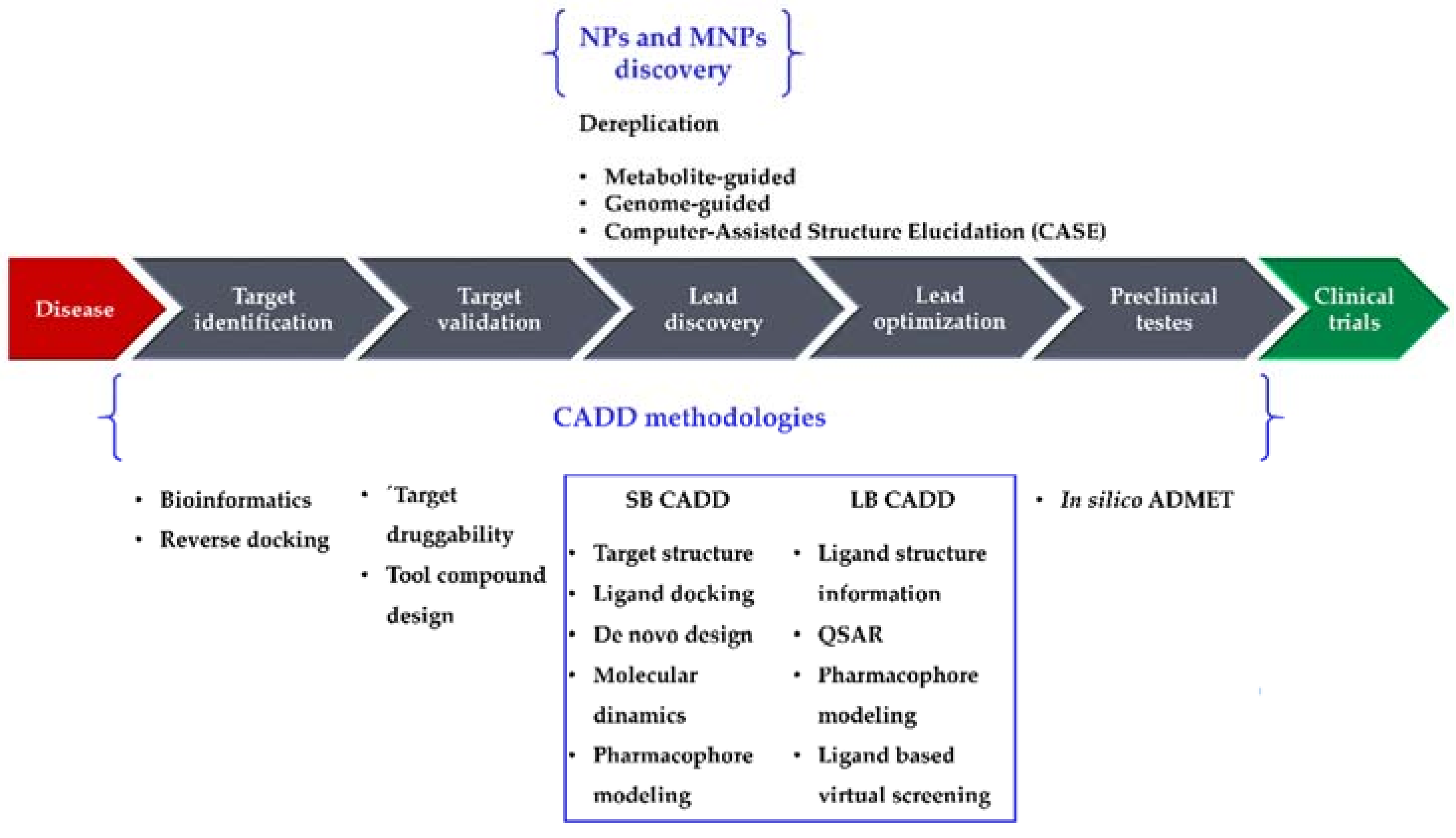
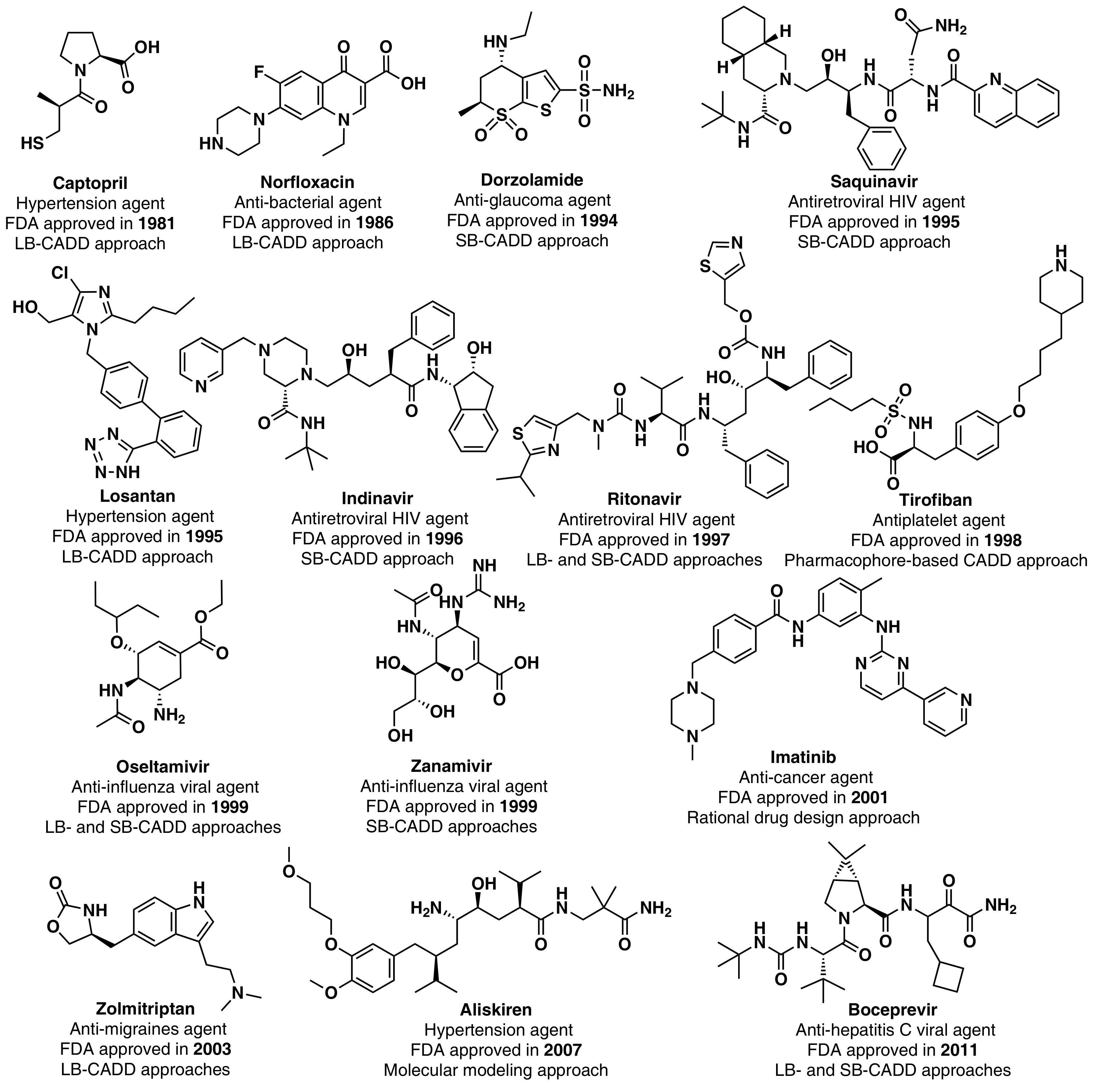
4. Computer-Aided Drug Design (CADD)
4.1. Ligand-Based (LB)
4.2. Structure-Based (SB)
Funding
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Nosengo, N. New tricks for old drugs. Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [PubMed]
- Nantasenamat, C.; Prachayasittikul, V. Maximizing computational tools for successful drug discovery. Expert Opin. Drug Discov. 2015, 10, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J. Chemoinformatics: Achievements and challenges, a personal view. Molecules 2016, 21, 151. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.; Dawson, E.S.; Meiler, J.; Rodriguez, A.L.; Chauder, B.A.; Bates, B.S.; Felts, A.S.; Lamb, J.P.; Menon, U.N.; Jadhav, S.B.; et al. Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu5): From an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem 2012, 7, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.-T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.L.; Grier, M.D.; Jones, C.K.; Herman, E.J.; Kane, A.S.; Smith, R.L.; Williams, R.; Zhou, Y.; Marlo, J.E.; Days, E.L.; et al. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 2010, 78, 1105–1123. [Google Scholar] [CrossRef] [PubMed]
- Gaudencio, S.P.; Pereira, F. Dereplication: Racing to speed up the natural products discovery process. Nat. Prod. Rep. 2015, 32, 779–810. [Google Scholar] [CrossRef] [PubMed]
- Perez-Victoria, I.; Martin, J.; Reyes, F. Combined LC/UV/MS and NMR strategies for the dereplication of marine natural products. Planta Med. 2016, 82, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “State of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishnan, R. Structure-based drug design and modern medicine. J. Postgrad. Med. 2009, 55, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Van Drie, J.H. Computer-aided drug design: The next 20 years. J. Comput. Aided Mol. Des. 2007, 21, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discov. 2006, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Geysen, H.M.; Schoenen, F.; Wagner, D.; Wagner, R. Combinatorial compound libraries for drug discovery: An ongoing challenge. Nat. Rev. Drug Discov. 2003, 2, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Dolle, R.E. Historical overview of chemical library design. Methods Mol. Biol. 2011, 685, 3–25. [Google Scholar] [PubMed]
- Sanger, F. Sequences, sequences, and sequences. Annu. Rev. Biochem. 1988, 57, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Joachimiak, A. High-throughput crystallography for structural genomics. Curr. Opin. Struct. Biol. 2009, 19, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Fuerst, P. The future of high-throughput screening. J. Biomol. Screen. 2008, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Rodriguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2009–2011: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2013, 11, 2510–2573. [Google Scholar] [PubMed]
- Ruiz-Torres, V.; Antonio Encinar, J.; Herranz-Lopez, M.; Perez-Sanchez, A.; Galiano, V.; Barrajon-Catalan, E.; Micol, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Naughton, L.M.; Montanchez, I.; Dobson, A.D.W.; Rai, D.K. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. USA 2005, 102, 17272–17277. [Google Scholar] [CrossRef] [PubMed]
- Berdy, J. Bioactive microbial metabolites-A personal view. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wohlleben, W.; Mast, Y.; Stegmann, E.; Ziemert, N. Antibiotic drug discovery. Microb. Biotechnol. 2016, 9, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Seo, C.H.; Park, Y. Marine peptides and their anti-infective activities. Mar. Drugs 2015, 13, 618–654. [Google Scholar] [CrossRef] [PubMed]
- Valliappan, K.; Sun, W.; Li, Z. Marine actinobacteria associated with marine organisms and their potentials in producing pharmaceutical natural products. Appl. Microbiol. Biotechnol. 2014, 98, 7365–7377. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.B.; Cheung, R.C.F.; Wong, J.H.; Bekhit, A.A.; Bekhit, A.E.-D. Antibacterial products of marine organisms. Appl. Microbiol. Biotechnol. 2015, 99, 4145–4173. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. Pubchem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kops, C.d.B.; Kirchmair, J. Data resources for the computer-guided discovery of bioactive natural products. J. Chem. Inf. Model. 2017, 57, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Klementz, D.; Doering, K.; Lucas, X.; Telukunta, K.K.; Erxleben, A.; Deubel, D.; Erber, A.; Santillana, I.; Thomas, O.S.; Bechthold, A.; et al. StreptomeDB 2.0-an extended resource of natural products produced by streptomycetes. Nucleic Acids Res. 2016, 44, D509–D514. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Cho, S.Y.; Pak, H.J.; Kim, Y.; Choi, J.-Y.; Lee, Y.J.; Gong, B.H.; Kang, Y.S.; Han, T.; Choi, G.; et al. NPCARE: Database of natural products and fractional extracts for cancer regulation. J. Cheminformatics 2017, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. Chemspider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Jones, L.H.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. Zinc 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMBD 3.0-the human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Barneh, F.; Jafari, M.; Mirzaie, M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of drugbank database. Brief. Bioinform. 2016, 17, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDB: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Tang, K.; Liu, Q.; Sun, Y.; Huang, Q.; Zhu, R.; Gao, J.; Zhang, D.; Huang, C.; Cao, Z. HIM-herbal ingredients in-vivo metabolism database. J. Cheminform. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Mangal, M.; Sagar, P.; Singh, H.; Raghava, G.P.S.; Agarwal, S.M. NPACT: Naturally occurring plant-based anti-cancer compound-activity-target database. Nucleic Acids Res. 2013, 41, D1124–D1129. [Google Scholar] [CrossRef] [PubMed]
- Valli, M.; dos Santos, R.N.; Figueira, L.D.; Nakajima, C.H.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 2013, 76, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.-C. TCM database@Taiwan: The world’s largest traditional chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Canh Hao, N.; Mamitsuka, H. Current status and prospects of computational resources for natural product dereplication: A review. Brief. Bioinform. 2016, 17, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Scheubert, K.; Bocker, S. New kids on the block: Novel informatics methods for natural product discovery. Nat. Prod. Rep. 2014, 31, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Qiao, Y.; Ang, E.L.; Zhao, H. Using natural products for drug discovery: The impact of the genomics era. Expert Opin. Drug Discov. 2017, 12, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Chanana, S.; Thomas, C.S.; Braun, D.R.; Hou, Y.; Wyche, T.P.; Bugni, T.S. Natural product discovery using planes of principal component analysis in R (POPCAR). Metabolites 2017, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.A.; Hou, Y.; Braun, D.R.; Wyche, T.P.; Adnani, N.; Vazquez-Rivera, E.; Bugni, T.S. LC/MS untargeted metabolomics for prioritizing marine invertebrate-associated bacteria for discovery of natural products. Planta Med. 2013, 79, 844–844. [Google Scholar] [CrossRef]
- Macintyre, L.; Zhang, T.; Viegelmann, C.; Martinez, I.J.; Cheng, C.; Dowdells, C.; Abdelmohsen, U.R.; Gernert, C.; Hentschel, U.; Edrada-Ebel, R. Metabolomic tools for secondary metabolite discovery from marine microbial symbionts. Mar. Drugs 2014, 12, 3416–3448. [Google Scholar] [CrossRef] [PubMed]
- Tawfike, A.F.; Viegelmann, C.; Edrada-Ebel, R. Metabolomics and dereplication strategies in natural products. Methods Mol. Biol. 2013, 1055, 227–244. [Google Scholar] [PubMed]
- Abdelmohsen, U.R.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication strategies for targeted isolation of new antitrypanosomal actinosporins A and B from a marine sponge associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220–1244. [Google Scholar] [CrossRef] [PubMed]
- Sidebottom, A.M.; Johnson, A.R.; Karty, J.A.; Trader, D.J.; Carlson, E.E. Integrated metabolomics approach facilitates discovery of an unpredicted natural product suite from streptomyces coelicolor M145. ACS Chem. Biol. 2013, 8, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Tawfike, A.F.; Tate, R.; Abbott, G.; Young, L.; Viegelmann, C.; Schumacher, M.; Diederich, M.; Edrada-Ebel, R.A. Metabolomic tools to assess the chemistry and bioactivity of endophytic Aspergillus strain. Chem. Biodivers. 2017, 14, e1700040. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Roullier, C.; Guitton, Y.; Valery, M.; Amand, S.; Prado, S.; du Pont, T.R.; Grovel, O.; Pouchus, Y.F. Automated detection of natural halogenated compounds from LC-MS profiles-application to the isolation of bioactive chlorinated compounds from marine-derived fungi. Anal. Chem. 2016, 88, 9143–9150. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Don Duy, N.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Crusemann, M.; O’Neill, E.C.; Larson, C.B.; Melnik, A.V.; Floros, D.J.; da Silva, R.R.; Jensen, P.R.; Dorrestein, P.C.; Moore, B.S. Prioritizing natural product diversity in a collection of 146 bacterial strains based on growth and extraction protocols. J. Nat. Prod. 2017, 80, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Parrot, D.; Intertaglia, L.; Jehan, P.; Grube, M.; Suzuki, M.T.; Tomasi, S. Chemical analysis of the alphaproteobacterium strain MOLA1416 associated with the marine lichen Lichina pygmaea. Phytochemistry 2018, 145, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kerber, A.; Laue, R.; Meringer, M.; Rucker, C. Molecules in silico: The generation of structural formulae and its applications. J. Comput. Chem. Jpn. 2004, 3, 85–96. [Google Scholar] [CrossRef]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.J.; Henry, C.S. Mines: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. Lipidblast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Boecker, S. Mining molecular structure databases: Identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom. Rev. 2017, 36, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Kangas, L.J.; Metz, T.O.; Isaac, G.; Schrom, B.T.; Ginovska-Pangovska, B.; Wang, L.; Tan, L.; Lewis, R.R.; Miller, J.H. In silico identification software (ISIS): A machine learning approach to tandem mass spectral identification of lipids. Bioinformatics 2012, 28, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Wolf, S.; Schmidt, S.; Mueller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, M.; Neumann, S. Metfusion: Integration of compound identification strategies. J. Mass Spectrom. 2013, 48, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. Midas: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S.; de Vos, R.C.H.; van Schaik, R.; Vervoort, J. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 2012, 26, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.J.; Verhoeven, S. Automatic compound annotation from mass spectrometry data using MAGMa. Mass Spectrom. 2014, 3, S0033. [Google Scholar] [CrossRef] [PubMed]
- Verdegem, D.; Lambrechts, D.; Carmeliet, P.; Ghesquiere, B. Improved metabolite identification with MIDAS and MAGMa through MS/MS spectral dataset-driven parameter optimization. Metabolomics 2016, 12, 98. [Google Scholar] [CrossRef]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Duhrkop, K.; Bocker, S.; Rousu, J. Metabolite identification through multiple kernel learning on fragmentation trees. Bioinformatics 2014, 30, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Boecker, S.; Rasche, F. Towards de novo identification of metabolites by analyzing tandem mass spectra. Bioinformatics 2008, 24, I49–I55. [Google Scholar] [CrossRef] [PubMed]
- Duehrkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Boecker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Fischbach, M.A. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Shinose, M.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M.; Omura, S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003, 21, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Udwary, D.W.; Zeigler, L.; Asolkar, R.N.; Singan, V.; Lapidus, A.; Fenical, W.; Jensen, P.R.; Moore, B.S. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. USA 2007, 104, 10376–10381. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.J.; Donia, M.S.; Siqueira-Neto, J.L.; Ray, D.; Raskatov, J.A.; Green, R.E.; McKerrow, J.H.; Fischbach, M.A.; Linington, R.G. Genome-directed lead discovery: Biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem. Biol. 2015, 10, 2373–2381. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Liu, L.W.; Jokela, J.; Wahlsten, M.; Gugger, M.; Calteau, A.; Permi, P.; Kerfeld, C.A.; Sivonen, K.; Fewer, D.P. Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem. Biol. 2013, 20, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Kim, H.U. The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Syst. Synth. Biotechnol. 2016, 1, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. Hmmer web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, A.; Zucko, J.; Simunkovic, J.; Long, P.F.; Cullum, J.; Hranueli, D. Clustscan: An integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 2008, 36, 6882–6892. [Google Scholar] [CrossRef] [PubMed]
- Khaldi, N.; Seifuddin, F.T.; Turner, G.; Haft, D.; Nierman, W.C.; Wolfe, K.H.; Fedorova, N.D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010, 47, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.W.; Schwalen, C.J.; Kautsar, S.A.; Duran, H.G.S.; Santos, E.; Kim, H.U.; Nave, M.; et al. AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Brown, L.C.W.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Morales, P.; Kopp, J.F.; Martinez-Guerrero, C.; Alfonso Yanez-Guerra, L.; Selem-Mojica, N.; Ramos-Aboites, H.; Feldmann, J.; Barona-Gomez, F. Phylogenomic analysis of natural products biosynthetic gene clusters allows discovery of arseno-organic metabolites in model Streptomycetes. Genome Biol. Evol. 2016, 8, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Takeda, I.; Umemura, M.; Koike, H.; Asai, K.; Machida, M. Motif-independent prediction of a secondary metabolism gene cluster using comparative genomics: Application to sequenced genomes of aspergillus and ten other filamentous fungal species. DNA Res. 2014, 21, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Johnston, C.W.; Li, H.; Webster, A.L.H.; Wyatt, M.A.; Magarvey, N.A. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015, 43, 9645–9662. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.Y.; Li, J.; Millan-Aguinaga, N.; Zhang, J.J.; O’Neill, E.C.; Ugalde, J.A.; Jensen, P.R.; Mantovani, S.M.; Moore, B.S. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem. Biol. 2015, 10, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Shelest, V.; Nath, N.; Shelest, E. CASSIS and SMIPS: Promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics 2016, 32, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C. Seneca: A platform-independent, distributed, and parallel system for computer-assisted structure elucidation in organic chemistry. J. Chem. Inf. Comput. Sci. 2001, 41, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Jayaseelan, K.V.; Steinbeck, C. Building blocks for automated elucidation of metabolites: Natural product-likeness for candidate ranking. BMC Bioinform. 2014, 15, 234. [Google Scholar] [CrossRef] [PubMed]
- Blinov, K.A.; Carlson, D.; Elyashberg, M.E.; Martin, G.E.; Martirosian, E.R.; Molodtsov, S.; Williams, A.J. Computer-assisted structure elucidation of natural products with limited 2D NMR data: Application of the struceluc system. Magn. Reson. Chem. 2003, 41, 359–372. [Google Scholar] [CrossRef]
- Elyashberg, M.E.; Blinov, K.A.; Williams, A.J.; Molodtsov, S.G.; Martin, G.E.; Martirosian, E.R. Structure elucidator: A versatile expert system for molecular structure elucidation from 1D and 2D NMR data and molecular fragments. J. Chem. Inf. Comput. Sci. 2004, 44, 771–792. [Google Scholar] [CrossRef] [PubMed]
- Molodtsov, S.G.; Elyashberg, M.E.; Blinov, K.A.; Williams, A.J.; Martirosian, E.E.; Martin, G.E.; Lefebvre, B. Structure elucidation from 2D NMR spectra using the struceluc expert system: Detection and removal of contradictions in the data. J. Chem. Inf. Comput. Sci. 2004, 44, 1737–1751. [Google Scholar] [CrossRef] [PubMed]
- Plainchont, B.; de Paulo Emerenciano, V.; Nuzillard, J.-M. Recent advances in the structure elucidation of small organic molecules by the LSD software. Magn. Reson. Chem. 2013, 51, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Troche-Pesqueira, E.; Anklin, C.; Gil, R.R.; Navarro-Vazquez, A. Computer-assisted 3D structure elucidation of natural products using residual dipolar couplings. Angew. Chem. Int. Ed. 2017, 56, 3660–3664. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sauri, J.; Mevers, E.; Peczuh, M.W.; Hiemstra, H.; Clardy, J.; Martin, G.E.; Williamson, R.T. Unequivocal determination of complex molecular structures using anisotropic NMR measurements. Science 2017, 356, 43. [Google Scholar] [CrossRef] [PubMed]
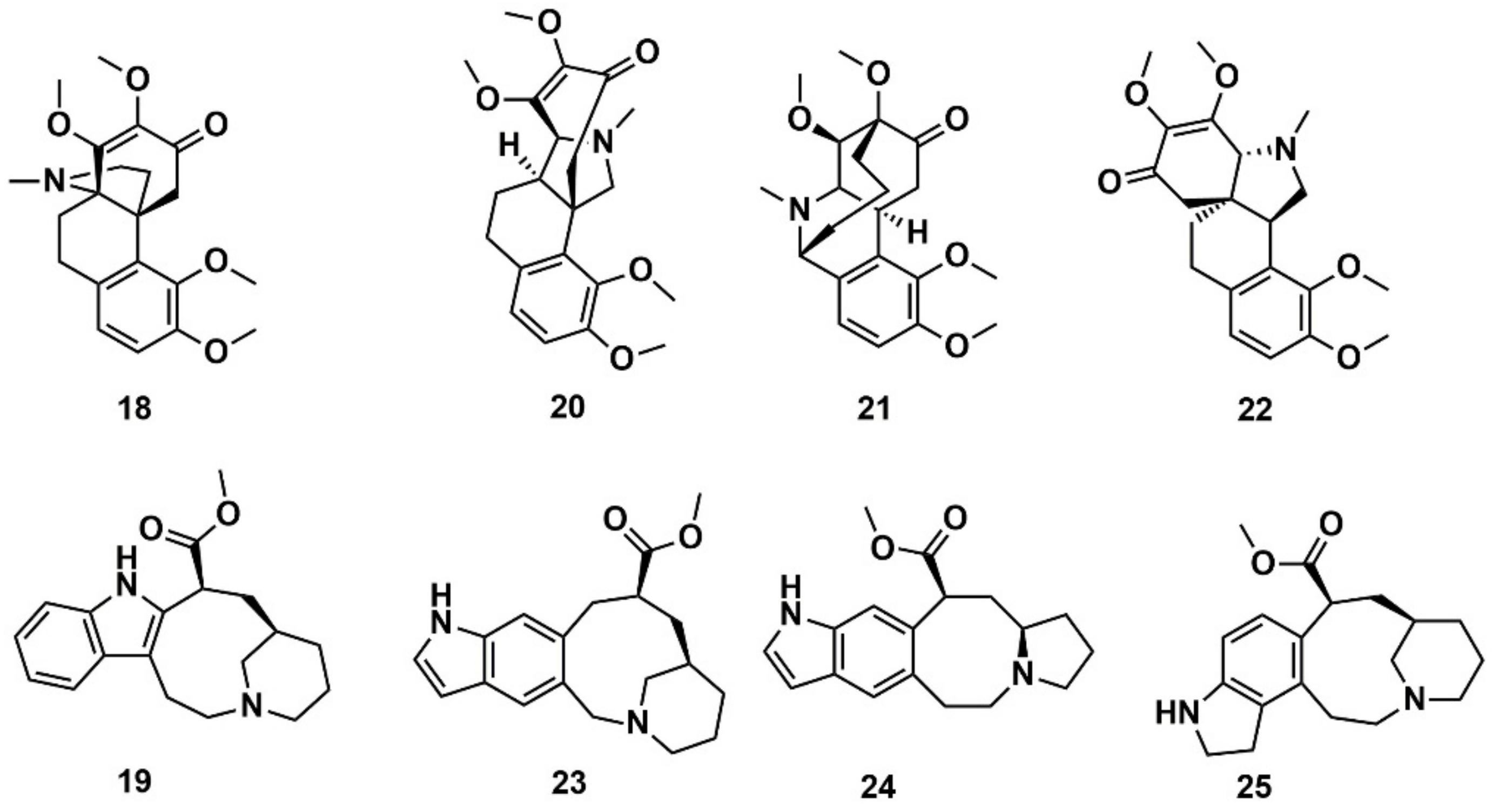
- Feliciano, A.S.; Medarde, M.; Delcorral, J.M.M.; Aramburu, A.; Gordaliza, M.; Barrero, A.F. Aquatolide—A new type of humulane-related sesquiterpene lactone. Tetrahedron Lett. 1989, 30, 2851–2854. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The correct structure of aquatolide-experimental validation of a theoretically-predicted structural revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef] [PubMed]
- Saya, J.M.; Vos, K.; Kleinnijenhuis, R.A.; van Maarseveen, J.H.; Ingemann, S.; Hiemstra, H. Total synthesis of aquatolide. Org. Lett. 2015, 17, 3892–3894. [Google Scholar] [CrossRef] [PubMed]
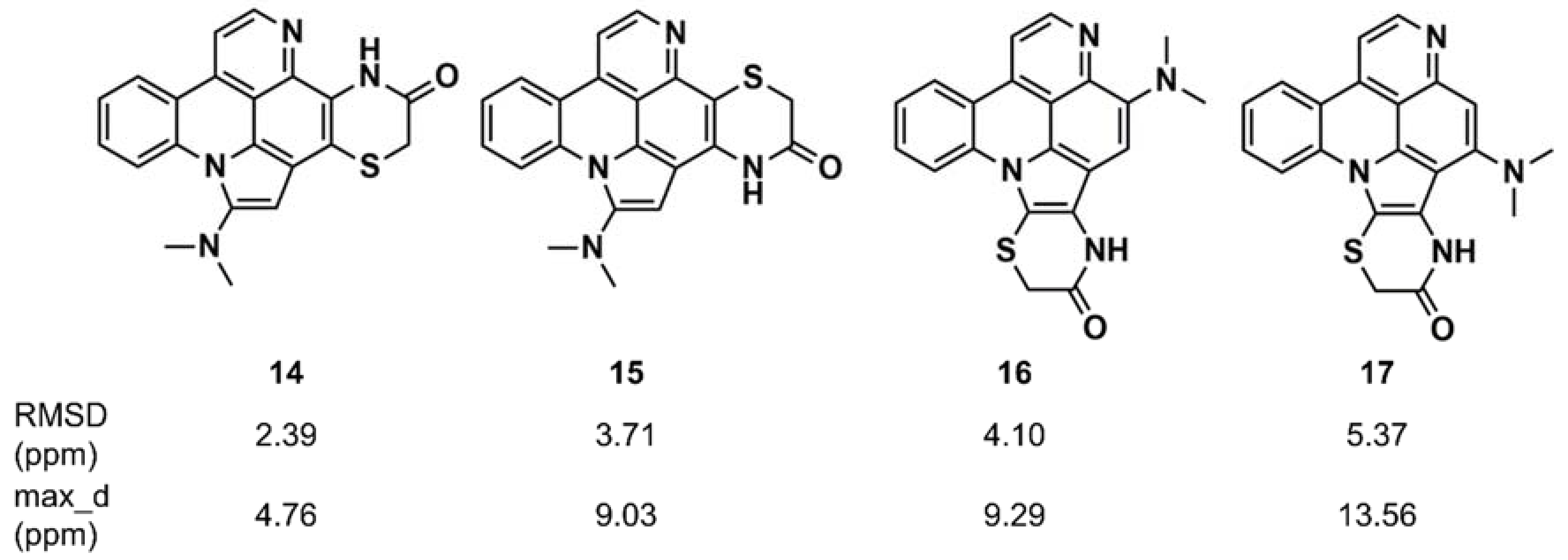
- Buevich, A.V.; Elyashberg, M.E. Synergistic combination of CASE algorithms and DFT chemical shift predictions: A powerful approach for structure elucidation, verification, and revision. J. Nat. Prod. 2016, 79, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.M.; Tantillo, D.J. Application of computational chemical shift prediction techniques to the cereoanhydride structure problem-carboxylate complications. Mar. Drugs 2017, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Buevich, A.V.; Elyashberg, M.E. Towards unbiased and more versatile NMR-based structure elucidation: A powerful combination of CASE algorithms and DFT calculations. Magn. Reson. Chem. 2017, 56, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Koren-Goldshlager, G.; Aknin, M.; Kashman, Y. Cycloshermilamine D, a new pyridoacridine from the marine tunicate Cystodytes violatinctus. J. Nat. Prod. 2000, 63, 830–831. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wu, Z.; Cai, C.; Wang, Q.; Tang, Y.; Cheng, F. Quantitative and systems pharmacology. 1. In silico prediction of drug-target interactions of natural products enables new targeted cancer therapy. J. Chem. Inf. Model. 2017, 57, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cheng, F.; Li, J.; Li, W.; Liu, G.; Tang, Y. Sdtnbi: An integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Brief. Bioinform. 2017, 18, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.-Y.; Matsuoka, Y.; Asai, Y.; Kamiyoshi, K.; Watanabe, T.; Kawaoka, Y.; Kitano, H. Systemsdock: A web server for network pharmacology-based prediction and analysis. Nucleic Acids Res. 2016, 44, W507–W513. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Moraga, M.; Njuguna, N.M.; Mugumbate, G.; Caballero, J.; Chibale, K. In silico comparison of antimycobacterial natural products with known antituberculosis drugs. J. Chem. Inf. Model. 2013, 53, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, L.-X.; Gong, J.-X.; Jiang, C.-S.; Yao, L.-G.; Li, J.-Y.; Li, J.; Xiao, W.; Guo, Y.-W. Design and synthesis of novel 1,2-dithiolan-4-yl benzoate derivatives as PTP1B inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2211–2216. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, D.; Makarieva, T.; Utkina, N.; Santalova, E.; Kryukova, E.; Methfessel, C.; Tsetlin, V.; Stonik, V.; Kasheverov, I. Marine natural products acting on the acetylcholine-binding protein and nicotinic receptors: From computer modeling to binding studies and electrophysiology. Mar. Drugs 2014, 12, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-C.; Xiao, X.; Zhang, Y.-K.; Talele, T.T.; Salim, A.A.; Chen, Z.-S.; Capon, R.J. Lamellarin O, a pyrrole alkaloid from an Australian marine sponge, Ianthella sp., reverses BCRP mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, W.; Xu, Y.; Ren, S.; Zhang, W.; Li, Y. Design, synthesis and biological evaluation of tasiamide B derivatives as BACE1 inhibitors. Bioorg. Med. Chem. 2015, 23, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Huebner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, H.Y.; El Sayed, K.A. Discovery of novel antiangiogenic marine natural product scaffolds. Mar. Drugs 2016, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Skariyachan, S.; Acharya, A.B.; Subramaniyan, S.; Babu, S.; Kulkarni, S.; Narayanappa, R. Secondary metabolites extracted from marine sponge associated Comamonas testosteroni and Citrobacter freundii as potential antimicrobials against MDR pathogens and hypothetical leads for VP40 matrix protein of Ebola virus: An in vitro and in silico investigation. J. Biomol. Struct. Dyn. 2016, 34, 1865–1883. [Google Scholar] [PubMed]
- Fang, J.; Yang, R.; Gao, L.; Zhou, D.; Yang, S.; Liu, A.-L.; Du, G.-H. Predictions of BuChE inhibitors using support vector machine and naïve bayesian classification techniques in drug discovery. J. Chem. Inf. Model. 2013, 53, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.D.J.; Vasanthi, A.H.R. QSAR based docking studies of marine algal anticancer compounds as inhibitors of protein kinase B (PKB). Eur. J. Pharm. Sci. 2015, 76, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Knight, N.J.; Hernando, E.; Haynes, C.J.E.; Busschaert, N.; Clarke, H.J.; Takimoto, K.; Garcia-Valverde, M.; Frey, J.G.; Quesada, R.; Gale, P.A. QSAR analysis of substituent effects on tambjamine anion transporters. Chem. Sci. 2016, 7, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Aswathy, L.; Jisha, R.S.; Masand, V.H.; Gajbhiye, J.M.; Shibi, I.G. Computational strategies to explore antimalarial thiazine alkaloid lead compounds based on an Australian marine sponge Plakortis lita. J. Biomol. Struct. Dyn. 2017, 35, 2407–2429. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.C.; Marquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-aided drug design applied to marine drug discovery: Meridianins as Alzheimer’s disease therapeutic agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Dineshkumar, K.; Aparna, V.; Madhuri, K.Z.; Hopper, W. Biological activity of sporolides A and B from salinispora tropica: In silico target prediction using ligand-based pharmacophore mapping and in vitro activity validation on HIV-1 reverse transcriptase. Chem. Biol. Drug Des. 2014, 83, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Dineshkumar, K.; Aparna, V.; Hopper, W. Ligand based-pharmacophore modeling and extended bioactivity prediction for salinosporamide A, B and C from marine actinomycetes Salinispora tropica. Comb. Chem. High Throughput Screen. 2017, 20, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. A chemoinformatics approach to the discovery of lead-like molecules from marine and microbial sources en route to antitumor and antibiotic drugs. Mar. Drugs 2014, 12, 757–778. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Hu, B.; Wang, J.; Zhu, F.; Kang, Y.; Li, D.; Sun, H.; Kong, D.-X.; Hou, T. A cheminformatic insight into the differences between terrestrial and marine originated natural products. J. Chem. Inf. Model. 2018. [Google Scholar] [CrossRef] [PubMed]
- Visini, R.; Arus-Pous, J.; Awale, M.; Reymond, J.-L. Virtual exploration of the ring systems chemical universe. J. Chem. Inf. Model. 2017, 57, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sciabola, S.; Barreiro, G.; Hou, X.; Bai, G.; Shapiro, M.J.; Koehn, F.; Villalobos, A.; Jacobson, M.P. Dihedral angle-based sampling of natural product polyketide conformations: Application to permeability prediction. J. Chem. Inf. Model. 2016, 56, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Tatonetti, N.P.; Liu, T.; Altman, R.B. Predicting drug side-effects by chemical systems biology. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Vanhaelen, Q.; Mamoshina, P.; Aliper, A.M.; Artemov, A.; Lezhnina, K.; Ozerov, I.; Labat, I.; Zhavoronkov, A. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 2017, 22, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Reker, D.; Rodrigues, T.; Schneider, P.; Schneider, G. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Reker, D.; Chen, T.; Hauenstein, K.; Schneider, P.; Altmann, K.-H. Deorphaning the macromolecular targets of the natural anticancer compound doliculide. Angew. Chem. Int. Ed. 2016, 55, 12408–12411. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Latino, D.A.R.S.; Gaudencio, S.P. QSAR-assisted virtual screening of lead-like molecules from marine and microbial natural sources for antitumor and antibiotic drug discovery. Molecules 2015, 20, 4848–4873. [Google Scholar] [CrossRef] [PubMed]
- Botic, T.; Defant, A.; Zanini, P.; Zuzek, M.C.; Frangez, R.; Janussen, D.; Kersken, D.; Knez, Z.; Mancini, I.; Sepcic, K. Discorhabdin alkaloids from Antarctic latrunculia spp. Sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 136, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Perez Manriquez, C.; Souto, L.M.; Norte, M.; Javier Fernandez, J.; Hernandez Daranas, A. Marine longilenes, oxasqualenoids with Ser-Thr protein phosphatase 2A inhibition activity. Mar. Drugs 2018, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.G.; Martinez Leal, J.F.; Hernandez Daranas, A.; Perez, M.; Cuevas, C. On the mechanism of action of dragmacidins I and J, two new representatives of a new class of protein phosphatase 1 and 2A inhibitors. ACS Omega 2018, 3, 3760–3767. [Google Scholar] [CrossRef]
- Xin, L.-T.; Liu, L.; Shao, C.-L.; Yu, R.-L.; Chen, F.-L.; Yue, S.-J.; Wang, M.; Guo, Z.-L.; Fan, Y.-C.; Guan, H.-S.; et al. Discovery of DNA topoisomerase I inhibitors with low-cytotoxicity based on virtual screening from natural products. Mar. Drugs 2017, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Qi, X.; Mo, X.; Yu, G.; Wang, Q.; Zhu, T.; Gu, Q.; Liu, M.; Li, J.; Li, D. Structure-based discovery of cytotoxic dimeric tetrahydroxanthones as potential topoisomerase I inhibitors from a marine-derived fungus. Eur. J. Med. Chem. 2018, 148, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Kim, D.H.; Seong, S.H.; Kim, H.-R.; Jung, H.A.; Choi, J.S. Alpha-glucosidase and protein tyrosine phosphatase 1B inhibitory activity of plastoquinones from marine brown alga Sargassum serratifolium. Mar. Drugs 2017, 15, 368. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Luo, J.; Wu, N.; Zhang, R.; Shi, D. BPN, a marine-derived PTP1B inhibitor, activates insulin signaling and improves insulin resistance in C2C12 myotubes. Int. J. Biol. Macromol. 2018, 106, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Z.; Wang, C.; Xu, Q.; Liang, J.; Xu, X.; Yang, J.; Wang, C.; Jiang, T.; Yu, R. Application of reverse docking for target prediction of marine compounds with anti-tumor activity. J. Mol. Graph. Model. 2017, 77, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, Y.-Y.; Lan, R.-F.; Du, L.; Wang, B.-S.; Zhou, T.; Li, Y.-P.; Zhang, Q.-Q.; Ying, M.-G.; Zheng, Q.-H.; et al. Dicitrinone D, an antimitotic polyketide isolated from the marine-derived fungus Penicillium citrinum. Tetrahedron 2017, 73, 5900–5911. [Google Scholar] [CrossRef]
- Yu, G.; Wang, Y.; Yu, R.; Feng, Y.; Wang, L.; Che, Q.; Gu, Q.; Li, D.; Li, J.; Zhu, T. Chetracins E and F, cytotoxic epipolythiodioxopiperazines from the marine-derived fungus Acrostalagmus luteoalbus HDN13-530. RSC Adv. 2018, 8, 53–58. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, D.; Sharma, M.; Sharma, N.; Bidve, P.; Prajapati, N.; Kalia, K.; Tiwari, V. Astaxanthin ameliorates behavioral and biochemical alterations in in-vitro and in-vivo model of neuropathic pain. Neurosci. Lett. 2018, 674, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-C.; Jang, J.; Ye, B.-R.; Kim, M.-S.; Choi, I.-W.; Park, W.-S.; Heo, S.-J.; Jung, W.-K. Purification and molecular docking study of angiotensin I-converting enzyme (ACE) inhibitory peptides from hydrolysates of marine sponge Stylotella aurantium. Process Biochem. 2017, 54, 180–187. [Google Scholar] [CrossRef]
- Pereira, R.C.C.; Lourenco, A.L.; Terra, L.; Abreu, P.A.; Laneuville Teixeira, V.; Castro, H.C. Marine diterpenes: Molecular modeling of thrombin inhibitors with potential biotechnological application as an antithrombotic. Mar. Drugs 2017, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, R.; Luo, Z.; Wang, W.; Chen, J. Antimicrobial activity and molecular docking studies of a novel anthraquinone from a marine-derived fungus Aspergillus versicolor. Nat. Prod. Res. 2018, 32, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Deplazes, E. Molecular simulations of disulfide-rich venom peptides with ion channels and membranes. Molecules 2017, 22, 362. [Google Scholar] [CrossRef] [PubMed]
- Mohyeldin, M.M.; Akl, M.R.; Siddique, A.B.; Hassan, H.M.; El Sayed, K.A. The marine-derived pachycladin diterpenoids as novel inhibitors of wild-type and mutant EGFR. Biochem. Pharmacol. 2017, 126, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Murcia, P.A.; Cortes-Cabrera, A.; Gago, F. Structural rationale for the cross-resistance of tumor cells bearing the A399V variant of elongation factor eEF1A1 to the structurally unrelated didemnin B, ternatin, nannocystin A and ansatrienin B. J. Comput. Aided Mol. Des. 2017, 31, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Roy, A.; Jung, J.H.; Choi, J.S. Evaluation of the inhibitory effects of eckol and dieckol isolated from edible brown alga Eisenia bicyclis on human monoamine oxidases A and B. Arch. Pharm. Res. 2017, 40, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Naine, S.J.; Devi, C.S.; Mohanasrinivasan, V.; Doss, C.G.P. Bioactivity of marine Streptomyces sp VITJS4: Interactions of cytotoxic phthalate derivatives with human topoisomerase II alpha: An in silico molecular docking analysis. Interdiscip. Sci. 2018, 10, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ai, X.; Hou, J.; Ye, X.; Liu, R.; Shen, S.; Lia, Z.; Lu, S. Integrated discovery of FOXO1-DNA stabilizers from marine natural products to restore chemosensitivity to anti-EGFR-based therapy for metastatic lung cancer. Mol. BioSyst. 2017, 13, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, P.L.; Pedersen, S.E.; Briggs, J.M. Interactions of acetylcholine binding site residues contributing to nicotinic acetylcholine receptor gating: Role of residues Y93, Y190, K145 and D200. J. Mol. Graph. Model. 2013, 44, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Avram, S.; Pacureanu, L.M.; Seclaman, E.; Bora, A.; Kurunczi, L. PLS-DA-docking optimized combined energetic terms (PLSDA-DOCET) protocol: A brief evaluation. J. Chem. Inf. Model. 2011, 51, 3169–3179. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Compounds 6 | Taxo. 7 | Bioact. 8 | Targets 9 | Spec. Data 10 | |
|---|---|---|---|---|---|---|
| Total | NPs | |||||
| CAS/SciFinder 1 | 9.0 × 107 | >283,000 | + | + | - | - |
| ChemSpider 2 | 5.9 × 107 | >13,800 | - | + | - | - |
| PubChem 2 | 9.3 × 107 | 4.4 × 105 | - | + | + | + 10 |
| ChEMBL 2 | 1.7 × 106 | >75,000 | - | + | + | - |
| REAXYS 1,2 | 1.1 × 108 | >215,000 | + | + | - | - |
| ZINC 2,5 | 1.2 × 108 | >44,000 | - | + | + | - |
| LOPAC 3,5 | 1280 | ? | - | + | + | - |
| Prestwick 3,5 | 1280 | ? | - | + | + | - |
| ACD/NMR DB 4 | >322,000 | >50,000 | - | - | - | + 10.2 |
| NMRShiftDB 4 | 43,440 | ? | - | - | - | + 10.2 |
| Massbank 4 | >15,000 | >2500 | - | - | - | + 10.3 |
| ReSpect 4 | - | >3595 | - | - | - | + 10.3 |
| METLIN 4 | - | 75,000 | - | - | - | + 10.3 |
| GNPS 4 | 22,644 | >3000 | + | - | - | + 10.3 |
| NaprAlert 4 | - | >155,000 12 | + | + | - | + 10.1 |
| DNP 4 | - | >270,000 | + | + | - | + 10.1 |
| DMNP 4 | - | >30,000 | + | + | - | + 10.1 |
| MarinLit 4 | - | >29,000 | + | + | - | + 10 |
| AntiBase 4 | - | 43,743 | + | + | - | + 10 |
| StreptomeDB 4 | - | 3991 | + | + | - | + 11 |
| NPCARE 2,4 | - | 6578 12 | + | + | + | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, F.; Aires-de-Sousa, J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Mar. Drugs 2018, 16, 236. https://doi.org/10.3390/md16070236
Pereira F, Aires-de-Sousa J. Computational Methodologies in the Exploration of Marine Natural Product Leads. Marine Drugs. 2018; 16(7):236. https://doi.org/10.3390/md16070236
Chicago/Turabian StylePereira, Florbela, and Joao Aires-de-Sousa. 2018. "Computational Methodologies in the Exploration of Marine Natural Product Leads" Marine Drugs 16, no. 7: 236. https://doi.org/10.3390/md16070236
APA StylePereira, F., & Aires-de-Sousa, J. (2018). Computational Methodologies in the Exploration of Marine Natural Product Leads. Marine Drugs, 16(7), 236. https://doi.org/10.3390/md16070236