Seco-Tetracenomycins from the Marine-Derived Actinomycete Saccharothrix sp. 10-10

 ,
,
Abstract
:
1. Introduction
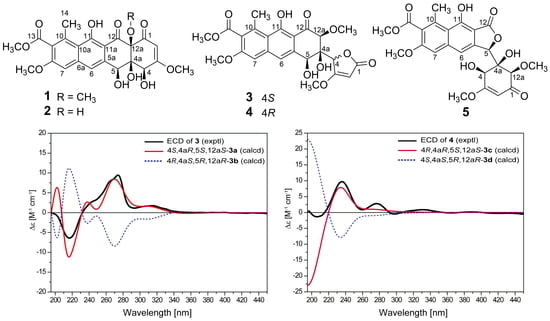
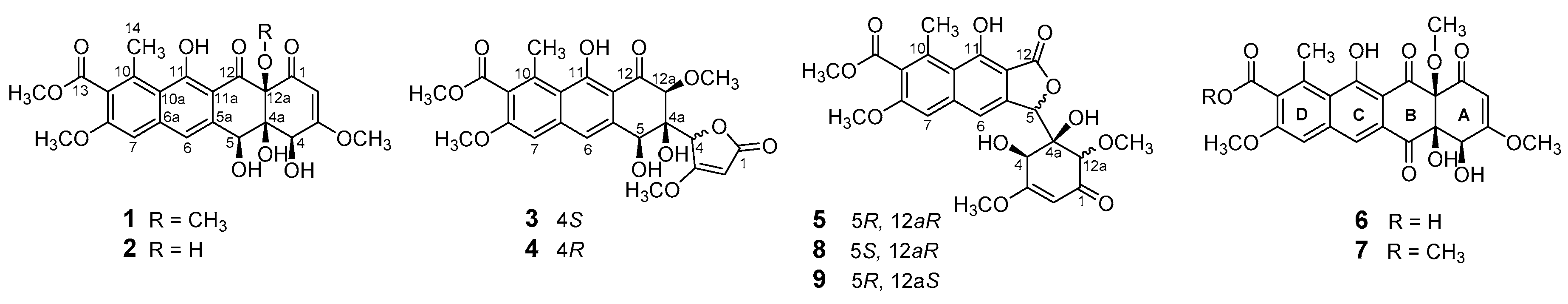
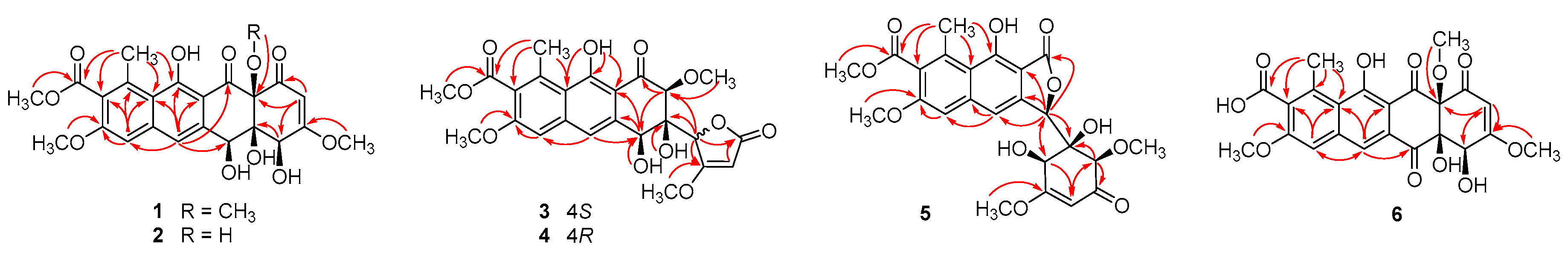
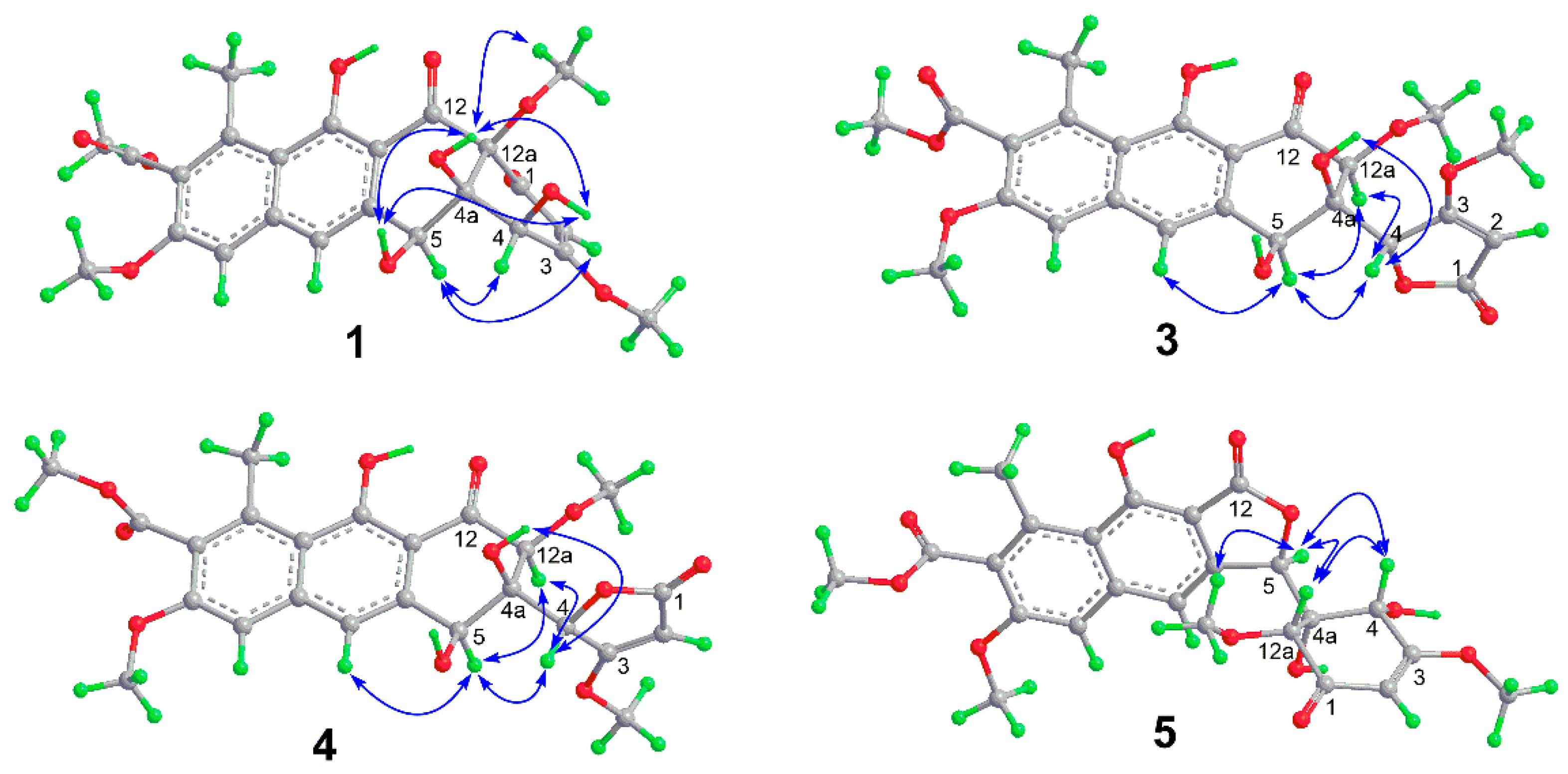
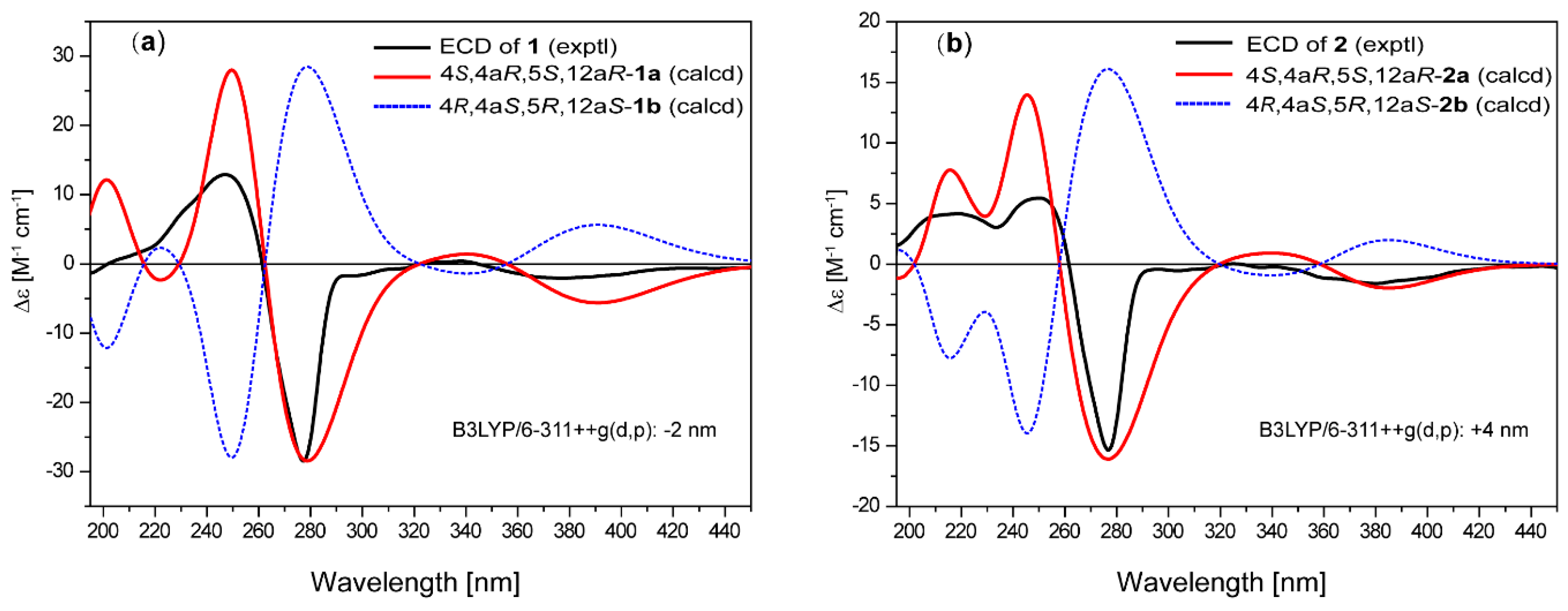
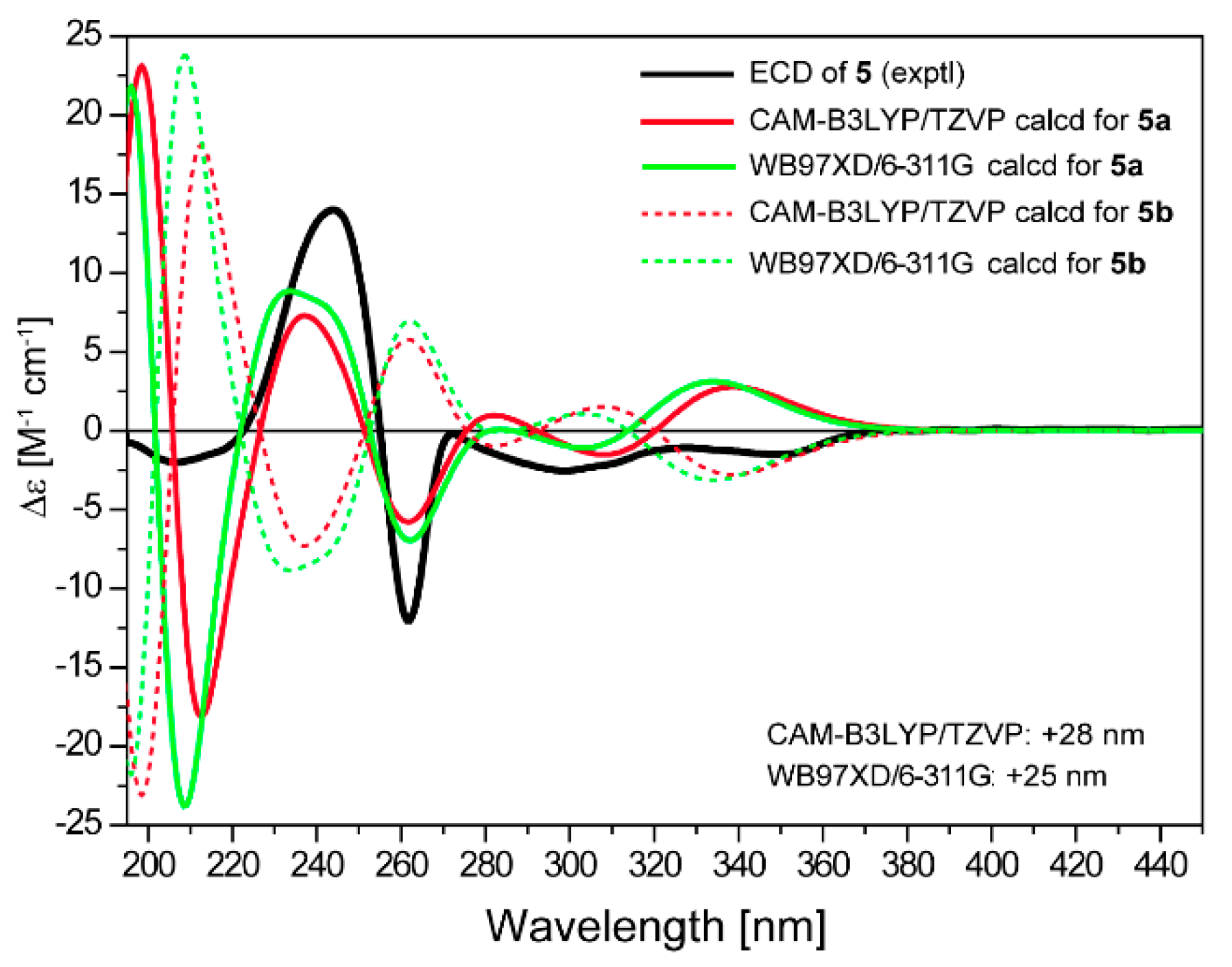
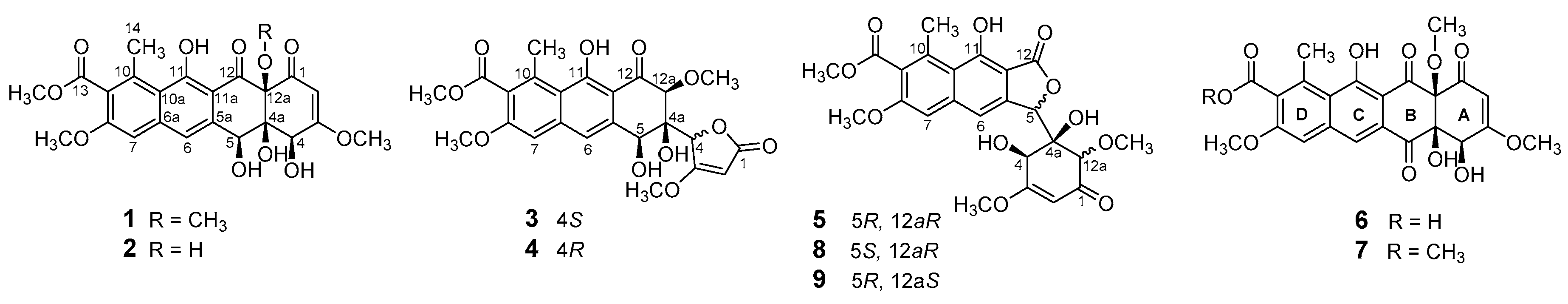
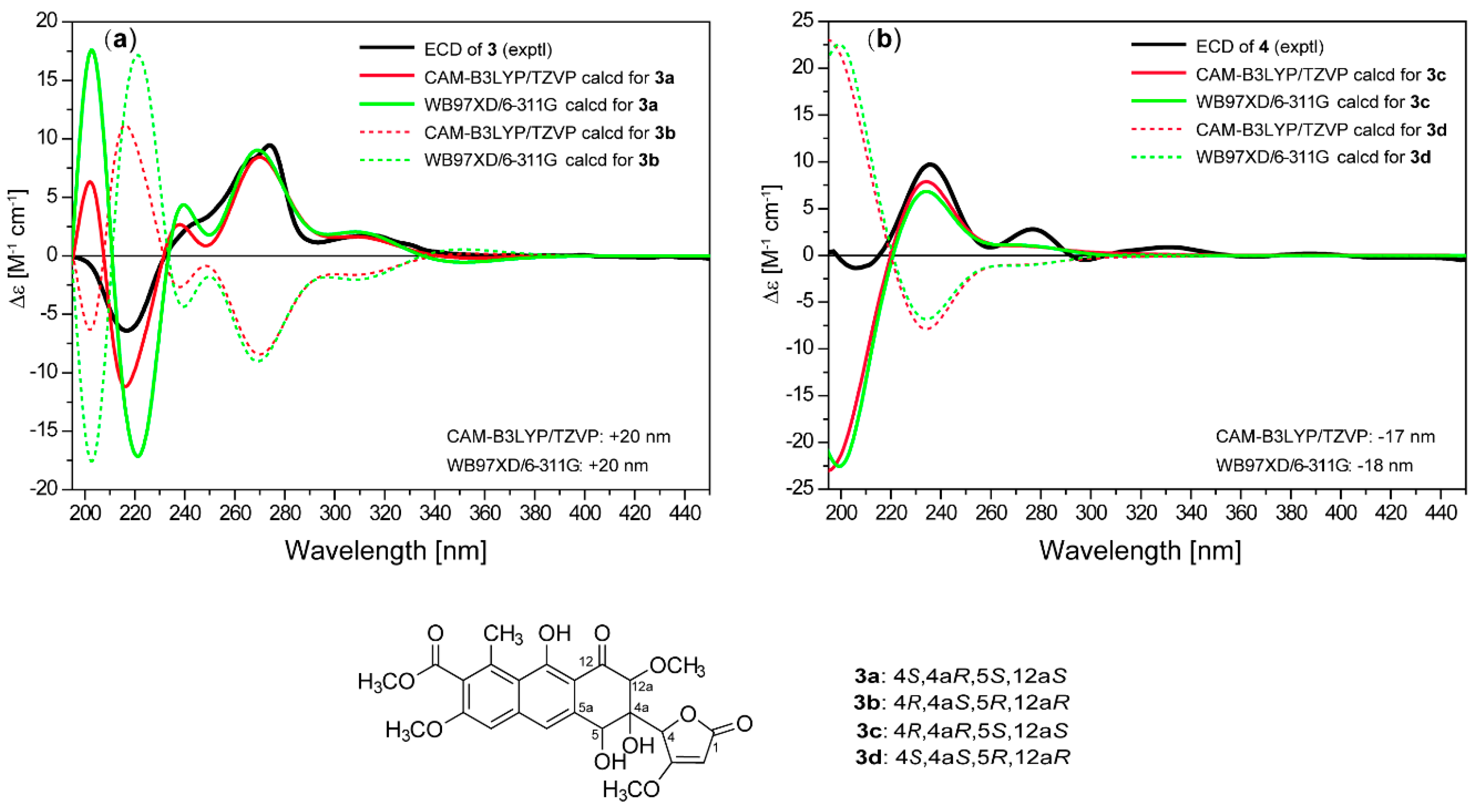
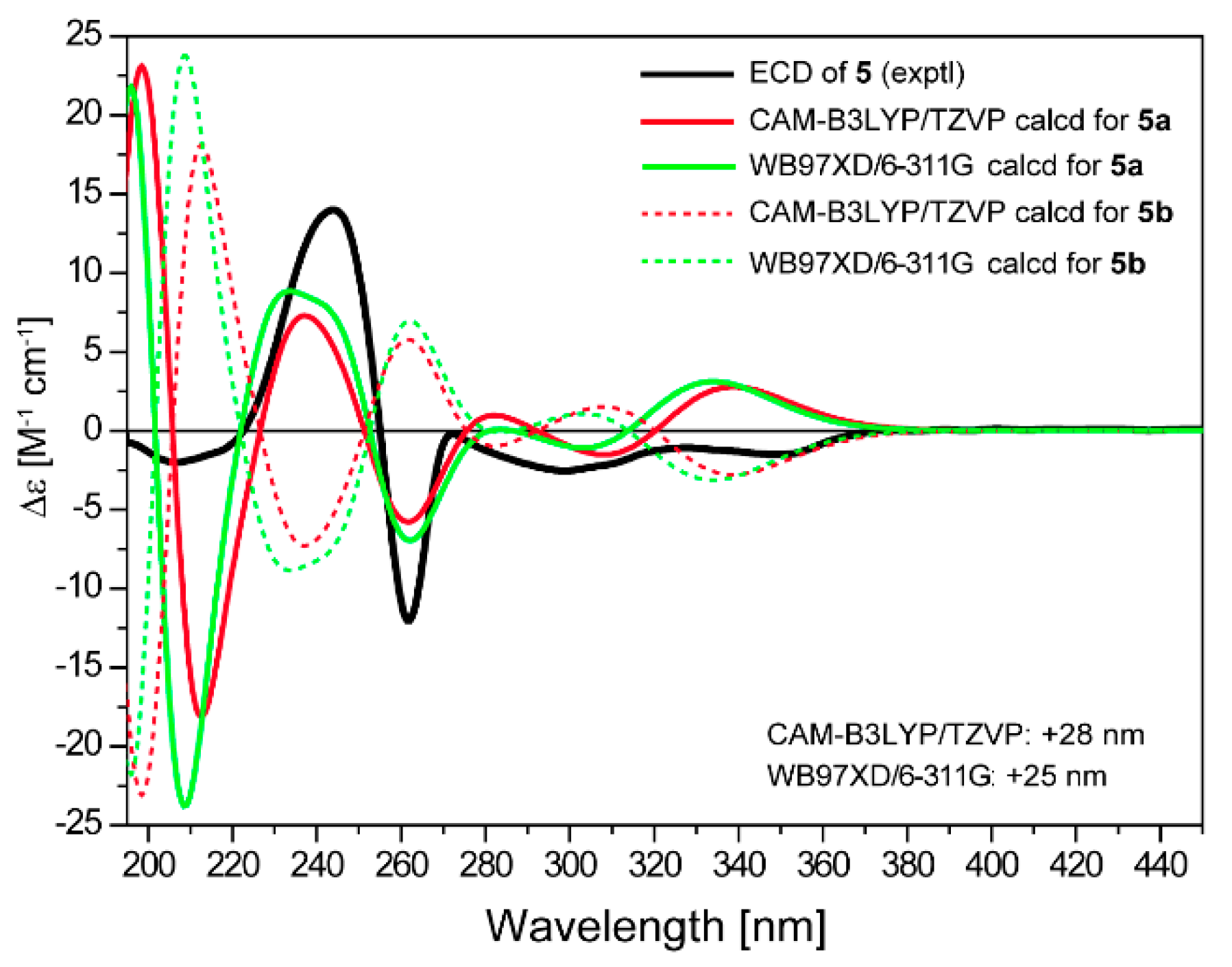
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fermentation and Isolation
3.3. ECD Calculations
3.4. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Das, A.; Khosla, C. Biosynthesis of aromatic polyketides in bacteria. Acc. Chem. Res. 2009, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. The Polyketide Metabolites; Elliis Horwood: Chichester, UK, 1991; ISBN 01368326959780136832690. [Google Scholar]
- Cragg, G.M.; Kingston, D.G.I.; Newman, D.J. Anticancer Agents from Natural Products; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-0-8493-1863-4. [Google Scholar]
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef] [PubMed]
- Egert, E.; Noltemeyer, M.; Siebers, J.; Rohr, J.; Zeeck, A. The structure of tetracenomycin C. J. Antibiot. 1992, 45, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C.; Luzhetskyy, A.; Rebets, Y.; Bechthold, A. Type II polyketide synthases: Gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007, 24, 162–190. [Google Scholar] [CrossRef] [PubMed]
- Gontang, E.A.; Gaudêncio, S.P.; Fenical, W.; Jensen, P.R. Sequence-based analysis of secondary-metabolite biosynthesis in marine actinobacteria. Appl. Environ. Microbiol. 2010, 76, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Z.; Li, S.; Yang, T.; Zhang, Q.; Ma, L.; Tian, X.; Zhang, H.; Huang, C.; Zhang, S.; et al. Spiroindimicins A–D: New bisindole alkaloids from a deep-sea-derived actinomycete. Org. Lett. 2012, 14, 3364–3367. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, M.; Wu, C.; Tan, Y.; Li, J.; Hao, X.; Duan, Y.; Guan, Y.; Shang, X.; Wang, Y.; et al. Identification and proposed relative and absolute configurations of niphimycins C–E from the marine-derived Streptomyces sp. IMB7-145 by genomic analysis. J. Nat. Prod. 2018, 81, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wang, M.; Tan, Y.; Hu, X.; He, H.; Xiao, C.; You, X.; Wang, Y.; Gan, M. Neo-actinomycins A and B, natural actinomycins bearing the 5H-oxazolo[4,5-b]phenoxazine chromophore, from the marine-derived streptomyces sp. IMB094. Sci. Rep. 2017, 7, 3591. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Tan, Y.; Gan, M.; Wang, Y.; Guan, Y.; Hu, X.; Zhou, H.; Shang, X.; You, X.; Yang, Z.; et al. Identification of elaiophylin derivatives from the marine-derived actinomycete Streptomyces sp. 7-145 using PCR-based screening. J. Nat. Prod. 2013, 76, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Liu, B.; Tan, Y.; Wang, Q.; Zhou, H.; He, H.; Ping, Y.; Yang, Z.; Wang, Y.; Xiao, C. Saccharothrixones A–D, tetracenomycin-type polyketides from the marine-derived actinomycete Saccharothrix sp. 10-10. J. Nat. Prod. 2015, 78, 2260–2265. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tan, Y.; Gan, M.; Zhou, H.; Wang, Y.; Ping, Y.; Li, B.; Yang, Z.; Xiao, C. Identification of tetracenomycin X from a marine-derived Saccharothrix sp. guided by genes sequence analysis. Acta Pharm. Sin. 2014, 49, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Khoo, C.L.; Rickards, R.W. Oxidation processes in the biosynthesis of the tetracenomycin and elloramycin antibiotics. J. Antibiot. 1989, 42, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 17, 2717–2727. [Google Scholar] [CrossRef]
- Legrand, M.; Rougier, M.J. Stereochemistry: Fundamentals and Methods; Kagan, H.B., Ed.; Georg Thieme: Stuttgart, Germany, 1977; pp. 123–127. ISBN 3-13-132601-8. [Google Scholar]
- Rohr, J.; Zeeck, A. Structure-activity relationships of elloramycin and tetracenomycin C. J. Antibiot. 1990, 43, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), version 2009.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2009.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision a.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. Specdis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 (acetone-d6) | 2 (acetone-d6) | 3 (acetone-d6) | 4 (acetone-d6) | 5 (acetone-d6) | 6 (CD3OD) |
|---|---|---|---|---|---|---|
| 2 | 5.61, s | 5.51, s | 5.29, d (1.2) | 5.30, d (1.2) | 5.32, d (1.0) | 5.53, d (1.8) |
| 4 | 4.62, brs | 4.93, s | 5.27, s | 5.44, d (1.2) | 4.65, d (1.0) | 4.85, brs |
| 5 | 4.77, brs | 5.32, s | 5.01, d (1.2) | 5.41, d (1.8) | 5.98, s | |
| 6 | 7.44, s | 7.46, s | 7.49, d (1.2) | 7.51, d (18) | 7.61, s | 8.01, s |
| 7 | 7.20, s | 7.24, s | 7.20, s | 7.24, s | 7.27, s | 7.46, s |
| 12a | 4.33, s | 3.70, s | 3.93, s | |||
| 14 | 2.79, s | 2.79, s | 2.79, s | 2.78, s | 2.81, s | 2.88, s |
| 3-OCH3 | 3.91, s | 3.88, s | 3.97, s | 3.96, s | 3.72, s | 3.80, s |
| 8-OCH3 | 3.94, s | 3.96, s | 3.95, s | 3.96, s | 3.93, s | 4.01, s |
| 12a-OCH3 | 3.73, s | 3.74, s | 3.34, s | 3.51, s | 3.56, s | |
| 13-OCH3 | 3.90, s | 3.90, s | 3.90, s | 3.89, s | 3.90, s |
| No. | 1 (acetone-d6) | 2 (acetone-d6) | 3 (acetone-d6) | 4 (acetone-d6) | 5 (acetone-d6) | 6 (CD3OD) |
|---|---|---|---|---|---|---|
| 1 | 193.6, C | 192.8, C | 172.2, C | 172.3, C | 194.9, C | 193.9, C |
| 2 | 101.5, CH | 100.4, CH | 89.5, CH | 89.1, CH | 100.3, CH | 101.9, CH |
| 3 | 177.0, C | 176.6, C | 183.9, C | 182.0, C | 173.1, C | 174.8, C |
| 4 | 70.1, CH | 74.2, CH | 77.8, CH | 80.3, CH | 68.3, CH | 70.9, CH |
| 4a | 78.6, C | 79.3, C | 82.0, C | 78.8, C | 80.3, C | 86.1, C |
| 5 | 69.6, CH | 70.5, CH | 68.4, CH | 68.0, CH | 81.0, CH | 194.4, C |
| 5a | 142.2, C | 139.9, C | 140.8, C | 140.8, C | 143.0, C | 141.9, C |
| 6 | 118.2, CH | 118.8, CH | 118.0, CH | 117.7, CH | 115.0, CH | 121.8, CH |
| 6a | 139.8, C | 142.3, C | 142.1, C | 142.4, C | 143.3, C | 128.7, C |
| 7 | 105.9, CH | 105.9, CH | 105.7, CH | 105.9, CH | 106.2, CH | 108.7, CH |
| 8 | 158.1, C | 158.0, C | 157.7, C | 158.0, C | 156.9, C | 159.3, C |
| 9 | 127.8, C | 127.9, C | 127.7, C | 127.6, C | 127.7, C | 132.8, C |
| 10 | 137.8, C | 137.5, C | 137.4, C | 137.5, C | 136.1, C | 138.0, C |
| 10a | 118.2, C | 117.9, C | 117.9, C | 118.0, C | 118.0, C | 121.9, C |
| 11 | 166.7, C | 165.5, C | 165.2, C | 167.1, C | 158.5, C | 167.5, C |
| 11a | 110.1, C | 109.6, C | 109.4, C | 109.0, C | 106.2, C | 110.5, C |
| 12 | 202.2, C | 200.2, C | 203.0, C | 199.1, C | 173.4, C | 197.6, C |
| 12a | 87.8, C | 82.5, C | 84.2, CH | 82.8, CH | 84.1, CH | 89.0, C |
| 13 | 168.6, C | 168.6, C | 168.7, C | 168.7, C | 168.6, C | 171.8, C |
| 14 | 21.0, CH3 | 21.0, CH3 | 21.0, CH3 | 21.0, CH3 | 20.4, CH3 | 21.1, CH3 |
| 3-OMe | 57.4, CH3 | 57.4, CH3 | 60.2, CH3 | 60.2, CH3 | 56.8, CH3 | 57.5, CH3 |
| 8-OMe | 56.4, CH3 | 56.4, CH3 | 56.4, CH3 | 56.4, CH3 | 56.3, CH3 | 56.7, CH3 |
| 12a-OMe | 56.6, CH3 | 61.5, CH3 | 58.8, CH3 | 60.6, CH3 | 56.7, CH3 | |
| 13-OMe | 52.5, CH3 | 52.5, CH3 | 52.5, CH3 | 52.5, CH3 | 52.5, CH3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Li, J.; Chen, M.; Hao, X.; Cao, F.; Tan, Y.; Ping, Y.; Wang, Y.; Xiao, C.; Gan, M. Seco-Tetracenomycins from the Marine-Derived Actinomycete Saccharothrix sp. 10-10. Mar. Drugs 2018, 16, 345. https://doi.org/10.3390/md16100345
Liu B, Li J, Chen M, Hao X, Cao F, Tan Y, Ping Y, Wang Y, Xiao C, Gan M. Seco-Tetracenomycins from the Marine-Derived Actinomycete Saccharothrix sp. 10-10. Marine Drugs. 2018; 16(10):345. https://doi.org/10.3390/md16100345
Chicago/Turabian StyleLiu, Bin, Jiao Li, Minghua Chen, Xiaomeng Hao, Fei Cao, Yi Tan, Yuhui Ping, Yiguang Wang, Chunling Xiao, and Maoluo Gan. 2018. "Seco-Tetracenomycins from the Marine-Derived Actinomycete Saccharothrix sp. 10-10" Marine Drugs 16, no. 10: 345. https://doi.org/10.3390/md16100345
APA StyleLiu, B., Li, J., Chen, M., Hao, X., Cao, F., Tan, Y., Ping, Y., Wang, Y., Xiao, C., & Gan, M. (2018). Seco-Tetracenomycins from the Marine-Derived Actinomycete Saccharothrix sp. 10-10. Marine Drugs, 16(10), 345. https://doi.org/10.3390/md16100345