
Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#


 ,
,
Abstract
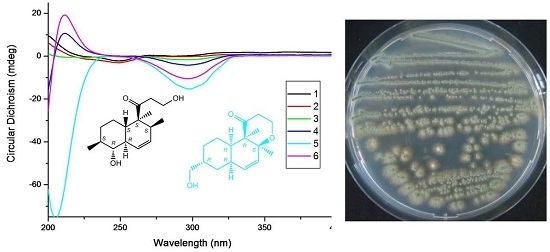
:
1. Introduction

2. Results and Discussion
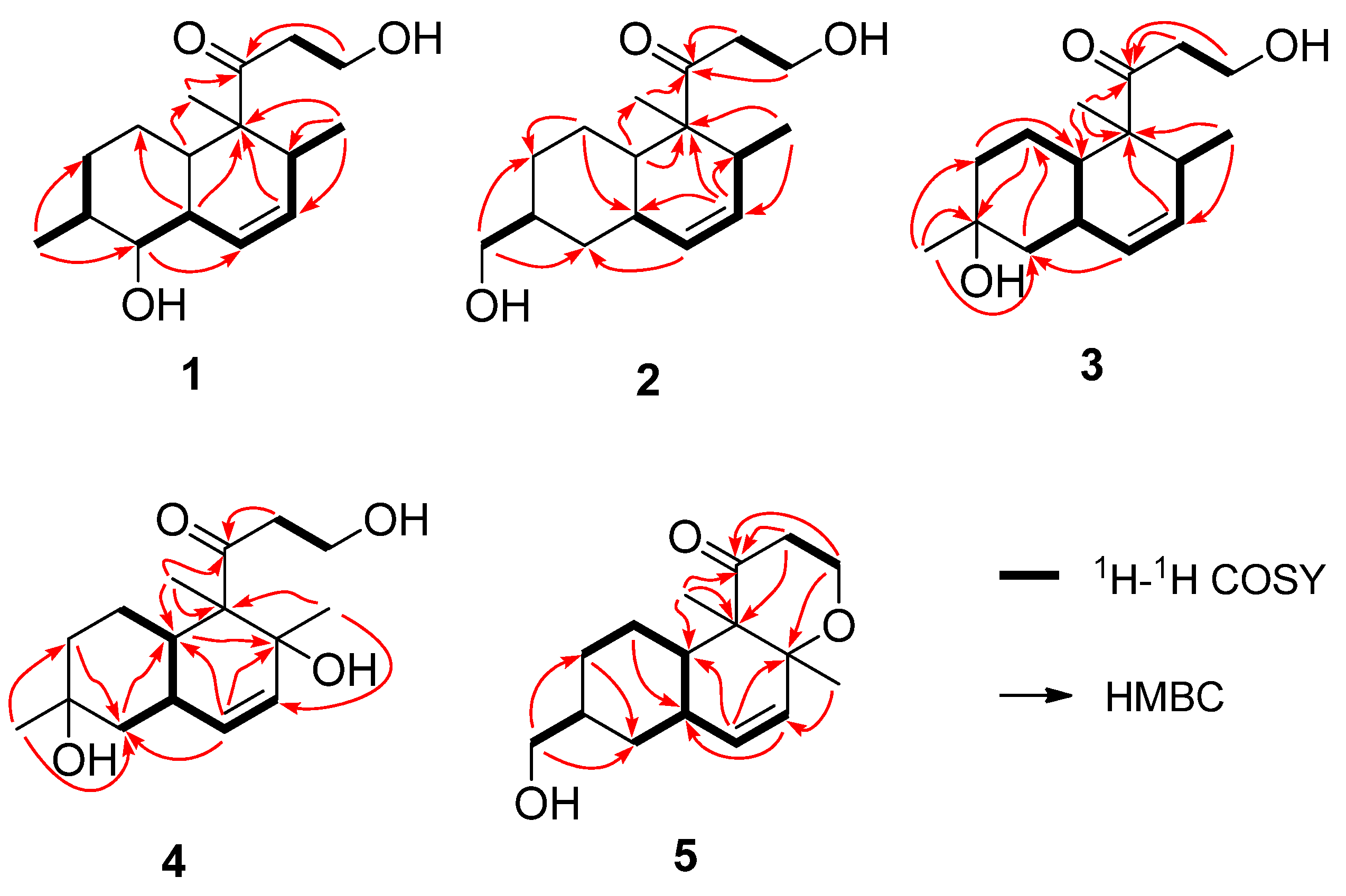

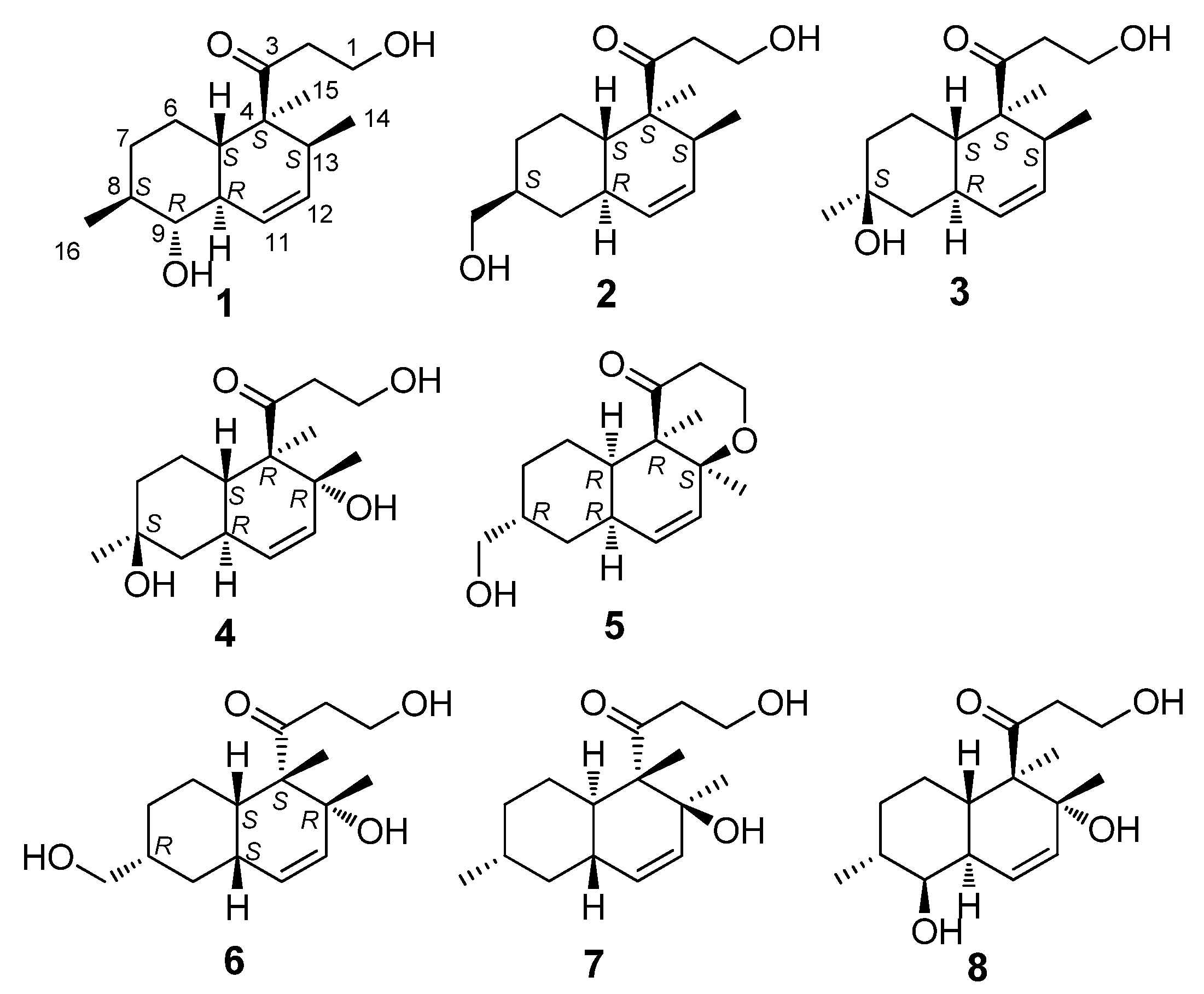
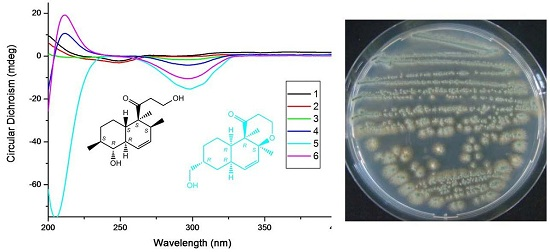{kind=link}
{kind=link}
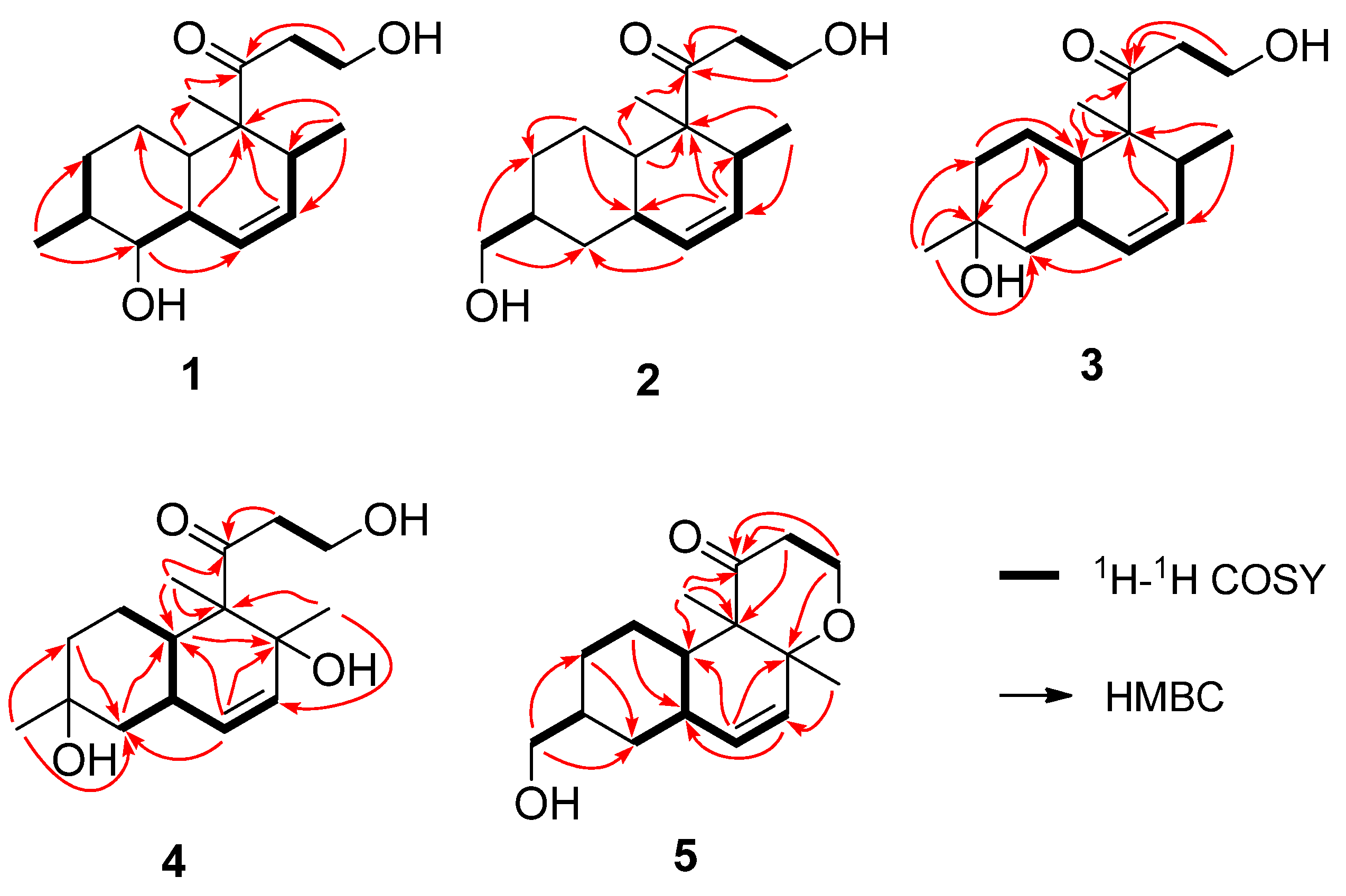{kind=link}
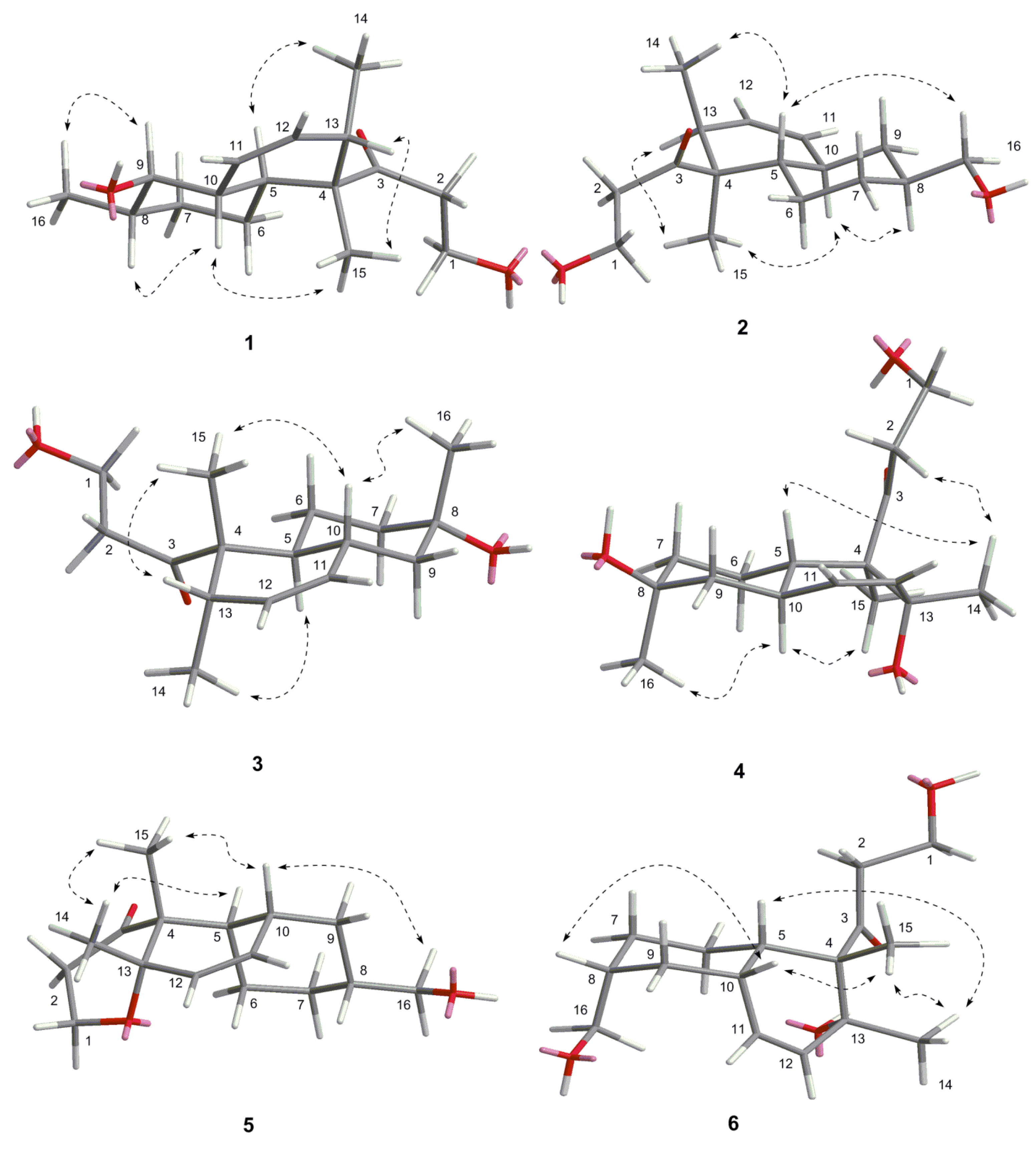{kind=link}
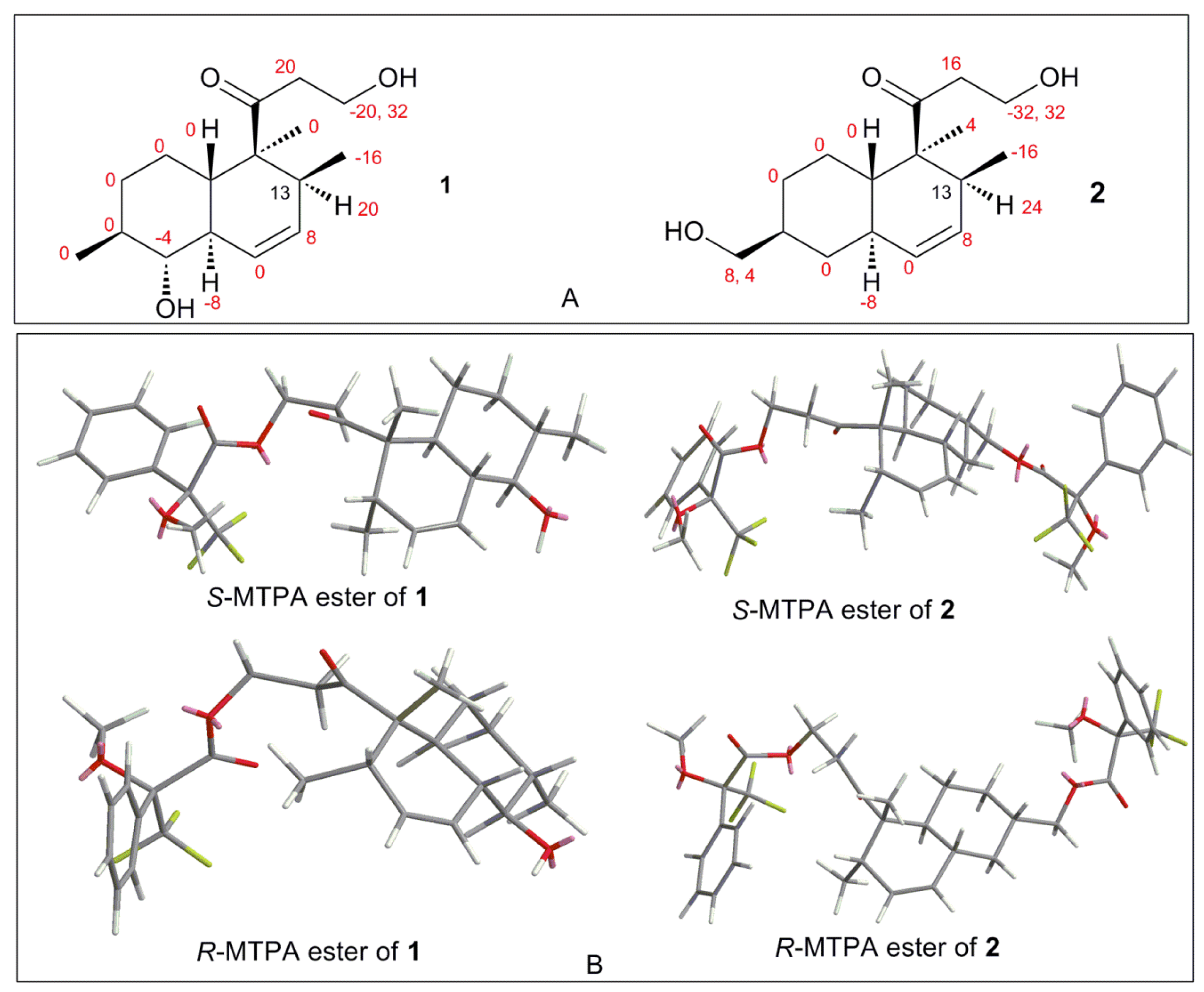{kind=link}
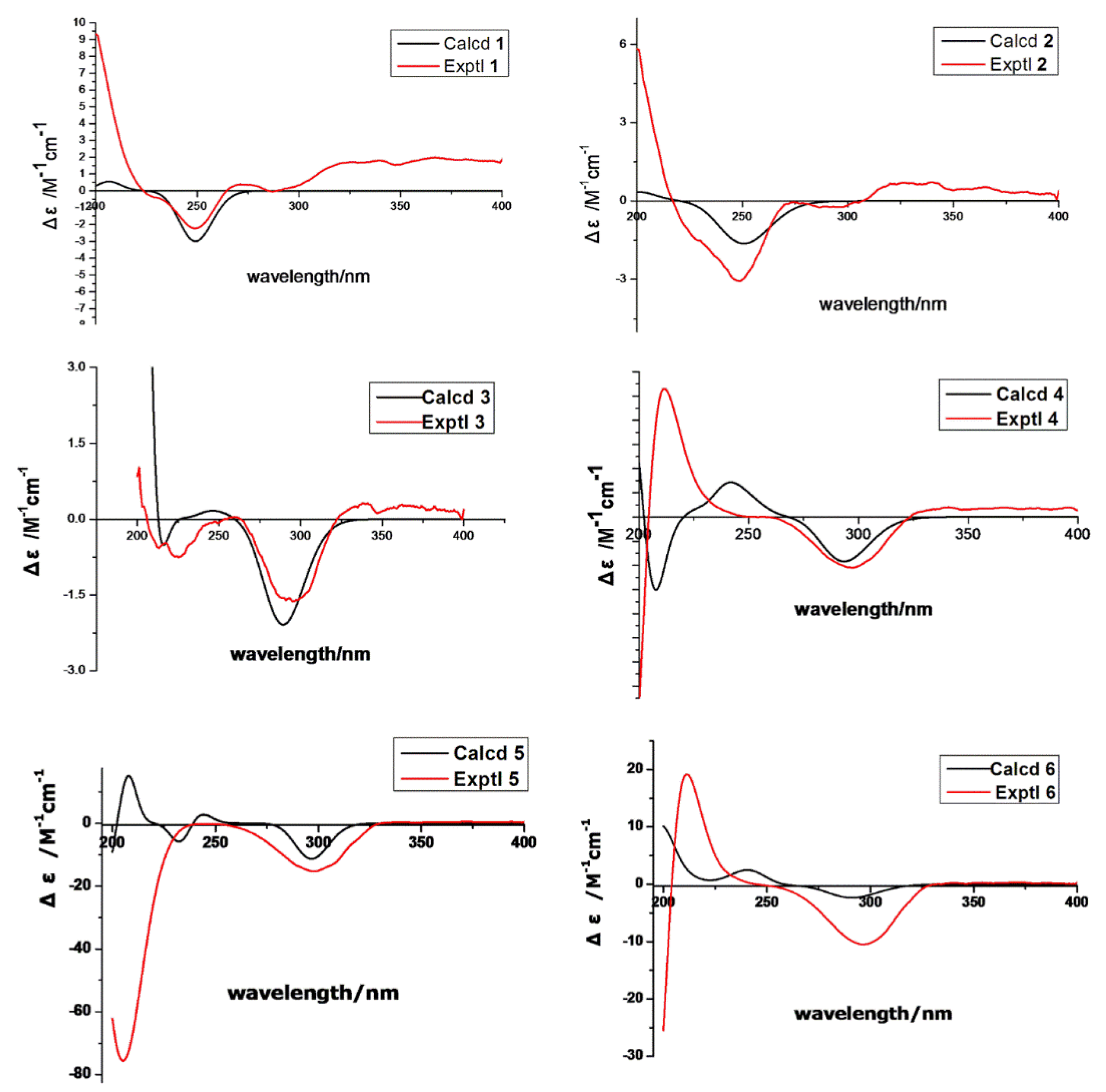{kind=link}
| 1 α | 2 | 3 | 4 | 5 | ||||||
| 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | |
| 1 | 58.0 t | 3.82 m | 57.9 t | 3.80 m | 58.2 t | 3.82 m | 58.4 t | 3.82 m | 61.2 t | 4.08 dd 12.0, 8.0 |
| 3.87 dt 12.0, 2.8 | ||||||||||
| 2 | 41.0 t | 2.64 ddd 18.6, 6.6, 4.2 | 41.0 t overlapped | 2.68 ddd 18.4, 6.0, 4.8 | 40.9 t | 2.66 q 4.4 | 44.1 t | 3.11 ddd 18.8, 6.4, 3.6 | 39.5 t | 2.79 ddd 14.0, 12.8, 8.0 |
| 2.63 ddd 18.6, 6.6, 4.2 | 2.61 ddd 18.4, 6.0, 4.8 | 2.67 ddd 18.8, 6.4, 3.6 | 2.19 ddd 14.0, 12.8, 8.0 | |||||||
| 3 | 215.4 s | 215.6 s | 215.8 s | 216.1 s | 212.6 s | |||||
| 4 | 52.3 s | 52.3 s | 52.4 s | 57.2 s | 57.1 s | |||||
| 5 | 45.3 d | 1.66 m | 39.0 d | 1.59 m | 38.7 d | 1.58 m | 43.3 d | 1.78 m | 43.0 d | 2.23 m |
| 6 | 26.8 t | 1.54 m | 26.7 t | 1.68 m | 23.0 t | 1.53 m | 23.1 t | 1.42 m | 25.8 t | 1.14 m |
| 0.91 m | 0.91 m | 1.26 m | 1.31 m | |||||||
| 7 | 33.5 t | 1.16 m, | 29.8 t | 1.80 m | 45.8 t | 1.71 m | 39.5 t | 1.67 m | 29.6 t | 1.83 m |
| 1.71 m | 1.08 m | 1.27 m | 1.50 m | 1.02 m | ||||||
| 8 | 41.0 d | 1.36 m | 41.0 d overlapped | 1.58 m | 70.2 s | 70.1 s | 40.8 d | 1.62 m | ||
| 9 | 79.3 d | 2.89 t 9.6 | 36.3 t | 1.84 m | 39.7 t | 1.65 m | 45.3 t | 1.74 m | 35.4 t | 1.94 m |
| 0.86 m | 1.53 m | 1.25 m | 1.03 m | |||||||
| 10 | 36.6 d | 1.67 m | 37.9 d | 1.69 m | 33.6 d | 2.13 m | 33.7 d | 2.24 tt 11.8, 2.8 | 37.4 d | 1.82 m |
| 11 | 125.0 d | 5.91 d 10.6 | 129.6 d | 5.32 d 10.0 | 129.6 d | 5.32 d 9.6 | 131.0 d | 5.34, s | 134.3 d | 5.66 dd 9.6, 1.2 |
| 12 | 130.6 d | 5.58 ddd 10.6, 4.8, 2.4 | 129.7 d | 5.45 ddd 10.0, 4.8, 2.4 | 130.0 d | 5.52 ddd 9.6, 4.8, 2.8 | 133.6 d | 5.34, s | 130.6 d | 5.52 dd 9.6, 2.8 |
| 13 | 39.5 d | 2.01 m | 39.9 d | 2.06 m | 40.0 d | 2.09 m | 74.0 s | 78.5 s | ||
| 14 | 18.6 q | 0.75 d 8.4 | 18.7 q | 0.72 d 7.2 | 18.8 q | 0.75 d 7.2 | 27.5 q | 1.13 s | 20.5 q | 1.18 s |
| 15 | 17.5 q | 1.19 s | 17.4 q | 1.17 s | 17.7 q | 1.22 s | 12.1 q | 1.33 s | 11.2 q | 0.88 s |
| 16 | 18.7 q | 1.00 d 9.6 | 68.3 t | 3.44 dd 10.8, 6.4 | 31.8 q | 1.22 s | 31.8 q | 1.25 s | 68.3 t | 3.48 m |
| 3.41 dd 10.8, 6.4 | ||||||||||
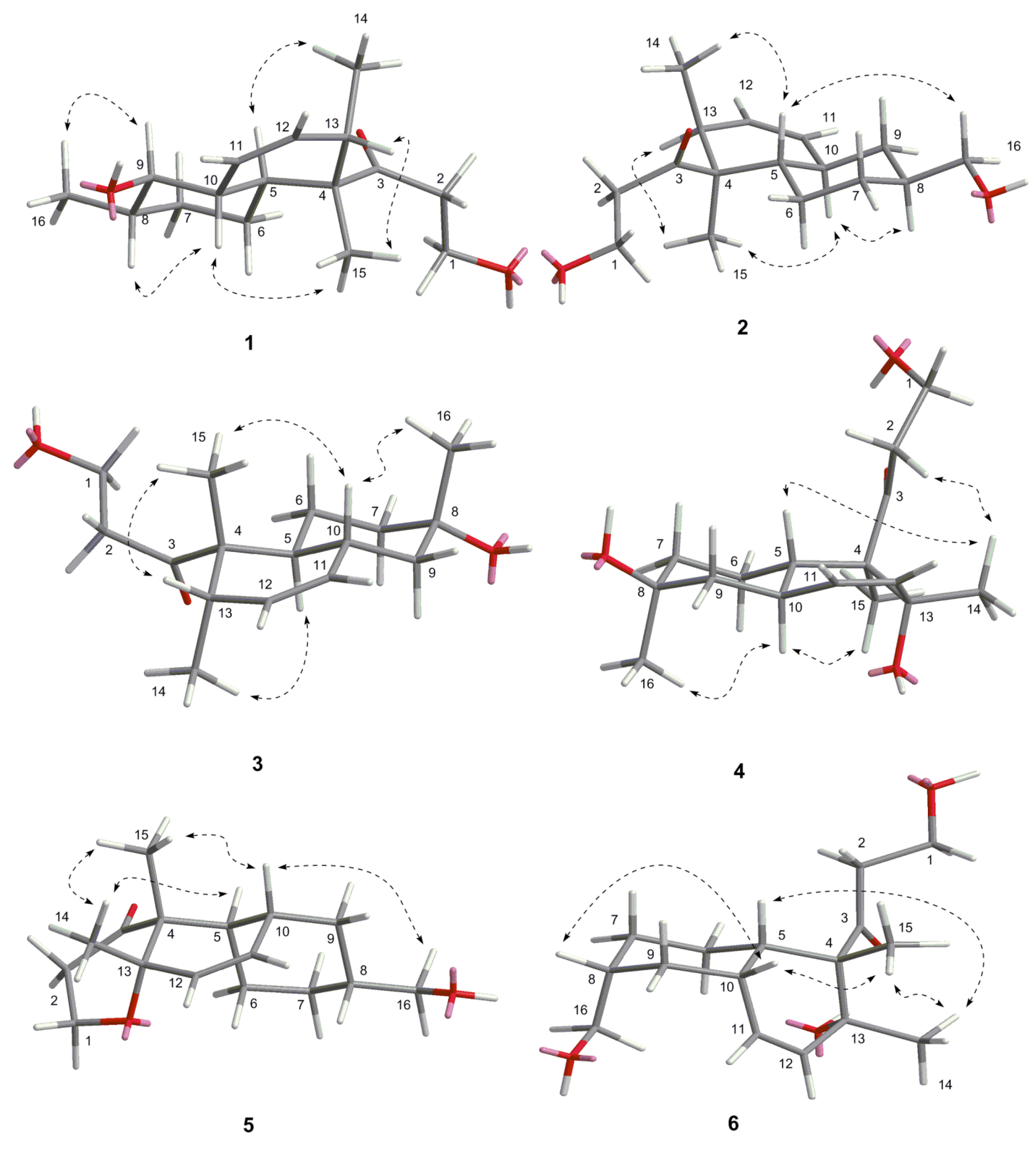
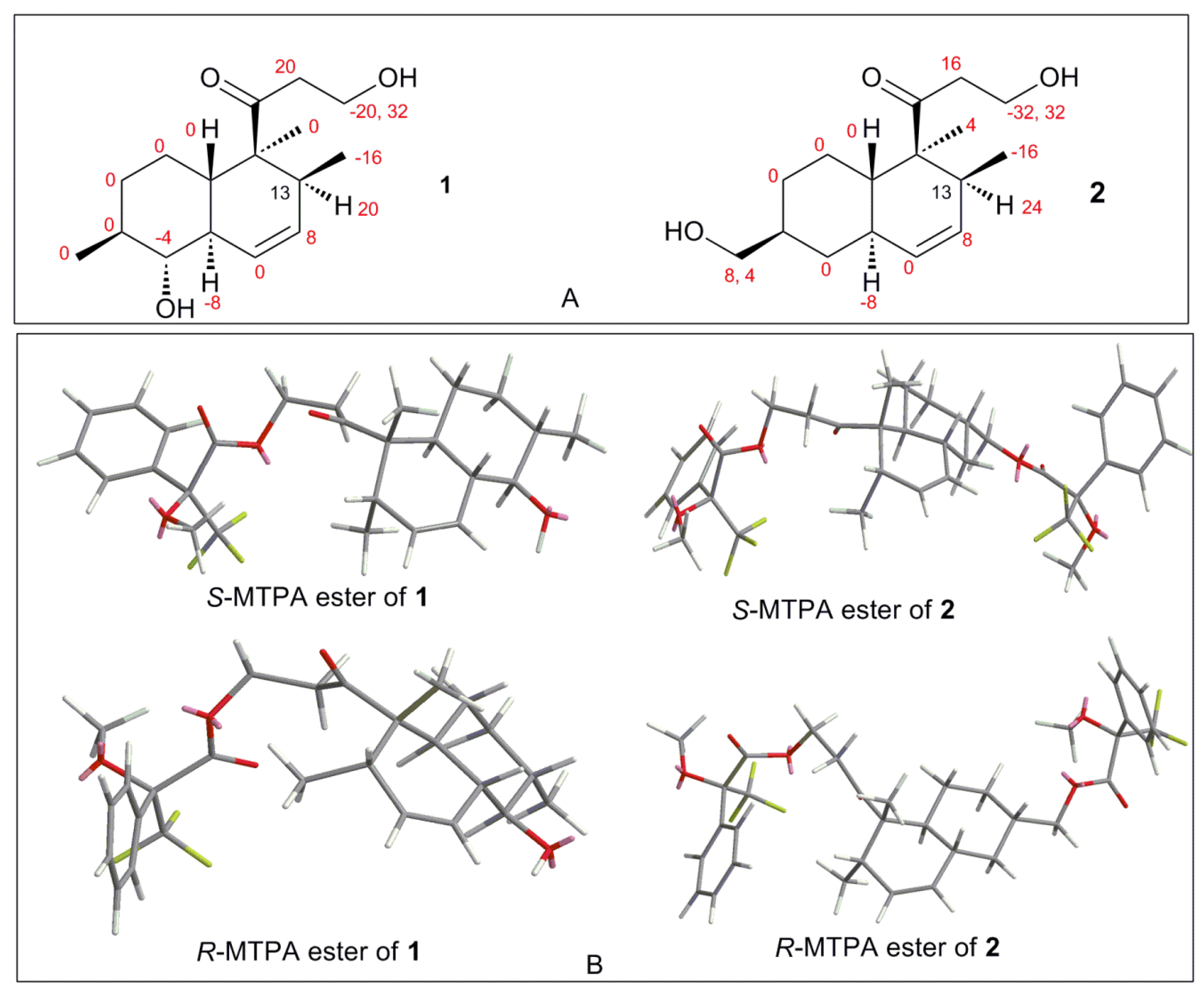
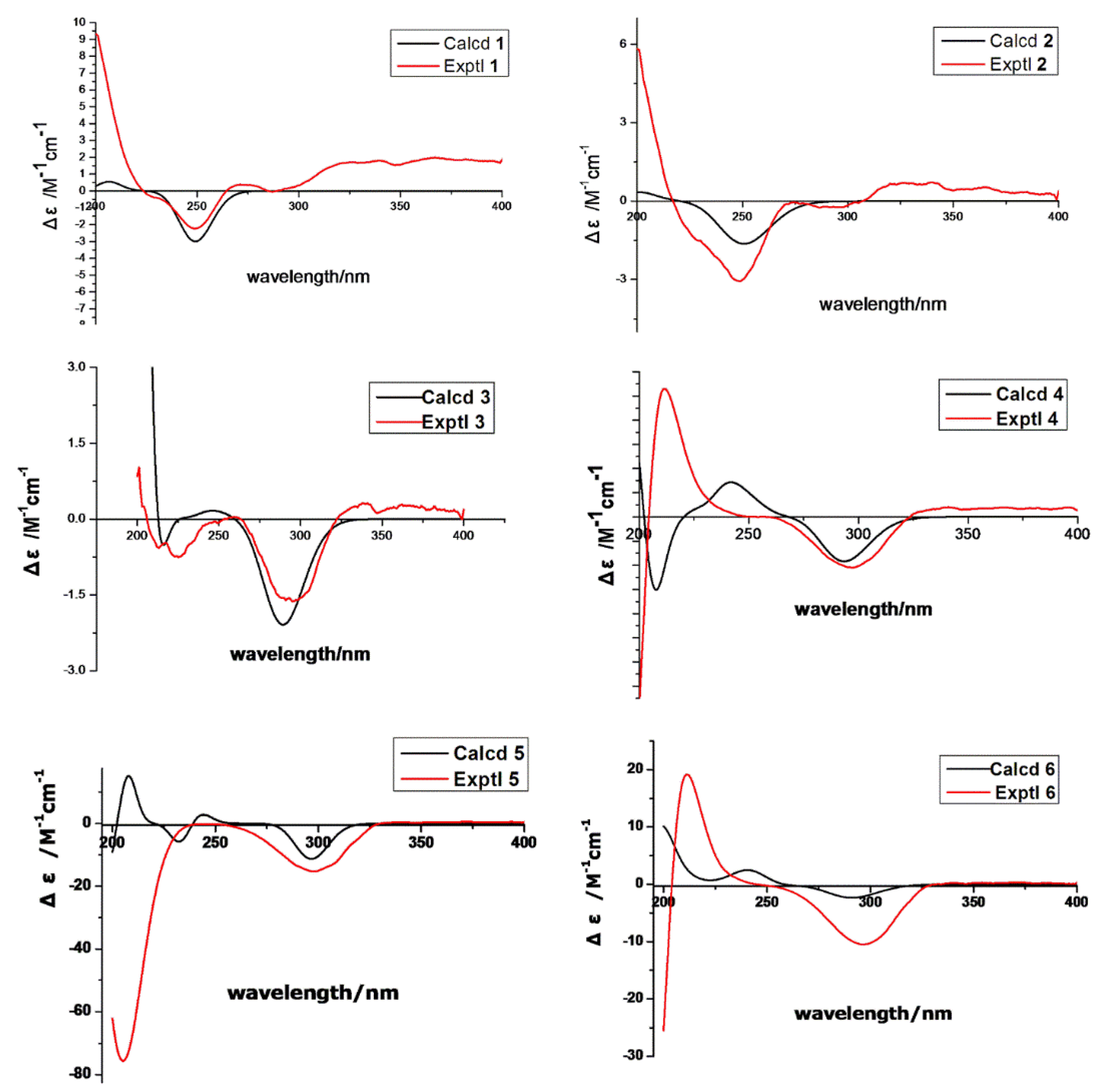
| Key Atoms | NOE Correlation | Main 3D Conformers | ||
|---|---|---|---|---|
| Me-aaa-Cis | Me-eee-Cis | Me-eea-Trans | ||
| Distance (Å) | Distance(Å) | Distance(Å) | ||
| CH3-14 | H-5 | 3.347715 | 4.256464 | 3.471439 |
| CH3-15 | H-10 | 3.608014 | 2.503497 | 1.862233 |
| CH3-16 | H-10 | 2.310649 | 5.322337 | 1.826166 |
| CH3-14 | H-2 | 5.109479 | 4.964076 | 2.470492 |
| CH-5 | H-10 | 2.265362 | 2.503497 | 3.087856 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction and Isolation
3.4. Spectral Data
3.5. Computational Analyses
3.6. Esterification Procedure
3.7. Inhibition of Aldose Reductase
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.X.; Wang, J.J.; Huang, H.B.; Ma, L.; Wang, J.; Gu, Y.C.; Liu, L.; Lin, Y.C. Four Eremophilane Sesquiterpenes from the mangrove endophytic fungus Xylaria sp. BL321. Mar. Drugs 2012, 10, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Cai, X.; Xu, F.; She, Z.; Chan, W.L.; Vrijmoed, L.L.; Jones, E.B.; Lin, Y. Three metabolites from the mangrove endophytic fungus Sporothrix sp. (#4335) from the South China Sea. J. Org. Chem. 2009, 74, 1093–1098. [Google Scholar] [PubMed]
- Li, J.; Yang, X.; Lin, Y.; Yuan, J.; Lu, Y.; Zhu, X.; Li, J.; Li, M.; Lin, Y.; He, J.; et al. Meroterpenes and azaphilones from marine mangrove endophytic fungus Penicillium 303#. Fitoterapia 2014, 97, 241–246. [Google Scholar] [PubMed]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Freire, F.; Seco, J.M.; Quiñoá, E.; Riguera, R. Chiral 1,2-diols: The assignment of their absolute configuration by NMR made easy. Org. Lett. 2010, 12, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Feng, T.; Li, Z.H.; Liu, J.K. Five new polyketides from the basidiomycete Craterellus odoratus. Nat. Prod. Bioprospect. 2012, 2, 170–173. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Miura, S.; Yamashita, Y.; Ohta, T. Aspermytin A: A new neurotrophic polyketide isolated from a marine-derived fungus of the genus Aspergillus. Bioorg. Med. Chem. Lett. 2004, 14, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, L.; Zhai, J.; Chen, Y.; Luo, H.; Hu, X. The molecular basis for inhibition of sulindac and its metabolites towards human aldose reductase. FEBS Lett. 2012, 586, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Qiu, Z.; You, J.; Tan, H.; Zhou, S. Isolation and characterization of endophytic streptomycete antagonists of fusarium wilt pathogen from surface-sterilized banana roots. FEMS Microbiol. Lett. 2005, 247, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision a.02; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Van Zandt, M.C.; Jones, M.L.; Gunn, D.E.; Geraci, L.S.; Jones, J.H.; Sawicki, D.R.; Sredy, J.; Jacot, J.L.; Dicioccio, A.T.; Petrova, T.; et al. Discovery of 3-[(4,5,7-trifluorobenzothiazol-2-yl)methyl]indole-N-acetic acid (lidorestat) and congeners as highly potent and selective inhibitors of aldose reductase for treatment of chronic diabetic complications. J. Med. Chem. 2005, 48, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Umebayashi, Y.; Kawashima, M.; Sugiura, Y.; Kikuchi, T.; Tanaka, R. Determination of the chemical structures of tandyukisins B–D, isolated from a marine sponge-derived fungus. Mar. Drugs 2015, 13, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Jeerapong, C.; Phupong, W.; Bangrak, P.; Intana, W.; Tuchinda, P. Trichoharzianol, a new antifungal from Trichoderma harzianum F031. J. Agric. Food Chem. 2015, 63, 3704–3708. [Google Scholar] [CrossRef] [PubMed]
- Nakadate, S.; Nozawa, K.; Horie, H.; Fujii, Y.; Nagai, M.; Hosoe, T.; Kawai, K.; Yaguchi, T.; Fukushima, K. Eujavanicols A–C, decalin derivatives from Eupenicillium javanicum. J. Nat. Prod. 2007, 70, 1510–1512. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Li, J.; Huang, M.; Liu, L.; Wang, J.; Lin, Y. Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#. Mar. Drugs 2015, 13, 6306-6318. https://doi.org/10.3390/md13106306
Ma Y, Li J, Huang M, Liu L, Wang J, Lin Y. Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#. Marine Drugs. 2015; 13(10):6306-6318. https://doi.org/10.3390/md13106306
Chicago/Turabian StyleMa, Yanhong, Jing Li, Meixiang Huang, Lan Liu, Jun Wang, and Yongcheng Lin. 2015. "Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#" Marine Drugs 13, no. 10: 6306-6318. https://doi.org/10.3390/md13106306
APA StyleMa, Y., Li, J., Huang, M., Liu, L., Wang, J., & Lin, Y. (2015). Six New Polyketide Decalin Compounds from Mangrove Endophytic Fungus Penicillium aurantiogriseum 328#. Marine Drugs, 13(10), 6306-6318. https://doi.org/10.3390/md13106306