Abstract
Paired box (PAX) genes encode a family of nine transcription factors that function as master regulators of embryogenesis, organogenesis, and lineage specification. Their tightly regulated spatial and temporal expression is essential for the development of multiple organ systems, including the central nervous system, eyes, kidneys, immune system, musculoskeletal system, and endocrine organs. Germline mutations of PAX genes result in a broad and often pleiotropic spectrum of human disease, reflecting the developmental programs governed by each family member. Pathogenic variants in PAX genes underlie diverse congenital disorders such as aniridia (PAX6), renal coloboma syndrome (PAX2), otofaciocervical syndrome with immunodeficiency (PAX1), Waardenburg syndrome (PAX3), maturity-onset diabetes of the young (PAX4), and tooth agenesis (PAX9). These conditions frequently demonstrate variable expressivity, incomplete penetrance, and overlapping phenotypes, which make it challenging to be clinically recognized. Beyond embryogenesis and embryologic development, emerging evidence indicates that several PAX proteins remain active in postnatal tissue maintenance, adult stem cell regulation, immune function, and regenerative responses (particularly PAX7 in skeletal muscle satellite cells and PAX5 in B-cell homeostasis), further expanding their clinical relevance. This review provides a synopsis of the major, clinically relevant, germline PAX gene mutations, emphasizing genotype–phenotype correlations, developmental mechanisms, and disease classification across the organ systems. By integrating molecular genetics with human pathology, we highlight the diagnostic implications of PAX genes as central determinants of congenital disease and provide a framework for understanding how alterations in the developmental transcriptional networks translate into human pathology.
1. Introduction
Paired box (PAX) genes are considered master regulators in both vertebrates and invertebrates, as they govern organ development and are essential for lineage-specific differentiation [1]. Classification within this gene family requires the presence of a paired domain, while the addition of a homeodomain and/or an octapeptide further divides it into four structural subclasses: group I (PAX1 and PAX9), group II (PAX2, PAX5, and PAX8), group III (PAX3 and PAX7), and group IV (PAX4 and PAX6) (Figure 1) [1]. This group of nine transcription factors plays a key role in numerous developmental processes during early growth, as demonstrated in studies of both human and mouse genes [2]. Furthermore, embryogenesis and organogenesis depend heavily on these evolutionarily conserved genes [2]. Despite their crucial role in human development, PAX genes are susceptible to mutations like any other gene, with alterations linked to a range of diseases, including lysosomal storage disorders, autoimmune conditions, and cancer [1].
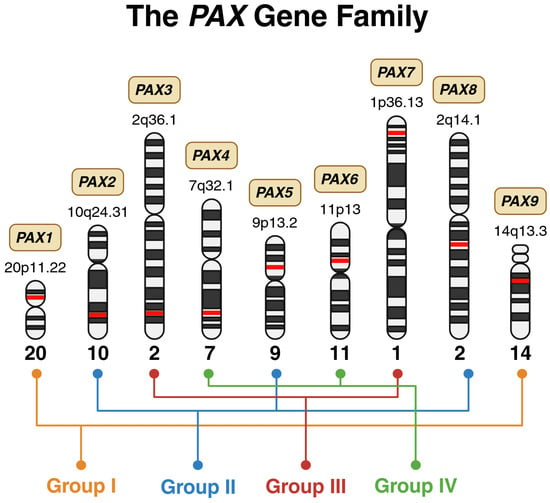
Figure 1.
Chromosomal distribution and structural grouping of the human PAX gene family. Schematic representation of the nine PAX genes mapped to their respective chromosomal loci, illustrated using ideograms of human chromosomes. The precise cytogenetic band for each PAX member is indicated adjacent to its chromosomal position, highlighted in red. The PAX genes are organized into four structural subclasses: Group I (PAX1, PAX9), Group II (PAX2, PAX5, PAX8), Group III (PAX3, PAX7), and Group IV (PAX4, PAX6). Created in BioRender. Bahmad, H. (2026) Available online: https://www.BioRender.com/gv0gwu7 (accessed on 31 January 2026).
The structural classification of PAX genes into four groups carries significant functional and clinical implications beyond mere molecular architecture. This grouping reflects evolutionarily conserved developmental programs and exhibits remarkable topographical coherence in disease manifestation [1,3]. Members within the same PAX group tend to cause disorders affecting anatomically related structures, reflecting their shared roles in specific developmental domains. For example, Group I genes (PAX1 and PAX9) both regulate pharyngeal arch derivatives and axial skeletal structures, with mutations causing overlapping phenotypes involving vertebral anomalies, thymic hypoplasia, and craniofacial defects. Group II genes (PAX2, PAX5, and PAX8) share expression in the developing urogenital system and are each associated with renal and/or reproductive tract malformations, though PAX5 additionally governs B-cell lineage commitment. Group III genes (PAX3 and PAX7) exhibit the most striking functional overlap, both serving as master regulators of neural crest development and skeletal myogenesis; their mutations produce related syndromes involving pigmentary abnormalities, hearing loss, and musculoskeletal defects, while their oncogenic fusions drive myogenic sarcomas. Group IV genes (PAX4 and PAX6), despite structural similarity, have diverged functionally—PAX6 governing eye and brain development while PAX4 specializes in pancreatic islet differentiation—yet both influence neuroendocrine lineages. This structure–function relationship provides a practical clinical framework. The affected PAX group often predicts the anatomical distribution of disease, aiding differential diagnosis and guiding targeted genetic testing strategies.
PAX genes are essential for organogenesis and lineage-specific differentiation, acting as key regulators in organ systems such as the renal, ophthalmic, respiratory, muscular, and central nervous system (CNS) [3]. They are also critical for pattern formation, likely determining the timing and location of organ development. These genes are tightly regulated during organogenesis, and mutations during this period are linked to major developmental defects [4]. Because PAX proteins function through DNA binding, mutations are also associated with an increased risk of cancer development [4].
Group I genes (PAX1 and PAX9) regulate sclerotomes, axial skeletal patterning, and pharyngeal pouch derivatives, including the thymus [5]. PAX9 specifically maintains tooth structure, and its absence can result in tooth agenesis [6]. Both also contribute to the proliferation of neural crest cells [6]. Group II genes (PAX2, PAX5, and PAX8) are important for the development of the renal system, CNS, and the thyroid gland [7]. Group III genes (PAX3 and PAX7) act as transcription factors for neural crest development, including induction, migration, and cell differentiation [8]. They influence proliferation early in development, determining multiple lineages such as cardiac, sensory, and teeth [8]. Group IV genes (PAX4 and PAX6) are involved in the development of the gastrointestinal and endocrine systems, eyes, and CNS [7]. PAX genes are themselves regulated through mechanisms such as posttranslational modifications, degradation, and partner protein interactions, including miRNAs and alternative splicing [7]. Although designed to guide organogenesis and tissue differentiation, mutations in PAX genes can result in both neoplastic and non-neoplastic diseases that are debilitating and potentially life-threatening [7].
While prior reviews have addressed PAX genes in cancer and developmental disorders, substantial advances have emerged over the past two years that warrant an updated comprehensive review [7,9,10,11]. These include: (i) newly identified germline and somatic PAX variants with expanded genotype–phenotype correlations across multiple organ systems; (ii) advances in understanding epigenetic regulation of PAX genes, particularly promoter methylation patterns in PAX1, PAX2, PAX5, PAX6, and PAX9 and their roles in tumorigenesis; (iii) emerging roles of PAX proteins in tumor biology and lineage plasticity, including novel fusion partners and metabolic reprogramming mechanisms; (iv) improved diagnostic and molecular testing implications for both germline disorders and somatic alterations [9,12,13,14,15,16,17,18,19,20]. The present review integrates these recent discoveries with established knowledge to provide clinicians and pathologists with a current, comprehensive framework for understanding PAX-related human pathology.
2. Germline PAX Gene Variations and Functional Consequences
Given the diversity of the PAX gene family, understanding the pathological spectrum linked to these transcription factors and their variations is essential for elucidating their biological and clinical significance. The various neoplastic and congenital conditions arising from PAX dysregulation highlight their central role in developmental biology and tumorigenesis. Here, we systematically examine the major diseases associated with the PAX family, including disorders driven by germline mutations, somatic alterations, gene fusions, and epigenetic dysregulation (Figure 2).
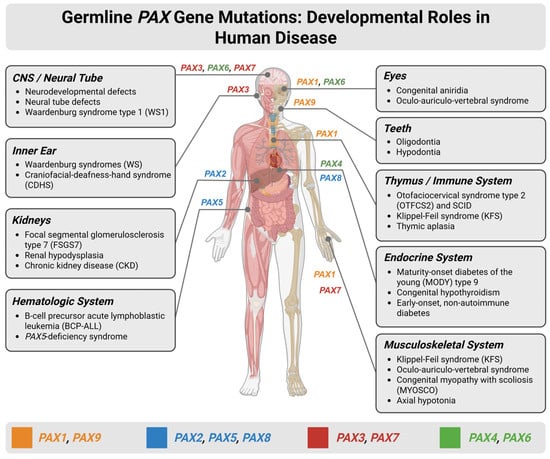
Figure 2.
Developmental roles and disease associations of germline PAX gene mutations across human organ systems. Schematic overview illustrating the expression patterns of PAX genes during human development and the major congenital disorders and disease phenotypes associated with germline mutations. The human body diagram highlights organ-specific involvement of individual PAX family members, including the central nervous system (CNS)/neural tube, eyes, inner ear, teeth, thymus and immune system, kidneys, endocrine organs, hematologic system, and musculoskeletal system. Disorders linked to PAX gene dysfunction—such as Waardenburg syndromes (WSs), congenital aniridia, renal hypodysplasia, focal segmental glomerulosclerosis, B-cell precursor acute lymphoblastic leukemia, maturity-onset diabetes of the young (MODY), thymic aplasia, and Klippel–Feil syndrome (KFS)—are listed by affected system. WS encompasses four clinical subtypes: WS1 and WS3 result from PAX3 mutations and feature dystopia canthorum; WS2 (typically MITF, SOX10, or SNAI2 mutations) and WS4 (SOX10, EDN3, or EDNRB mutations) lack dystopia canthorum. Inner ear involvement with sensorineural hearing loss occurs across all WS subtypes. Created in BioRender. Bahmad, H. (2026) Available online: https://www.BioRender.com/8dixrhs (accessed on 31 January 2026).
Germline mutations can be found through genetic family trees as they are heritable mutations leading to different congenital and neoplastic conditions. Given the distinct developmental functions of each PAX gene, germline mutations typically give rise to disorders affecting the same organs and tissues in which that gene normally plays a critical developmental role.
2.1. PAX1 Germline Mutations
PAX1 plays a central role in skeletal and thymic development during embryogenesis. Consequently, germline mutations in this gene can result in a spectrum of developmental disorders involving skeletal malformations and metabolic dysfunction. Consistent with its role in axial skeletogenesis and pharyngeal derivatives, rare germline PAX1 variants have been described across a spectrum of congenital malformations. In a series of patients with congenital vertebral malformations, Giampietro et al. identified heterozygous missense substitutions in exon 4 of PAX1 in two males, including one with complex thoracic vertebral defects, rib anomalies, polydactyly, and a ventricular septal defect, suggesting that an altered PAX1 gene (CCC (Pro) to CTC (Leu) change at amino acid 410) may have contributed to combined axial and cardiac malformations [21]. In Klippel–Feil syndrome, characterized by cervical vertebral fusion, short neck, and low posterior hairline, sequencing of 63 affected individuals uncovered several rare PAX1 coding and intronic variants not observed in controls, supporting a contribution of PAX1 haploinsufficiency in at least a subset of cases [22]. Similarly, in two fetuses with Jarcho–Levin syndrome and a “crab-like” thorax, Bannykh et al. demonstrated markedly reduced PAX1 and PAX9 protein expression in vertebral chondrocytes, implicating dysregulated PAX1/PAX9 signaling in severe costovertebral segmentation defects [23]. More recently, biallelic loss-of-function PAX1 mutations have been shown to underlie autosomal recessive otofaciocervical syndrome type 2 (OTFCS2), a multisystem disorder with craniofacial dysmorphism, vertebral and shoulder-girdle anomalies, thymic aplasia, and profound T-cell immunodeficiency, further delineating the pleiotropic effects of germline PAX1 disruption [24,25,26] (Table 1).

Table 1.
Commonly reported pathogenic/likely pathogenic germline PAX1 variants and their clinical consequences.
2.2. PAX2 Germline Mutations
PAX2 is a paired-domain transcription factor that is crucial for normal development of the kidney, urinary tract, and optic nerve, and germline loss-of-function variants underlie a wide spectrum now grouped as PAX2-related disorder, classically presenting as renal-coloboma (papillorenal) syndrome (RCS) (Table 2). Heterozygous truncating mutations (nonsense, frameshift, splice-site) clustered in exons 2-4, which encode the paired domain, represent the most common pathogenic variants and typically produce renal hypodysplasia/dysplasia, vesicoureteral reflux, and optic nerve coloboma or dysplasia, with marked intra- and interfamilial variability [29,30,31].
Table 2.
Commonly reported pathogenic/likely pathogenic germline PAX2 variants and their clinical consequences.
Beyond classic RCS, germline PAX2 mutations have been increasingly reported in patients with isolated renal disease, including congenital anomalies of the kidney and urinary tract (CAKUT), oligomeganephronia, steroid-resistant nephrotic syndrome, and focal segmental glomerulosclerosis (FSGS). The recurrent paired-domain frameshift c.76dupG (p.Val26fs/Val27fs) is now recognized as the most prevalent PAX2 pathogenic allele and has been described in families with renal hypodysplasia, RCS, and in twins with FSGS, reflecting the broad phenotypic expressivity of a single variant [20,32,33,39,45]. Larger deletions involving one or more exons, as well as whole-gene deletions, can also cause PAX2-related disorder and may present with renal hypodysplasia in the absence of ocular findings [31,41,42,43].
Missense mutations affecting conserved residues in the paired domain generally act as hypomorphic alleles and have been reported in patients with “forme fruste” papillorenal phenotypes or isolated CKD, sometimes without optic nerve anomalies [46,47]. Adult-onset FSGS with little or no structural renal malformation has been linked to heterozygous PAX2 missense variants that disrupt DNA binding, transactivation, or interactions with corepressors, suggesting both haploinsufficiency and dominant-negative mechanisms [33,37,48,49].
2.3. PAX3 Germline Mutations
In humans, germline PAX3 mutations give rise to Waardenburg syndrome (WS) subtypes and craniofacial-deafness-hand syndrome (CDHS) (Table 3). WS is classified into four clinical subtypes (WS1-4) based on the presence or absence of specific features. WS type 1 (WS1) and WS type 3 (WS3) are both caused by PAX3 mutations and are characterized by hearing loss, pigmentary disturbances of the hair, skin, and eyes, and dystopia canthorum (lateral displacement of the inner canthi); WS3 (Klein–Waardenburg syndrome) additionally presents with upper limb abnormalities including contractures and hypoplasia. WS type 2 (WS2) lacks dystopia canthorum and is typically caused by mutations in MITF, SOX10, or SNAI2 rather than PAX3. WS type 4 (WS4 or Waardenburg–Shah syndrome) combines features of WS2 with Hirschsprung disease and results from mutations in SOX10, EDN3, or EDNRB. Sensorineural hearing loss and pigmentary abnormalities affect all WS subtypes, with inner ear involvement (cochlear melanocyte deficiency) representing a common pathogenic mechanism across types [50,51,52,53,54,55,56,57]. Most pathogenic PAX3 mutations occur within exons 2-6, encoding the paired domain and homeodomain [58,59]. Reports of germline mosaicism and de novo mutations further highlight the variable inheritance patterns [55,60]. CDHS, a related condition, presents with craniofacial anomalies, profound sensorineural deafness, and ulnar deviation with finger contractures, and has been linked to mutations in PAX3 exon 2 [61].
Table 3.
Commonly reported pathogenic/likely pathogenic germline PAX3 variants and their clinical consequences.
2.4. PAX4 Germline Mutations
PAX4 encodes a paired-domain transcription factor that is essential for pancreatic islet development and β-cell survival. Germline loss-of-function or hypomorphic PAX4 variants have been linked to a monogenic form of early-onset diabetes designated maturity-onset diabetes of the young type 9 (MODY9), as well as to increased susceptibility to type 2 diabetes in East Asian populations [72,73,74,75,76,77] (Table 4).
Table 4.
Commonly reported pathogenic/likely pathogenic germline PAX4 variants and their clinical consequences.
In Thai MODY probands, Plengvidhya et al. identified two rare PAX4 variants—p.Arg164Trp (R164W) and a splice-site mutation IVS7-1G>A—that were absent in several hundred controls; R164W segregated with diabetes and impaired repression of insulin and glucagon promoters in vitro, while IVS7-1G>A produced aberrant splicing consistent with loss of function. A Japanese MODY family carrying a 39 bp deletion (c.374-412 del39) in exon 3 showed a truncated protein lacking most of the homeodomain, with early-onset, non-autoimmune diabetes in multiple affected relatives [72]. More recently, a heterozygous missense variant c.487C>T in exon 7 was reported in a 19-month-old Chinese boy with MODY9 [77].
Beyond classical MODY, the missense variant p.Arg121Trp (R121W) is enriched in Japanese patients with type 2 diabetes and functionally reduces PAX4 transcriptional repressor activity [78]. The ethnic-specific variant p.Arg192His (R192H) is associated with earlier age at diabetes onset and increased type 2 diabetes risk [73]. A more recent report describes a novel heterozygous PAX4 variant c.61C>T (p.Gln21*) in a child with MODY9 and neurodevelopmental impairment, underscoring that PAX4 dysfunction may extend beyond isolated β-cell failure [79].
2.5. PAX5 Germline Mutations
PAX5 encodes the B-cell-specific activator protein (BSAP), a master regulator of B-cell lineage commitment and maintenance. Germline loss-of-function or hypomorphic PAX5 variants define a rare, but increasingly recognized, inherited predisposition syndrome that is largely restricted to B-cell precursor acute lymphoblastic leukemia (BCP-ALL), with a separate spectrum of biallelic variants causing immunodeficiency and neurodevelopmental disease (Table 5).
The first description of a PAX5 leukemia-predisposition allele identified a recurrent heterozygous c.547G>A missense variant (p.Gly183Ser) in the octapeptide domain in two unrelated kindreds with autosomal dominant pre-B-ALL; leukemic blasts consistently showed 9p loss of heterozygosity with retention of the mutant allele, and functional assays demonstrated reduced transactivation capacity, establishing a hypomorphic mechanism [80]. Subsequent reports confirmed germline c.547G>A in additional families and sporadic cases, consolidating this variant as a prototypical PAX5-related leukemia predisposition allele with incomplete penetrance [81,82]. Additional germline missense changes at the same hotspot (c.547G>C; p.Gly183Arg) and within the paired domain (c.113G>A; p.Arg38His) have been described in familial BCP-ALL, again behaving as hypomorphic alleles that require secondary somatic events—typically 9p deletion, a second PAX5 hit, or lesions in JAK-STAT/RAS signaling—for overt leukemogenesis [82,83,84].
More recently, a germline splice-site variant (c.1013-2A>G) resulting in a single-exon deletion of PAX5 was reported in a child with BCP-ALL, broadening the allelic spectrum to include structural and truncating germline lesions [85]. Aggregated cohort analyses indicate that most PAX5 leukemia-predisposition alleles are heterozygous, cluster within functional domains, and confer an autosomal dominant syndrome with variable expressivity, in which unaffected carriers are common but at increased lifetime risk of B-lineage malignancy [82].
Table 5.
Commonly reported pathogenic/likely pathogenic germline PAX5 variants and their clinical consequences.
Table 5.
Commonly reported pathogenic/likely pathogenic germline PAX5 variants and their clinical consequences.
| Variant | Variant Type and Domain | Zygosity | Main Phenotype/Diagnosis | Inheritance | References |
|---|---|---|---|---|---|
| c.547G>A (p.Gly183Ser) | Missense in octapeptide domain (exon 6); hypomorphic allele with reduced transcriptional activity | Heterozygous | Familial pre-B/BCP-ALL; leukemic cells with 9p LOH retaining mutant allele | Autosomal dominant (incomplete penetrance) | [80,81,82] |
| c.547G>C (p.Gly183Arg) | Missense in octapeptide domain (same codon 183 hotspot); hypomorphic/loss-of-function effect | Heterozygous | Familial BCP-ALL and sporadic pediatric BCP-ALL; often with secondary 9p/PAX5 lesions | Autosomal dominant (incomplete penetrance) | [82,85] |
| c.113G>A (p.Arg38His) | Missense in paired DNA-binding domain (exon 2); hypomorphic allele with reduced DNA | Heterozygous | Familial BCP-ALL with multiple affected relatives; additional sporadic B-ALL cases | Autosomal dominant leukemia predisposition (incomplete penetrance) | [82,83,84] |
| c.1013-2A>G (PAX5 exon 6 deletion) | Canonical splice-acceptor variant at intron 8; predicted in-frame skipping of exon 9 → altered C-terminal region (hypomorphic) | Heterozygous (de novo) | Pediatric BCP-ALL; immunophenotype similar to other PAX5-mutated BCP-ALL | Autosomal dominant leukemia predisposition (incomplete penetrance; evidence still limited) | [85,86] |
2.6. PAX6 Germline Mutations
PAX6 is a master transcription factor for ocular development, required for proper formation of the optic cup, iris, lens, fovea, and optic nerve, and it remains expressed in select neural and ocular tissues throughout life. Germline PAX6 variants underlie a broad phenotypic spectrum now grouped as “PAX6-related aniridia,” ranging from classic pan-ocular aniridia to isolated foveal hypoplasia, anterior segment dysgenesis (including Peters anomaly), microphthalmia, coloboma, and more complex neurodevelopmental phenotypes [87,88,89,90,91,92,93,94,95,96,97,98,99] (Table 6).
Most pathogenic germline variants are heterozygous loss-of-function changes—nonsense, frameshift, canonical splice-site variants, or multiexonic/whole-gene deletions—that result in PAX6 haploinsufficiency. These typically cause classic congenital aniridia with high penetrance but variable expressivity. Larger 11p13 deletions that encompass PAX6 and neighboring WT1 produce WAGR (Wilms tumor–aniridia–genitourinary anomalies–intellectual disability) syndrome, highlighting the need for cytogenetic or copy-number evaluation in children with aniridia and systemic features [87,91,96].
More than 500 distinct PAX6 variants have been catalogued, distributed across the paired domain, linker region, homeodomain, and C-terminal proline/serine/threonine-rich (PST) transactivation region. Truncating variants in the DNA-binding domains generally produce typical aniridia, whereas certain missense substitutions in highly conserved residues or in the alternative exon 5a splice region are enriched in atypical phenotypes such as Peters anomaly, anterior segment malformations, or relatively iris-sparing phenotypes (e.g., isolated foveal hypoplasia). Recurrent nonsense and frameshift alleles (e.g., c.265C>T p.Gln89*, c.718C>T p.Arg240*, c.112del p.Arg38Glyfs16, c.278_281del p.Glu93Alafs30) have been reported across multiple cohorts and illustrate the predominance of haploinsufficient mechanisms [70,94,96,98,99,100,101].
Although ocular disease dominates the clinical picture, a subset of individuals with PAX6 haploinsufficiency or compound heterozygosity exhibit brain malformations (e.g., agenesis of the pineal gland, polymicrogyria), olfactory dysfunction, neurocognitive or psychiatric features, and, rarely, glucose intolerance or diabetes [92,96,102,103].
Table 6.
Commonly reported pathogenic/likely pathogenic germline PAX6 variants and their clinical consequences.
Table 6.
Commonly reported pathogenic/likely pathogenic germline PAX6 variants and their clinical consequences.
| Variant | Variant Type and Domain | Zygosity | Main Phenotype/Diagnosis | Inheritance | References |
|---|---|---|---|---|---|
| 11p13 contiguous deletion including PAX6 and WT1 | Large heterozygous deletion of PAX6 plus neighboring genes at 11p13 | Heterozygous | WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, intellectual disability) | Usually de novo; autosomal dominant when familial | [91,95,96] |
| Intragenic multiexon or whole-gene PAX6 deletion | Heterozygous deletion of one or more coding exons or entire gene (often detected by MLPA/CNV analysis) | Heterozygous | Classic aniridia ± foveal hypoplasia, cataract, glaucoma; occasionally neurodevelopmental anomalies | Autosomal dominant; often de novo | [90,91] |
| c.265C>T (p.Gln89Ter) | Nonsense; premature stop in exon 6 within the paired domain → truncated protein/functional haploinsufficiency | Heterozygous | Congenital aniridia with bilateral congenital cataracts (familial) | Autosomal dominant (familial) | [97] |
| c.718C>T (p.Arg240*) | Nonsense (premature stop-gain) in exon 9; truncates the homeodomain/linker region | Heterozygous | Classic congenital aniridia with nystagmus; reported in multiple families across ethnicities; often associated with congenital cataract or keratopathy | Autosomal dominant; de novo or familial | [97,98,100] |
| c.949C>T (p.Arg317*) | Nonsense (premature stop-gain) in exon 8 | Heterozygous | Classic/congenital ocular malformation (e.g., Aniridia—pan-ocular: iris hypoplasia/absence, foveal hypoplasia, possible cataract, glaucoma, keratopathy) | Autosomal dominant (haploinsufficiency) | [87,99] |
| c.781C>T (p.Arg261*) | Nonsense (premature stop-gain) in exon 10 | Heterozygous | PAX6-related aniridia/eye malformation likely (given truncation and established mutational spectrum) | Autosomal dominant (haploinsufficiency) | [87,99] |
| c.607C>T (p.Arg203*) | Nonsense (premature stop-gain) in exon 11 | Heterozygous | Likely classic aniridia/PAX6-associated ocular developmental disorder (consistent with many truncating PAX6 mutations) | Autosomal dominant (haploinsufficiency) | [87,99] |
| c.112del (p.Arg38Glyfs*16) | Frameshift deletion in exon 5′ region of the paired domain → premature termination | Heterozygous | Familial aniridia with nystagmus; haploinsufficiency due to truncated paired-domain protein | Autosomal dominant | [100] |
| c.299G>A (p.Trp100*) | Nonsense mutation in exon 6 within paired domain → early stop | Heterozygous | Congenital aniridia with lens opacities, foveal hypoplasia; classic haploinsufficiency phenotype | Autosomal dominant | [100] |
| c.278_281del (p.Glu93Alafs*30) | 4 bp deletion causing frameshift in paired domain (exon 6) → truncated protein | Heterozygous | Aniridia with spontaneous anterior lens capsule rupture, cataract, and foveal hypoplasia | Autosomal dominant (familial) | [100] |
| c.76A>G (p.Arg26Gly) | Missense in N-terminal paired domain; affects DNA-binding surface and reduces transcriptional activation | Heterozygous | Iris hypoplasia, variable aniridia, anterior segment dysgenesis, sometimes Peters anomaly | Autosomal dominant; familial | [88,89,101,104] |
| c.106G>C (p.Gly36Arg) | Missense in PAX6 paired domain; structurally disruptive at a conserved DNA-contact residue | Heterozygous | Anterior segment dysgenesis; iris hypoplasia; reported in families with atypical aniridia | Autosomal dominant | [88,89,93] |
| c.20T>A, exon 5a (p.Val54Asp) | Missense variant in the alternatively spliced exon 5a affecting N-terminal paired-domain structure | Heterozygous | Anterior segment dysgenesis spectrum, including Peters anomaly, Axenfeld anomaly, congenital cataract, microcornea/microphthalmos, and foveal hypoplasia; variable severity across families | Autosomal dominant (familial or sporadic); possible regional founder effect in Japan | [105] |
| c.170_174delTGGGC (p.Leu57fs*17) | 5 bp deletion in exon 7 → frameshift with premature stop ~17 codons downstream (N-terminal paired domain; predicted loss-of-function, haploinsufficiency via NMD) | Heterozygous | Familial autosomal dominant aniridia with almost complete iris absence, corneal pannus, foveal hypoplasia, markedly reduced visual acuity; in index case, absent pineal gland and severe hypoplasia of anterior commissure on brain MRI | Autosomal dominant (familial) | [92] |
| c.475delC (p.Arg159fs*47) | Single-base deletion in exon 8 → frameshift with premature stop ~47 codons downstream (paired-domain/central region; predicted loss-of-function, haploinsufficiency via NMD) | Heterozygous | Familial autosomal dominant aniridia with foveal hypoplasia, corneal pannus, anterior/posterior polar cataracts, lens dislocation; in index case, absent pineal gland and absent posterior commissure on brain MRI | Autosomal dominant (familial) | [92] |
| c.117_128del | In-frame 12 bp deletion in the 5′ coding region of PAX6 → loss of four amino acids within the paired domain; predicted disruption of DNA binding → loss-of-function | Heterozygous in the father; compound heterozygous in both fetuses | Father: aniridia (sporadic) with midline brain anomalies on CT (aplasia of pineal gland, hypoplasia of corpus callosum and anterior commissure). Fetuses (with both mutations): severe brain malformations, anophthalmia/microphthalmia, agenesis of corpus callosum, massive germinal matrix overgrowth, disorganized cortex and cerebellum, absent pyramidal tracts—lethal PAX6-null-like phenotype | In the father: autosomal dominant PAX6-related aniridia/CNS midline defects. In fetuses: effectively autosomal recessive/PAX6-null state due to compound heterozygosity. | [103] |
| c.112del | 1 bp deletion near the N-terminal paired domain → frameshift and premature stop; predicted truncated, non-functional protein → loss-of-function | Heterozygous in the mother; compound heterozygous in both fetuses | Mother: aniridia (sporadic) with similar midline brain anomalies on CT (pineal aplasia, corpus callosum and anterior commissure hypoplasia). Fetuses: same severe, early-lethal brain malformation phenotype when combined with c.117_128del12 | In the mother: autosomal dominant PAX6-related aniridia/CNS anomalies. In fetuses: autosomal recessive/PAX6-null phenotype when in trans with c.117_128del | [103] |
2.7. PAX7 Germline Mutations
PAX7 encodes a paired-box transcription factor that is indispensable for neural crest formation and maintenance of skeletal muscle satellite cells. Consistent with its role in postnatal myogenesis, human germline PAX7 variants are now recognized as the cause of a distinctive autosomal recessive “satellite cell-opathy” spectrum, ranging from severe neurodevelopmental syndromes to progressive congenital myopathy with scoliosis (MYOSCO) [106] (Table 7).
The first human PAX7-related disorder was described by Proskorovski-Ohayon et al., who reported a consanguineous family with an autosomal recessive syndrome of failure to thrive, severe global developmental delay, microcephaly, axial hypotonia, pyramidal signs, dystonia, seizures, irritability, and self-mutilation due to a biallelic splice-disrupting mutation in PAX7 isoform 3 [107].
Subsequently, Feichtinger et al. identified biallelic loss-of-function PAX7 variants (nonsense, frameshift, and splice-site changes) in multiple unrelated families with progressive congenital myopathy, axial and limb-girdle weakness, early scoliosis, ptosis, and markedly reduced satellite cell numbers on muscle biopsy, establishing PAX7-associated MYOSCO as a primary satellite cell disorder [108]. More recent work has expanded the clinical and imaging spectrum of PAX7-related myopathy, describing additional biallelic missense variants with characteristic whole-body muscle MRI patterns and variable scoliosis severity [109,110].
Beyond myopathy, rare heterozygous and compound heterozygous PAX7 variants have been reported in cohorts with congenital scoliosis, suggesting that impaired PAX7-mediated muscle development and paraspinal muscle dysfunction may contribute to vertebral malformations in at least a subset of cases [111].
Table 7.
Commonly reported pathogenic/likely pathogenic germline PAX7 variants and their clinical consequences.
Table 7.
Commonly reported pathogenic/likely pathogenic germline PAX7 variants and their clinical consequences.
| Variant | Variant Type and Domain | Zygosity | Main Phenotype/Diagnosis | Inheritance | References |
|---|---|---|---|---|---|
| Splice-site variant in PAX7 isoform 3 | Predicted loss-of-function splice-site change leading to aberrant splicing and reduced PAX7 protein; affects DNA-binding/activation of target genes | Homozygous | Severe neurodevelopmental syndrome with failure to thrive, microcephaly, axial hypotonia, pyramidal signs, dystonia, seizures, irritability, and self-mutilation | Autosomal recessive | [107] |
| Multiple biallelic truncating and splice-site variants across PAX7 (nonsense, frameshift, canonical splice; distributed across paired and C-terminal transactivation domains) | Loss-of-function variants causing markedly reduced or absent PAX7, satellite cell depletion, and impaired myogenesis | Mostly homozygous; some compound heterozygous | Progressive congenital myopathy with scoliosis: early-onset axial and limb-girdle weakness, ptosis, progressive scoliosis, restrictive respiratory failure; muscle biopsies show reduced satellite cells and dystrophic changes | Autosomal recessive | [108] |
| Novel biallelic PAX7 variants in congenital myopathy with scoliosis | Missense/splice-affecting variants; functionally consistent with PAX7 loss-of-function; characteristic whole-body MRI pattern | Homozygous | PAX7-related congenital myopathy with prominent axial weakness, early scoliosis, and distinctive whole-body muscle MRI | Autosomal recessive | [109] |
| Multiple rare heterozygous PAX7 missense variants enriched in congenital scoliosis cohort | Predicted deleterious missense variants, many clustering within or near DNA-binding domains | Heterozygous (in most probands; some complex inheritance) | Congenital scoliosis (vertebral formation/segmentation defects) with or without neuromuscular features; PAX7 variants proposed as risk alleles contributing to congenital scoliosis pathogenesis via impaired muscle development | Likely autosomal dominant or risk-modifying; segregation incomplete | [111] |
2.8. PAX8 Germline Mutations
PAX8 is a paired-domain transcription factor essential for thyroid organogenesis, as well as kidney and Müllerian tract development. Monoallelic loss-of-function variants in PAX8 are a well-established cause of congenital hypothyroidism due to thyroid dysgenesis, typically presenting with thyroid hypoplasia or ectopy (Table 8). Most pathogenic germline variants are missense, nonsense, or small indels clustered within the N-terminal paired domain (amino acids 9-133), where they impair DNA binding and transcriptional activation of thyroid-specific targets such as thyroglobulin and thyroperoxidase [112,113,114].
Early reports described heterozygous paired-domain substitutions (for example p.Arg31His, p.Gln40Pro, p.Ser54Gly, p.Cys57Tyr, p.Leu62Arg) that segregate with familial congenital hypothyroidism and show loss of transactivation in vitro, supporting a haploinsufficiency model with variable penetrance and intrafamilial expressivity [112,113,114,115]. Subsequent cohort studies across different ethnicities confirmed that pathogenic PAX8 variants account for roughly 1-3% of congenital hypothyroidism cases due to thyroid dysgenesis, with phenotypes ranging from overt neonatal hypothyroidism with severe hypoplasia to subclinical disease with a normal-appearing gland in situ [116,117,118,119].
Although PAX8 is also expressed in the kidney and urogenital tract, extrathyroid malformations are relatively uncommon, occurring in <10% of carriers; reported anomalies include renal agenesis, vesicoureteral reflux, and other CAKUT-spectrum defects [114,120]. More recent work has expanded the mutational spectrum beyond the paired domain to include truncating variants in the transactivation domain (p.Leu186Hisfs*22) and in-frame deletions (p.Glu90del) that retain partial transcriptional activity yet still cause congenital hypothyroidism with a gland in situ [116,118,119].
Table 8.
Commonly reported pathogenic/likely pathogenic germline PAX8 variants and their clinical consequences.
Table 8.
Commonly reported pathogenic/likely pathogenic germline PAX8 variants and their clinical consequences.
| Variant | Variant Type and Domain | Zygosity | Main Phenotype/Diagnosis | Inheritance | References |
|---|---|---|---|---|---|
| c.92G>A (p.Arg31His) | Missense in paired domain (DNA-binding) | Heterozygous | Congenital hypothyroidism with thyroid dysgenesis (ectopy or hypoplasia) | Autosomal dominant | [112,117] |
| c.119A>C (p.Gln40Pro) | Missense in paired domain | Heterozygous | Congenital hypothyroidism with thyroid hypoplasia; mother with normal-sized gland and mild adult-onset autoimmune hypothyroidism | Autosomal dominant | [113] |
| c.160A>G (p.Ser54Gly) | Missense in paired domain helix | Heterozygous | Congenital hypothyroidism with normal-sized eutopic thyroid at birth; one carrier with unilateral renal agenesis | Autosomal dominant | [114] |
| c.170G>A (p.Cys57Tyr) | Missense in paired domain | Heterozygous | Familial congenital hypothyroidism with thyroid hypoplasia | Autosomal dominant | [115] |
| c.185T>G (p.Leu62Arg) | Missense in paired domain | Heterozygous | Congenital hypothyroidism with thyroid dysgenesis (ectopy/hypoplasia) | Autosomal dominant | [112,121] |
| c.322C>T (p.Arg108*) | Nonsense, truncating within paired domain | Heterozygous | Congenital hypothyroidism with thyroid hypoplasia | Autosomal dominant | [112,120] |
| c.74C>G (p.Pro25Arg) | Missense near N-terminus/paired domain boundary | Heterozygous | Congenital hypothyroidism with thyroid hypoplasia across three generations; some carriers with urogenital malformations | Autosomal dominant | |
| c.116A>C (p.His39Pro) | Missense in paired domain | Heterozygous | Familial congenital hypothyroidism with thyroid hypoplasia | Autosomal dominant | [117,118] |
| c.122G>T (p.Gly41Val) | Missense in paired domain | Heterozygous | Congenital hypothyroidism with thyroid agenesis or small eutopic gland | Autosomal dominant | |
| c.280G>A (p.Asp94Asn) | Missense in paired domain | Heterozygous | Congenital hypothyroidism with thyroid dysgenesis; often gland in situ | Autosomal dominant | [116,117] |
| c.268_270delGAA (p.Glu90del) | In-frame deletion in paired domain | Heterozygous | Congenital hypothyroidism with gland in situ | Autosomal dominant | [116] |
| c.555dupA (p.Leu186Hisfs*22) | Frameshift truncation | Heterozygous | Congenital hypothyroidism with gland in situ | Autosomal dominant | [116] |
2.9. PAX9 Germline Mutations
PAX9 encodes a paired-box transcription factor essential for tooth morphogenesis, particularly posterior tooth development. Germline loss-of-function and missense variants in PAX9 are among the most frequent monogenic causes of non-syndromic tooth agenesis (selective tooth agenesis 3, STHAG3), usually presenting as autosomal dominant hypodontia or oligodontia with a molar-predominant pattern (Table 9).
Table 9.
Commonly reported pathogenic/likely pathogenic germline PAX9 variants and their clinical consequences.
The first description of PAX9-related oligodontia identified heterozygous truncating variants segregating with familial absence of posterior teeth, establishing the concept of PAX9 haploinsufficiency. Subsequent case series and functional studies have expanded the mutational spectrum to include nonsense, frameshift, initiation codon, and paired-domain missense variants, as well as regulatory changes, with 50–60 distinct pathogenic or likely pathogenic alleles now catalogued.
Systematic reviews and genotype–phenotype analyses consistently show that null (nonsense/frameshift) variants are associated with more severe oligodontia, particularly involving second molars, whereas some missense substitutions within the paired domain allow partial protein function and milder phenotypes. Rare large deletions encompassing PAX9 further support dosage sensitivity. Although most reported cases are non-syndromic, occasional families show co-occurring craniofacial or dental anomalies, suggesting possible interaction with other odontogenic pathways (e.g., MSX1, WNT signaling).
3. Somatic PAX Gene Variations and Functional Consequences
Across human cancers, PAX genes are recurrently altered at the somatic level, but the mode of alteration varies substantially between the nine family members. Rather than classic hotspot missense mutations in all genes, the landscape includes promoter hypermethylation (PAX1, PAX6, PAX9), copy-number gain and overexpression (PAX2, PAX8, PAX9), oncogenic gene fusions (PAX3, PAX7, PAX5, PAX8), and true coding “driver” mutations.
PAX1 behaves primarily as a tumor-suppressor gene in squamous epithelia. Dense promoter CpG island hypermethylation and transcript silencing are frequent in high-grade squamous intraepithelial lesions (HSIL) of the cervix (cervical intraepithelial neoplasia; CIN) and invasive cervical carcinoma [9,12,16], as well as in head-and-neck and oral squamous cell carcinomas, and correlate with higher grade and poorer outcome [13,146,147].
PAX2 is rarely mutated at the coding level in solid tumors, but copy-number gain and strong nuclear expression have been reported in renal cell carcinoma and subsets of ovarian and endometrial carcinomas, where PAX2 acts as a lineage-restricted survival factor and may cooperate with PAX8 [10,148,149,150,151]. In addition, although PAX2 loss is a frequent immunophenotypic abnormality in AH/EIN—including within endometrial polyps, where PAX2 loss occurs in approximately two-thirds of cases (64.8%)—this aberrancy reflects transcriptional silencing rather than coding mutations, underscoring that PAX2 inactivation in endometrial neoplasia may be epigenetic or regulatory rather than mutational in origin [152].
PAX3 and PAX7 are prototypically altered through balanced translocations that fuse their N-terminal paired domain to the C-terminal transactivation domain of FOXO1 (PAX3::FOXO1, PAX7::FOXO1) in alveolar rhabdomyosarcoma [153,154], and to MAML3 (PAX3::MAML3) as well as NCOA1, NCOA2, FOXO1, and WWTR1 (PAX3::NCOA1 associated with rhabdomyoblastic differentiation, PAX3::NCOA2, PAX3::FOXO1, and PAX3::WWTR1) in biphenotypic sinonasal sarcoma [155,156,157,158,159,160].
For PAX4, most pathogenic variants are germline alleles predisposing to MODY9 and type 2 diabetes, discussed in the previous section. PAX5 is one of the most frequently altered genes in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) [161]. Approximately 40% of pediatric BCP-ALL cases harbor PAX5 variants, including point mutations, intragenic deletions, and fusions [17,162]. A single recurrent missense change, p.Pro80Arg (c.239C>G), defines a distinct molecular subtype (“PAX5P80R”) with characteristic gene-expression profile and biallelic PAX5 inactivation [163]. PAX5 is also affected by fusions such as PAX5::JAK2 which exerts a distinct gene expression signature in B-ALL and is exclusively found in the Ph-like subtype [164,165,166].
PAX6 acts as a tumor suppressor in the body. In glioblastoma, non-small lung cancer, and liver cancer, reduced PAX6 expression—often associated with promoter methylation—is linked to higher grade and shorter survival, as demonstrated by in vitro and in vivo studies [15,102,167,168,169]. Somatic PAX6 mutations—mostly truncating or paired-domain missense—have also been reported in colorectal and pancreatic cancers [170,171,172].
PAX8 is a lineage-defining factor in thyroid, renal, and Müllerian carcinomas [173]. Somatically, it is altered by (i) PAX8::PPARγ and related fusions in follicular thyroid carcinoma [174]; (ii) non-coding mutations in distal regulatory elements that enhance PAX8 binding and drive proliferation in ovarian and renal cancer models [175,176,177,178]; (iii) a documented paired-domain missense mutation p.Arg49His (PAX8R49H) in intramucosal gastric carcinoma with lymph-node metastasis [179].
Lastly, PAX9 is frequently altered in squamous carcinomas. In esophageal squamous cell carcinoma, reduced PAX9 expression via promoter methylation or copy-number loss is associated with poor prognosis and reduced radiosensitivity [180,181,182], whereas in lung squamous cell carcinoma PAX9 resides within a 14q13.3 amplicon (with NKX2-1/NKX2-8) and appears to support tumor growth as a lineage-dependency factor [183,184].
4. Epigenetic PAX Gene Modifications
Epigenetic regulation of PAX genes represents a critical mechanism of transcriptional control in both normal development and disease pathogenesis. Unlike genetic mutations that alter DNA sequence, epigenetic modifications (including DNA methylation, histone modifications, and chromatin remodeling) reversibly modulate PAX gene expression and contribute to diverse pathological processes [2].
4.1. DNA Methylation of PAX Gene Promoters
Aberrant promoter hypermethylation is the most extensively characterized epigenetic alteration affecting PAX genes in human cancer. PAX1 promoter methylation occurs frequently in CIN3 and invasive cervical carcinoma, where specific CpG sites (notably cg16767801) serve as biomarkers for disease progression and screening triage in high-risk HPV-positive women [14]. Similarly, PAX1 methylation is observed in oral and head-and-neck squamous cell carcinomas, correlating with higher tumor grade and poorer outcomes [13].
PAX2 exhibits context-dependent epigenetic regulation. In endometrial cancer, a distinct mechanism involving replacement of active chromatin marks (H3K27ac, H3K4me3) with repressive marks (H3K27me3) silences PAX2 expression in more than 80% of cases, representing a novel “third mechanism” of tumor suppressor inactivation beyond mutation and promoter methylation [185]. Conversely, in some endometrial cancers, PAX2 promoter hypermethylation paradoxically promotes PAX2 expression by silencing the repressive transcription factor MZF1, illustrating the complexity of epigenetic regulation [186,187]. PAX2 methylation patterns also distinguish subtypes of renal cell carcinoma, with differential methylation observed in clear cell, papillary, and oncocytic variants [188].
PAX5 promoter methylation of both alpha and beta isoforms occurs in approximately 65% of breast and lung cancers, with dense methylation correlating with transcriptional silencing [189]. PAX5β encodes B-cell-specific activator protein (BSAP), which directly regulates CD19 (a negative regulator of cell growth) suggesting that PAX5 methylation contributes to neoplastic development through loss of growth control [189,190]. In non-small cell lung cancer, PAX5 methylation is associated with silencing and loss of its tumor-suppressor functions, including inhibition of β-catenin signaling and upregulation of GADD45G [190].
PAX6 promoter hypermethylation has been documented in glioblastoma, non-small cell lung cancer, and hepatocellular carcinoma, where it correlates with reduced expression, higher tumor grade, and shorter survival [15]. Experimental restoration of PAX6 expression through demethylation inhibits tumor growth and metastasis, confirming its tumor-suppressor role [15].
PAX9 methylation is frequent in esophageal squamous cell carcinoma, where it associates with poor prognosis and reduced radiosensitivity [191].
4.2. Histone Modification and Chromatin Remodeling
Beyond DNA methylation, PAX genes recruit chromatin-remodeling complexes that introduce activating or repressive histone marks [2]. PAX2 silencing in endometrial cancer involves spreading of repressive H3K27me3 marks in a “pearl necklace” pattern dictated by three-dimensional genome organization and cohesin loops, preventing transcriptional dysregulation of neighboring genes [185]. PAX7 exhibits pioneer transcription factor activity, uniquely capable of “opening” closed chromatin to initiate developmental programs [192]. In parathyroid adenomas, reduced histone H3K9 acetylation at the PAX1 promoter contributes to transcriptional downregulation [193].
4.3. Regulatory Feedback Loops
Epigenetic regulation of PAX genes often involves complex feedback mechanisms. In breast cancer, a PAX5-miR-142-DNMT1/ZEB1 negative feedback loop regulates tumor progression. PAX5 positively regulates miR-142-5p/3p, which in turn targets DNMT1 and ZEB1; these factors then promote PAX5 promoter methylation, creating a self-reinforcing circuit [194].
4.4. Clinical and Therapeutic Implications
The reversibility of epigenetic modifications makes them attractive therapeutic targets. Treatment with demethylating agents (5-aza-2′-deoxycytidine) restores PAX gene expression in multiple cancer models, suggesting potential for epigenetic therapies [187,189,190]. Furthermore, PAX gene methylation status shows promise as a diagnostic and prognostic biomarker, particularly PAX1 methylation in cervical cancer screening and PAX2 methylation patterns in endometrial and renal neoplasms.
5. Conclusions
The paired box (PAX) gene family occupies a central position at the intersection of developmental biology and human disease. Through highly conserved, lineage-defining transcriptional programs, PAX proteins orchestrate organogenesis, cellular differentiation, and tissue patterning across multiple systems, including the nervous, musculoskeletal, endocrine, immune, renal, and sensory organs. Germline alterations of these tightly regulated developmental pathways give rise to a broad spectrum of congenital and inherited disorders whose phenotypes closely mirror the embryologic roles of each PAX gene. The resulting conditions—ranging from aniridia, renal coloboma syndrome, and Waardenburg syndrome to otofaciocervical syndrome, congenital immunodeficiency, monogenic diabetes, and tooth agenesis—underscore the profound and pleiotropic consequences of PAX dysregulation.
From a pathology perspective, understanding germline PAX gene mutations provides critical insights into genotype–phenotype correlations, disease pathogenesis, and developmental origins of human pathology. Although PAX proteins are widely recognized for their diagnostic utility in surgical pathology through immunohistochemistry and molecular testing, their roles as drivers of congenital disease and tissue-specific vulnerability are equally important. Increasing recognition of variable expressivity, incomplete penetrance, and overlapping phenotypes further emphasizes the need for integrated clinicopathologic and genomic approaches when evaluating patients with suspected PAX-related disorders.
Recent advances have illuminated the multifaceted roles of PAX genes beyond classical germline disorders. Epigenetic silencing through promoter methylation and histone modifications represents a prevalent mechanism of PAX gene inactivation in cancer, offering reversible therapeutic targets [185,189]. Somatic PAX gene fusions (particularly PAX3/7::FOXO1 in rhabdomyosarcoma and PAX5::JAK2 in B-cell leukemia) define distinct molecular subtypes with specific therapeutic vulnerabilities [195]. Furthermore, the recognition that PAX proteins maintain adult stem cell populations and regulate tissue regeneration suggests broader roles in aging, tissue repair, and regenerative medicine [196].
Beyond development, studies suggest that select PAX genes remain active in postnatal tissue homeostasis, immune regulation, and regeneration, raising important questions about their broader biological functions and long-term disease implications. As genomic sequencing becomes increasingly embedded in clinical practice, systematic characterization of germline PAX variants will refine diagnostic precision, improve genetic counseling, and inform surveillance strategies. Continued integration of developmental genetics with human pathology will be essential to fully elucidate the lifelong impact of PAX gene dysregulation and to translate these insights into improved patient care.
Author Contributions
Conceptualization, H.F.B.; methodology, V.L.G., S.W., S.O., D.P., D.J., V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; software, V.L.G., S.W., S.O., D.P., D.J., V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; validation, H.F.B.; investigation, V.L.G., S.W., S.O., D.P., D.J., V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; resources, V.L.G., S.W., S.O., D.P., D.J., V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; data curation, V.L.G., S.W., S.O., D.P., D.J., V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; writing—original draft preparation, V.L.G., S.W., S.O., D.P., D.J. and H.F.B.; writing—review and editing, V.F., A.A., M.B., W.A.-K., T.K.E. and H.F.B.; visualization, W.A.-K. and H.F.B.; supervision, W.A.-K. and H.F.B.; project administration, H.F.B.; funding acquisition, H.F.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The figures were created with BioRender.com. All rights and ownership of BioRender content are reserved by BioRender. BioRender content included in the completed graphic is not licensed for any commercial uses beyond publication in a journal. For any commercial use of these figures, users may, if allowed, recreate it in BioRender under an Industry BioRender Plan.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Thompson, B.; Davidson, E.A.; Liu, W.; Nebert, D.W.; Bruford, E.A.; Zhao, H.; Dermitzakis, E.T.; Thompson, D.C.; Vasiliou, V. Overview of PAX gene family: Analysis of human tissue-specific variant expression and involvement in human disease. Hum. Genet. 2021, 140, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Mayran, A.; Pelletier, A.; Drouin, J. Pax factors in transcription and epigenetic remodelling. Semin. Cell Dev. Biol. 2015, 44, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Dahl, E.; Koseki, H.; Balling, R. Pax genes and organogenesis. BioEssays 1997, 19, 755–765. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, W.-H.; Krupinski, J.; Kumar, S.; Slevin, M.; Kumar, P. Pax genes in embryogenesis and oncogenesis. J. Cell. Mol. Med. 2008, 12, 2281–2294. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Kong, X.; Jia, Y.; Jia, Y.; Ou, W.; Dai, C.; Li, G.; Gao, R. An overview of PAX1: Expression, function and regulation in development and diseases. Front. Cell Dev. Biol. 2022, 10, 1051102. [Google Scholar] [CrossRef]
- Bhol, C.S.; Patil, S.; Sahu, B.B.; Patra, S.K.; Bhutia, S.K. The clinical significance and correlative signaling pathways of paired box gene 9 in development and carcinogenesis. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188561. [Google Scholar] [CrossRef]
- Shaw, T.; Barr, F.G.; Üren, A. The PAX Genes: Roles in Development, Cancer, and Other Diseases. Cancers 2024, 16, 1022. [Google Scholar] [CrossRef]
- Monsoro-Burq, A.H. PAX transcription factors in neural crest development. Semin. Cell Dev. Biol. 2015, 44, 87–96. [Google Scholar] [CrossRef]
- Huang, M.; Wang, T.; Li, M.; Qin, M.; Deng, S.; Chen, D. Evaluating PAX1 methylation for cervical cancer screening triage in non-16/18 hrHPV-positive women. BMC Cancer 2024, 24, 913. [Google Scholar] [CrossRef]
- Li, L.; Hossain, S.M.; Eccles, M.R. The Role of the PAX Genes in Renal Cell Carcinoma. Int. J. Mol. Sci. 2024, 25, 6730. [Google Scholar] [CrossRef]
- Chase, C.L.; Accardo, M.-L.; Greve, V.; Ames, E.G.; Quinonez, S.C.; Scott, A.; Hipp, L.; Damon, J.; Uhlmann, W.R.; Broome, D.T.; et al. Genetics Evaluation Outcomes From an Academic Multidisciplinary Atypical Diabetes Program. J. Endocr. Soc. 2025, 9, bvaf091. [Google Scholar] [CrossRef]
- Yin, M.; Dai, J.; Sun, R.; Wang, Y. Diagnostic performance of PAX1 methylation as a biomarker for cervical lesions: A clinical study and meta-analysis. Ann. Med. 2025, 57, 2583319. [Google Scholar] [CrossRef]
- Mao, Y.Q.; Sun, R.; Liu, S.; Zhang, W.B.; Yu, Y.; Jia, L.F.; Yu, G.Y.; Peng, X. Distribution of PAX1 and ZNF582 Hypermethylation in the Oral Exfoliated Cells of Oral Squamous Cell Carcinoma. Clin. Med. Insights Oncol. 2025, 19, 11795549251335172. [Google Scholar] [CrossRef] [PubMed]
- Bowden, S.J.; Bodinier, B.; Paraskevaidi, M.; Kalliala, I.; Nasioutziki, M.; Ellis, L.B.; Zuehlke, R.C.; Flanagan, J.M.; Kyrgiou, M.; Chadeau-Hyam, M. DNA methylation signatures of cervical pre-invasive and invasive disease: An epigenome-wide association study. Int. J. Cancer 2025, 157, 305–316. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Chen, R.-Y.; Wu, T.-H.; Chang, S.-Y.; Hsieh, T.-Y.; Shih, Y.-L.; Lin, Y.-W. Promoter hypermethylation-mediated downregulation of PAX6 promotes tumor growth and metastasis during the progression of liver cancer. Clin. Epigenet. 2024, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Bhavya; Rajaram, S.; Gupta, B.; Banerjee, B.D.; Arora, V.K.; Thakur, G.; Jain, S. PAX1 Methylation Status in Cervical Scrapes as Novel Diagnostic Biomarker in CIN 2/3 and Invasive Squamous Cell Carcinoma. J. Obs. Gynaecol. India 2022, 72, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Kaiser, F.M.P.; Gruenbacher, S.; Oyaga, M.R.; Nio, E.; Jaritz, M.; Sun, Q.; van der Zwaag, W.; Kreidl, E.; Zopf, L.M.; Dalm, V.; et al. Correction: Biallelic PAX5 mutations cause hypogammaglobulinemia, sensorimotor deficits, and autism spectrum disorder. J. Exp. Med. 2023, 220, e2022049812012022c. [Google Scholar] [CrossRef]
- Hu, X.; Lin, W.; Luo, Z.; Zhong, Y.; Xiao, X.; Tang, R. Frameshift Mutation in PAX2 Related to Focal Segmental Glomerular Sclerosis: A Case Report and Literature Review. Mol. Genet. Genom. Med. 2024, 12, e70006. [Google Scholar] [CrossRef]
- Kim, J.H.; Ahn, Y.H.; Jang, Y.; Park, E.; Lee, H.; Kim, S.H.; Song, J.Y.; Han, K.H.; Jung, J.; Lee, J.H.; et al. Genotype of PAX2-related disorders correlates with kidney and ocular manifestations. Eur. J. Hum. Genet. 2025, 33, 441–450. [Google Scholar] [CrossRef]
- Giampietro, P.F.; Raggio, C.L.; Reynolds, C.E.; Shukla, S.K.; McPherson, E.; Ghebranious, N.; Jacobsen, F.S.; Kumar, V.; Faciszewski, T.; Pauli, R.M.; et al. An analysis of PAX1 in the development of vertebral malformations. Clin. Genet. 2005, 68, 448–453. [Google Scholar] [CrossRef] [PubMed]
- McGaughran, J.M.; Oates, A.; Donnai, D.; Read, A.P.; Tassabehji, M. Mutations in PAX1 may be associated with Klippel-Feil syndrome. Eur. J. Hum. Genet. 2003, 11, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Bannykh, S.I.; Emery, S.C.; Gerber, J.K.; Jones, K.L.; Benirschke, K.; Masliah, E. Aberrant Pax1 and Pax9 expression in Jarcho-Levin syndrome: Report of two Caucasian siblings and literature review. Am. J. Med. Genet. A 2003, 120, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Paganini, I.; Sestini, R.; Capone, G.L.; Putignano, A.L.; Contini, E.; Giotti, I.; Gensini, F.; Marozza, A.; Barilaro, A.; Porfirio, B.; et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin. Genet. 2017, 92, 664–668. [Google Scholar] [CrossRef]
- Patil, S.J.; Das Bhowmik, A.; Bhat, V.; Satidevi Vineeth, V.; Vasudevamurthy, R.; Dalal, A. Autosomal recessive otofaciocervical syndrome type 2 with novel homozygous small insertion in PAX1 gene. Am. J. Med. Genet. A 2018, 176, 1200–1206. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Urrutia, R.; Franco, L.M.; Giliani, S.; Zhang, K.; Alazami, A.M.; Dobbs, A.K.; Masneri, S.; Joshi, A.; Otaizo-Carrasquero, F.; et al. PAX1 is essential for development and function of the human thymus. Sci. Immunol. 2020, 5, eaax1036. [Google Scholar] [CrossRef]
- Elbagoury, N.M.; Abdel-Aleem, A.F.; Sharaf-Eldin, W.E.; Ashaat, E.A.; Esswai, M.L. A Novel Truncating Mutation in PAX1 Gene Causes Otofaciocervical Syndrome Without Immunodeficiency. J. Mol. Neurosci. 2023, 73, 976–982. [Google Scholar] [CrossRef]
- Carter, S.; Fellows, B.J.; Gibson, K.; Bicknell, L.S. Extending the PAX1 spectrum: A dominantly inherited variant causes oculo-auriculo-vertebral syndrome. Eur. J. Hum. Genet. 2022, 30, 1178–1181. [Google Scholar] [CrossRef]
- Amiel, J.; Audollent, S.; Joly, D.; Dureau, P.; Salomon, R.; Tellier, A.L.; Augé, J.; Bouissou, F.; Antignac, C.; Gubler, M.C.; et al. PAX2 mutations in renal-coloboma syndrome: Mutational hotspot and germline mosaicism. Eur. J. Hum. Genet. 2000, 8, 820–826. [Google Scholar] [CrossRef]
- Eccles, M.R.; Schimmenti, L.A. Renal-coloboma syndrome: A multi-system developmental disorder caused by PAX2 mutations. Clin. Genet. 1999, 56, 1–9. [Google Scholar] [CrossRef]
- Bower, M.; Salomon, R.; Allanson, J.; Antignac, C.; Benedicenti, F.; Benetti, E.; Binenbaum, G.; Jensen, U.B.; Cochat, P.; DeCramer, S.; et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 2012, 33, 457–466. [Google Scholar] [CrossRef]
- Schimmenti, L.A. Renal coloboma syndrome. Eur. J. Hum. Genet. 2011, 19, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhang, Y.; Xiao, H.; Yao, Y.; Liu, X.; Su, B.; Zhang, H.; Xu, K.; Wang, S.; Wang, F.; et al. Diverse phenotypes in children with PAX2-related disorder. Mol. Genet. Genom. Med. 2019, 7, e701. [Google Scholar] [CrossRef]
- Schimmenti, L.A.; Cunliffe, H.E.; McNoe, L.A.; Ward, T.A.; French, M.C.; Shim, H.H.; Zhang, Y.H.; Proesmans, W.; Leys, A.; Byerly, K.A.; et al. Further delineation of renal-coloboma syndrome in patients with extreme variability of phenotype and identical PAX2 mutations. Am. J. Hum. Genet. 1997, 60, 869–878. [Google Scholar] [PubMed]
- Schimmenti, L.A.; Manligas, G.S.; Sieving, P.A. Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: Further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet. 2003, 24, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.-Y.; Shi, Y.-Q.; Zhong, C.; Yang, Q.; Zhang, G.; Yang, H.; Wu, D.; Chen, Y.; Li, Q.; Wang, M. Detection of De Novo PAX2 Variants and Phenotypes in Chinese Population: A Single-Center Study. Front. Genet. 2022, 13, 799562. [Google Scholar] [CrossRef]
- Barua, M.; Stellacci, E.; Stella, L.; Weins, A.; Genovese, G.; Muto, V.; Caputo, V.; Toka, H.R.; Charoonratana, V.T.; Tartaglia, M.; et al. Mutations in PAX2 associate with adult-onset FSGS. J. Am. Soc. Nephrol. 2014, 25, 1942–1953. [Google Scholar] [CrossRef]
- Rachwani Anil, R.; Rocha-de-Lossada, C.; Ayala, C.H.; Contreras, M.E. A new mutation in the PAX2 gene in a Papillorenal Syndrome patient. Am. J. Ophthalmol. Case Rep. 2019, 16, 100563. [Google Scholar] [CrossRef]
- Chang, Y.M.; Chen, C.C.; Lee, N.C.; Sung, J.M.; Chou, Y.Y.; Chiou, Y.Y. PAX2 Mutation-Related Renal Hypodysplasia: Review of the Literature and Three Case Reports. Front. Pediatr. 2021, 9, 765929. [Google Scholar] [CrossRef]
- Martinovic-Bouriel, J.; Benachi, A.; Bonnière, M.; Brahimi, N.; Esculpavit, C.; Morichon, N.; Vekemans, M.; Antignac, C.; Salomon, R.; Encha-Razavi, F.; et al. PAX2 mutations in fetal renal hypodysplasia. Am. J. Med. Genet. Part A 2010, 152, 830–835. [Google Scholar] [CrossRef]
- Hoefele, J.; Gabert, M.; Heinrich, U.; Benz, K.; Rompel, O.; Rost, I.; Klein, H.G.; Kunstmann, E. A novel interstitial deletion of 10q24.2q24.32 in a patient with renal coloboma syndrome. Eur. J. Med. Genet. 2012, 55, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Raca, G.; Jackson, C.A.; Kucinskas, L.; Warman, B.; Shieh, J.T.; Schneider, A.; Bardakjian, T.M.; Schimmenti, L.A. Array comparative genomic hybridization analysis in patients with anophthalmia, microphthalmia, and coloboma. Genet. Med. 2011, 13, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Artifoni, L.; Salviati, L.; Pinello, L.; Perrotta, S.; Zuffardi, O.; Zacchello, G.; Murer, L. Renal hypoplasia without optic coloboma associated with PAX2 gene deletion. Nephrol. Dial. Transpl. 2007, 22, 2076–2078. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Furuichi, K.; Higashide, T.; Sakurai, M.; Hashimoto, S.; Shinozaki, Y.; Hara, A.; Iwata, Y.; Sakai, N.; Sugiyama, K.; et al. Association of PAX2 and Other Gene Mutations with the Clinical Manifestations of Renal Coloboma Syndrome. PLoS ONE 2015, 10, e0142843. [Google Scholar] [CrossRef]
- Hu, S.; Gan, H.; Yang, F. Significance analysis of PAX8 expression in endometrial carcinoma. Medicine 2022, 101, e31159. [Google Scholar] [CrossRef]
- Oh, S.H.; Keum, C.; Her, M.; Chung, W.Y.; Kim, Y.H.; Kim, T. A Novel Missense Mutation (L44V) of PAX2 Associated with Adult-Onset End-Stage Renal Disease and No Syndromic Features. Ann. Clin. Lab. Sci. 2020, 50, 687–690. [Google Scholar]
- Forero-Delgadillo, J.M.; Ochoa, V.; Duque, N.; Restrepo, J.M.; Londoño, H.; Nastasi-Catanese, J.A.; Pachajoa, H. New PAX2 Mutation Associated with Polycystic Kidney Disease: A Case Report. Clin. Med. Insights Pediatr. 2021, 15, 1179556521992354. [Google Scholar] [CrossRef]
- Longaretti, L.; Trionfini, P.; Brizi, V.; Xinaris, C.; Mele, C.; Breno, M.; Romano, E.; Giampietro, R.; Remuzzi, G.; Benigni, A.; et al. Unravelling the Role of PAX2 Mutation in Human Focal Segmental Glomerulosclerosis. Biomedicines 2021, 9, 1808. [Google Scholar] [CrossRef]
- Muntean, C.; Chirtes, C.; Baczoni, B.; Banescu, C. PAX2 Gene Mutation in Pediatric Renal Disorders-A Narrative Review. Int. J. Mol. Sci. 2023, 24, 12737. [Google Scholar] [CrossRef]
- Huang, S.; Song, J.; He, C.; Cai, X.; Yuan, K.; Mei, L.; Feng, Y. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 2022, 29, 479–497. [Google Scholar] [CrossRef]
- Thongpradit, S.; Jinawath, N.; Javed, A.; Jensen, L.T.; Chunsuwan, I.; Rojnueangnit, K.; Tim-Aroon, T.; Lertsukprasert, K.; Shiao, M.S.; Sirachainan, N.; et al. Novel SOX10 Mutations in Waardenburg Syndrome: Functional Characterization and Genotype-Phenotype Analysis. Front. Genet. 2020, 11, 589784. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jiang, L.; Xie, Z.; Mei, L.; He, C.; Hu, Z.; Xia, K.; Feng, Y. Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem. Biophys. Res. Commun. 2010, 397, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Powell, S.K.; Plummer, R.S.; Young, K.P.; Ruggeri, B.A. PAX genes: Roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 2007, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Nickerson, S.J.; Al-Dahmash, S.; Shields, J.A. Waardenburg Syndrome: Iris and Choroidal Hypopigmentation: Findings on Anterior and Posterior Segment Imaging. JAMA Ophthalmol. 2013, 131, 1167–1173. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Xiao, X.; Wang, P.; Guo, X.; Zhang, Q. PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol. Vis. 2010, 16, 1146–1153. [Google Scholar]
- Wang, G.; Li, X.; Gao, X.; Su, Y.; Han, M.; Gao, B.; Guo, C.; Kang, D.; Huang, S.; Yuan, Y.; et al. Analysis of genotype-phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum. Genet. 2022, 141, 839–852. [Google Scholar] [CrossRef]
- Donnai, D.; Read, A.P. How clinicians add to knowledge of development. Lancet 2003, 362, 477–484. [Google Scholar] [CrossRef]
- Li, C.G.; Eccles, M.R. PAX Genes in Cancer; Friends or Foes? Front. Genet. 2012, 3, 6. [Google Scholar] [CrossRef]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef]
- Jang, M.-A.; Lee, T.; Lee, J.; Cho, E.-H.; Ki, C.-S. Identification of a novel de novo variant in the PAX3 gene in Waardenburg syndrome by diagnostic exome sequencing: The first molecular diagnosis in Korea. Ann. Lab. Med. 2015, 35, 362–365. [Google Scholar] [CrossRef][Green Version]
- Asher, J.H., Jr.; Sommer, A.; Morell, R.; Friedman, T.B. Missense mutation in the paired domain of PAX3 causes craniofacial-deafness-hand syndrome. Hum. Mutat. 1996, 7, 30–35. [Google Scholar] [CrossRef]
- Yang, S.Z.; Cao, J.Y.; Zhang, R.N.; Liu, L.X.; Liu, X.; Zhang, X.; Kang, D.Y.; Li, M.; Han, D.Y.; Yuan, H.J.; et al. Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients. Chin. Med. J. 2007, 120, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Zafiri, J.; Papageorgiou, E.; Pampanos, A.; Valla, O.; Sotiriou, S.; Papaioannou, G.K.; Psarris, A.; Siomou, E.; Manolakos, E. Association of Waardenburg syndrome with a new mutation in the PAX3 gene: A case report and literature review. Biomed. Rep. 2025, 23, 189. [Google Scholar] [CrossRef] [PubMed]
- Hazan, F.; Ozturk, A.T.; Adibelli, H.; Unal, N.; Tukun, A. A novel missense mutation of the paired box 3 gene in a Turkish family with Waardenburg syndrome type 1. Mol. Vis. 2013, 19, 196–202. [Google Scholar]
- Yoshida, Y.; Doi, R.; Adachi, K.; Nanba, E.; Kodani, I.; Ryoke, K. A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1. Hum. Genome Var. 2016, 3, 16005. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Xu, B.; Lin, D.; Wang, Y.; Bian, P. A novel frameshift variant of PAX3 in a Chinese Yugur family with Waardenburg syndrome type 1. Front. Genet. 2025, 16, 1679351. [Google Scholar] [CrossRef]
- Tekin, M.; Bodurtha, J.N.; Nance, W.E.; Pandya, A. Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: A simple variant or a true syndrome? Clin. Genet. 2001, 60, 301–304. [Google Scholar] [CrossRef]
- De Saxe, M.; Kromberg, J.G.; Jenkins, T. Waardenburg syndrome in South Africa. Part I. An evaluation of the clinical findings in 11 families. S. Afr. Med. J. 1984, 66, 256–261. [Google Scholar]
- Tassabehji, M.; Newton, V.E.; Liu, X.Z.; Brady, A.; Donnai, D.; Krajewska-Walasek, M.; Murday, V.; Norman, A.; Obersztyn, E.; Reardon, W.; et al. The mutational spectrum in Waardenburg syndrome. Hum. Mol. Genet. 1995, 4, 2131–2137. [Google Scholar] [CrossRef]
- Niu, Z.; Li, J.; Tang, F.; Sun, J.; Wang, X.; Jiang, L.; Mei, L.; Chen, H.; Liu, Y.; Cai, X.; et al. Identification and functional analysis of a novel mutation in the PAX3 gene associated with Waardenburg syndrome type I. Gene 2018, 642, 362–366. [Google Scholar] [CrossRef]
- Qin, W.; Shu, A.; Qian, X.; Gao, J.; Xing, Q.; Zhang, J.; Zheng, Y.; Li, X.; Li, S.; Feng, G.; et al. A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome. Mol. Vis. 2006, 12, 1001–1008. [Google Scholar]
- Jo, W.; Endo, M.; Ishizu, K.; Nakamura, A.; Tajima, T. A novel PAX4 mutation in a Japanese patient with maturity-onset diabetes of the young. Tohoku J. Exp. Med. 2011, 223, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kooptiwut, S.; Plengvidhya, N.; Chukijrungroat, T.; Sujjitjoon, J.; Semprasert, N.; Furuta, H.; Yenchitsomanus, P.T. Defective PAX4 R192H transcriptional repressor activities associated with maturity onset diabetes of the young and early onset-age of type 2 diabetes. J. Diabetes Complicat. 2012, 26, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Plengvidhya, N.; Kooptiwut, S.; Songtawee, N.; Doi, A.; Furuta, H.; Nishi, M.; Nanjo, K.; Tantibhedhyangkul, W.; Boonyasrisawat, W.; Yenchitsomanus, P.T.; et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J. Clin. Endocrinol. Metab. 2007, 92, 2821–2826. [Google Scholar] [CrossRef] [PubMed]
- Sujjitjoon, J.; Kooptiwut, S.; Chongjaroen, N.; Semprasert, N.; Hanchang, W.; Chanprasert, K.; Tangjittipokin, W.; Yenchitsomanus, P.T.; Plengvidhya, N. PAX4 R192H and P321H polymorphisms in type 2 diabetes and their functional defects. J. Hum. Genet. 2016, 61, 943–949. [Google Scholar] [CrossRef]
- Sujjitjoon, J.; Kooptiwut, S.; Chongjaroen, N.; Tangjittipokin, W.; Plengvidhya, N.; Yenchitsomanus, P.T. Aberrant mRNA splicing of paired box 4 (PAX4) IVS7-1G>A mutation causing maturity-onset diabetes of the young, type 9. Acta Diabetol. 2016, 53, 205–216. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, C.; Yang, W.; Piao, Y.; Ren, L.; Sang, Y. C.487C>T mutation in PAX4 gene causes MODY9: A case report and literature review. Medicine 2022, 101, e32461. [Google Scholar] [CrossRef]
- Shimajiri, Y.; Sanke, T.; Furuta, H.; Hanabusa, T.; Nakagawa, T.; Fujitani, Y.; Kajimoto, Y.; Takasu, N.; Nanjo, K. A missense mutation of Pax4 gene (R121W) is associated with type 2 diabetes in Japanese. Diabetes 2001, 50, 2864–2869. [Google Scholar] [CrossRef]
- Afsar, T.; Waqas, A.; Nayab, A.; Abbas, S.; Mahmood, A.; Umair, M.; Razak, S. A Novel Variant in the PAX4 Gene Causes Maturity-Onset Diabetes of the Young (MODY), Type IX with Neurodevelopmental Disorder. J. Disabil. Res. 2023, 2, 18–24. [Google Scholar] [CrossRef]
- Shah, S.; Schrader, K.A.; Waanders, E.; Timms, A.E.; Vijai, J.; Miething, C.; Wechsler, J.; Yang, J.; Hayes, J.; Klein, R.J.; et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1226–1231. [Google Scholar] [CrossRef]
- Auer, F.; Rüschendorf, F.; Gombert, M.; Husemann, P.; Ginzel, S.; Izraeli, S.; Harit, M.; Weintraub, M.; Weinstein, O.Y.; Lerer, I.; et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 2014, 28, 1136–1138. [Google Scholar] [CrossRef] [PubMed]
- Escudero, A.; Takagi, M.; Auer, F.; Friedrich, U.A.; Miyamoto, S.; Ogawa, A.; Imai, K.; Pascual, B.; Vela, M.; Stepensky, P.; et al. Clinical and immunophenotypic characteristics of familial leukemia predisposition caused by PAX5 germline variants. Leukemia 2022, 36, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Yazdanparast, S.; Khatami, S.R.; Galehdari, H.; Jaseb, K. One missense mutation in exon 2 of the PAX5 gene in Iran. Genet. Mol. Res. 2015, 14, 17768–17775. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Jamrog, L.A.; Fregona, V.; Hamelle, C.; Fenwarth, L.; Lejeune, S.; Helevaut, N.; Geffroy, S.; Caillault, A.; Marceau-Renaut, A.; et al. Germline PAX5 mutation predisposes to familial B-cell precursor acute lymphoblastic leukemia. Blood 2021, 137, 1424–1428. [Google Scholar] [CrossRef]
- van Engelen, N.; Roest, M.; van Dijk, F.; Sonneveld, E.; Bladergroen, R.; van Reijmersdal, S.V.; van der Velden, V.H.J.; Hoogeveen, P.G.; Kors, W.A.; Waanders, E.; et al. A novel germline PAX5 single exon deletion in a pediatric patient with precursor B-cell leukemia. Leukemia 2023, 37, 1908–1911. [Google Scholar] [CrossRef]
- Stasevich, I.; Inglott, S.; Austin, N.; Chatters, S.; Chalker, J.; Addy, D.; Dryden, C.; Ancliff, P.; Ford, A.; Williams, O.; et al. PAX5 alterations in genetically unclassified childhood Precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2015, 171, 263–272. [Google Scholar] [CrossRef]
- Moosajee, M.; Hingorani, M.; Moore, A.T. PAX6-Related Aniridia. In GeneReviews®; Adam, M.P., Bick, S., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hanson, I.M.; Fletcher, J.M.; Jordan, T.; Brown, A.; Taylor, D.; Adams, R.J.; Punnett, H.H.; van Heyningen, V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat. Genet. 1994, 6, 168–173. [Google Scholar] [CrossRef]
- Grønskov, K.; Rosenberg, T.; Sand, A.; Brøndum-Nielsen, K. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur. J. Hum. Genet. 1999, 7, 274–286. [Google Scholar] [CrossRef]
- Tzoulaki, I.; White, I.M.; Hanson, I.M. PAX6 mutations: Genotype-phenotype correlations. BMC Genet. 2005, 6, 27. [Google Scholar] [CrossRef]
- Robinson, D.O.; Howarth, R.J.; Williamson, K.A.; van Heyningen, V.; Beal, S.J.; Crolla, J.A. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am. J. Med. Genet. A 2008, 146, 558–569. [Google Scholar] [CrossRef]
- Abouzeid, H.; Youssef, M.A.; ElShakankiri, N.; Hauser, P.; Munier, F.L.; Schorderet, D.F. PAX6 aniridia and interhemispheric brain anomalies. Mol. Vis. 2009, 15, 2074–2083. [Google Scholar]
- Hingorani, M.; Williamson, K.A.; Moore, A.T.; van Heyningen, V. Detailed Ophthalmologic Evaluation of 43 Individuals with PAX6 Mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, M.S.; Chae, H.; Kim, Y.; Kim, M. Molecular analysis of the PAX6 gene for congenital aniridia in the Korean population: Identification of four novel mutations. Mol. Vis. 2012, 18, 488–494. [Google Scholar] [PubMed]
- Wawrocka, A.; Sikora, A.; Kuszel, L.; Krawczynski, M.R. 11p13 deletions can be more frequent than the PAX6 gene point mutations in Polish patients with aniridia. J. Appl. Genet. 2013, 54, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Thurm, A.; Golden Williams, C.; Joseph, L.A.; Zein, W.M.; Brooks, B.P.; Butman, J.A.; Brady, S.M.; Fuhr, S.R.; Hicks, M.D.; et al. Association of brain-derived neurotrophic factor (BDNF) haploinsufficiency with lower adaptive behaviour and reduced cognitive functioning in WAGR/11p13 deletion syndrome. Cortex 2013, 49, 2700–2710. [Google Scholar] [CrossRef]
- Chograni, M.; Derouiche, K.; Chaabouni, M.; Lariani, I.; Bouhamed, H.C. Molecular analysis of the PAX6 gene for aniridia and congenital cataracts in Tunisian families. Hum. Genome Var. 2014, 1, 14008. [Google Scholar] [CrossRef]
- Lin, Y.; Gao, H.; Zhu, Y.; Chen, C.; Li, T.; Liu, B.; Lyu, C.; Huang, Y.; Li, H.; Wu, Q.; et al. Two Paired Box 6 mutations identified in Chinese patients with classic congenital aniridia and cataract. Mol. Med. Rep. 2018, 18, 4439–4445. [Google Scholar] [CrossRef]
- Lima Cunha, D.; Arno, G.; Corton, M.; Moosajee, M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes 2019, 10, 1050. [Google Scholar] [CrossRef]
- Guo, R.; Zhang, X.; Liu, A.; Ji, J.; Liu, W. Novel clinical presentation and PAX6 mutation in families with congenital aniridia. Front. Med. 2022, 9, 1042588. [Google Scholar] [CrossRef]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous PAX6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA–protein interaction. Genet. Med. 2020, 22, 598–609. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Tan, F.; Hess, K.R.; Yung, W.K. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: Relationship to tumor grade and survival. Clin. Cancer Res. 2003, 9, 3369–3375. [Google Scholar] [PubMed]
- Schmidt-Sidor, B.; Szymańska, K.; Williamson, K.; van Heyningen, V.; Roszkowski, T.; Wierzba-Bobrowicz, T.; Zaremba, J. Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 2009, 47, 371–382. [Google Scholar]
- Hanson, I.M. PAX6 and congenital eye malformations. Pediatr. Res. 2003, 54, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Hayakawa, M.; Kanai, A.; Yamada, M. Missense Mutation in the Alternative Splice Region of the PAX6 Gene in Eye Anomalies. Am. J. Hum. Genet. 1999, 65, 656–663. [Google Scholar] [CrossRef]
- Ganassi, M.; Zammit, P.S. Involvement of muscle satellite cell dysfunction in neuromuscular disorders: Expanding the portfolio of satellite cell-opathies. Eur. J. Transl. Myol. 2022, 32, 10064. [Google Scholar] [CrossRef]
- Proskorovski-Ohayon, R.; Kadir, R.; Michalowski, A.; Flusser, H.; Perez, Y.; Hershkovitz, E.; Sivan, S.; Birk, O.S. PAX7 mutation in a syndrome of failure to thrive, hypotonia, and global neurodevelopmental delay. Hum. Mutat. 2017, 38, 1671–1683. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Mucha, B.E.; Hengel, H.; Orfi, Z.; Makowski, C.; Dort, J.; D’Anjou, G.; Nguyen, T.T.M.; Buchert, R.; Juenger, H.; et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet. Med. 2019, 21, 2521–2531. [Google Scholar] [CrossRef]
- Haliloğlu, G.; Donkervoort, S.; Yıldız, S.; Potticary, A.; Hu, Y.; Pais, L.; Carlier, R.Y.; Amthor, H.; Aydıngöz, Ü.; Bönnemann, C.G. Distinct whole-body muscle MRI imaging patterns in PAX7-congenital myopathy: A case report. J. Neuromuscul. Dis. 2024, 11, 1276–1282. [Google Scholar] [CrossRef]
- Onnée, M.; Malfatti, E. The widening genetic and myopathologic spectrum of congenital myopathies (CMYOs): A narrative review. Neuromuscul. Disord. 2025, 49, 105338. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Zhao, S.; Zheng, Z.; Wang, Y.; Qiu, G.; Wu, Z.; Wu, N.; Zhang, T.J.; Cai, S. The identification of PAX7 variants and a potential role of muscle development dysfunction in congenital scoliosis. Cell Regen. 2022, 11, 16. [Google Scholar] [CrossRef]
- Macchia, P.E.; Lapi, P.; Krude, H.; Pirro, M.T.; Missero, C.; Chiovato, L.; Souabni, A.; Baserga, M.; Tassi, V.; Pinchera, A.; et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat. Genet. 1998, 19, 83–86. [Google Scholar] [CrossRef]
- Congdon, T.; Nguyen, L.Q.; Nogueira, C.R.; Habiby, R.L.; Medeiros-Neto, G.; Kopp, P. A novel mutation (Q40P) in PAX8 associated with congenital hypothyroidism and thyroid hypoplasia: Evidence for phenotypic variability in mother and child. J. Clin. Endocrinol. Metab. 2001, 86, 3962–3967. [Google Scholar] [CrossRef] [PubMed]
- Meeus, L.; Gilbert, B.; Rydlewski, C.; Parma, J.; Roussie, A.L.; Abramowicz, M.; Vilain, C.; Christophe, D.; Costagliola, S.; Vassart, G. Characterization of a novel loss of function mutation of PAX8 in a familial case of congenital hypothyroidism with in-place, normal-sized thyroid. J. Clin. Endocrinol. Metab. 2004, 89, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Vilain, C.; Rydlewski, C.; Duprez, L.; Heinrichs, C.; Abramowicz, M.; Malvaux, P.; Renneboog, B.; Parma, J.; Costagliola, S.; Vassart, G. Autosomal dominant transmission of congenital thyroid hypoplasia due to loss-of-function mutation of PAX8. J. Clin. Endocrinol. Metab. 2001, 86, 234–238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Batjargal, K.; Tajima, T.; Fujita-Jimbo, E.; Yamaguchi, T.; Nakamura, A.; Yamagata, T. Functional analysis of PAX8 variants identified in patients with congenital hypothyroidism in situ. Clin. Pediatr. Endocrinol. 2022, 31, 234–241. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Zou, H.; Ge, Y.; Wang, F.; Wang, Y.; Yan, S.; Xia, H.; Xing, M. Identification and characterization of novel PAX8 mutations in Congenital Hypothyroidism(CH) in a Chinese population. Oncotarget 2017, 8, 8707–8716. [Google Scholar] [CrossRef]
- Iwahashi-Odano, M.; Fujisawa, Y.; Ogata, T.; Nakashima, S.; Muramatsu, M.; Narumi, S. Identification and functional characterization of a novel PAX8 mutation (p.His39Pro) causing familial thyroid hypoplasia. Clin. Pediatr. Endocrinol. 2020, 29, 173–178. [Google Scholar] [CrossRef]
- Iwahashi-Odano, M.; Nagasaki, K.; Fukami, M.; Nishioka, J.; Yatsuga, S.; Asakura, Y.; Adachi, M.; Muroya, K.; Hasegawa, T.; Narumi, S. Congenital Hypothyroidism Due to Truncating PAX8 Mutations: A Case Series and Molecular Function Studies. J. Clin. Endocrinol. Metab. 2020, 105, e4055–e4065. [Google Scholar] [CrossRef]
- Carvalho, A.; Hermanns, P.; Rodrigues, A.L.; Sousa, I.; Anselmo, J.; Bikker, H.; Cabral, R.; Pereira-Duarte, C.; Mota-Vieira, L.; Pohlenz, J. A new PAX8 mutation causing congenital hypothyroidism in three generations of a family is associated with abnormalities in the urogenital tract. Thyroid 2013, 23, 1074–1078. [Google Scholar] [CrossRef]
- Tell, G.; Pellizzari, L.; Esposito, G.; Pucillo, C.; Macchia, P.E.; Di Lauro, R.; Damante, G. Structural defects of a Pax8 mutant that give rise to congenital hypothyroidism. Biochem. J. 1999, 341, 89–93. [Google Scholar] [CrossRef]
- Nieminen, P.; Arte, S.; Tanner, D.; Paulin, L.; Alaluusua, S.; Thesleff, I.; Pirinen, S. Identification of a nonsense mutation in the PAX9 gene in molar oligodontia. Eur. J. Hum. Genet. 2001, 9, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Intarak, N.; Tongchairati, K.; Termteerapornpimol, K.; Chantarangsu, S.; Porntaveetus, T. Tooth agenesis patterns and variants in PAX9: A systematic review. Jpn. Dent. Sci. Rev. 2023, 59, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Gan, S.; Zheng, S.; Li, M.; An, Y.; Yuan, S.; Gu, X.; Zhang, L.; Hou, Y.; Du, Q.; et al. Genotype-phenotype pattern analysis of pathogenic PAX9 variants in Chinese Han families with non-syndromic oligodontia. Front. Genet. 2023, 14, 1142776. [Google Scholar] [CrossRef] [PubMed]
- Fauzi, N.H.; Ardini, Y.D.; Zainuddin, Z.; Lestari, W. A review on non-syndromic tooth agenesis associated with PAX9 mutations. Jpn. Dent. Sci. Rev. 2018, 54, 30–36. [Google Scholar] [CrossRef]
- Wong, S.W.; Han, D.; Zhang, H.; Liu, Y.; Zhang, X.; Miao, M.Z.; Wang, Y.; Zhao, N.; Zeng, L.; Bai, B.; et al. Nine Novel PAX9 Mutations and a Distinct Tooth Agenesis Genotype-Phenotype. J. Dent. Res. 2018, 97, 155–162. [Google Scholar] [CrossRef]
- Wang, J.; Jian, F.; Chen, J.; Wang, H.; Lin, Y.; Yang, Z.; Pan, X.; Lai, W. Sequence analysis of PAX9, MSX1 and AXIN2 genes in a Chinese oligodontia family. Arch. Oral Biol. 2011, 56, 1027–1034. [Google Scholar] [CrossRef]
- Lammi, L.; Halonen, K.; Pirinen, S.; Thesleff, I.; Arte, S.; Nieminen, P. A missense mutation in PAX9 in a family with distinct phenotype of oligodontia. Eur. J. Hum. Genet. 2003, 11, 866–871. [Google Scholar] [CrossRef]
- Jumlongras, D.; Lin, J.Y.; Chapra, A.; Seidman, C.E.; Seidman, J.G.; Maas, R.L.; Olsen, B.R. A novel missense mutation in the paired domain of PAX9 causes non-syndromic oligodontia. Hum. Genet. 2004, 114, 242–249. [Google Scholar] [CrossRef]
- Zhao, K.; Lian, M.; Zou, D.; Huang, W.; Zhou, W.; Shen, Y.; Wang, F.; Wu, Y. Novel mutations identified in patients with tooth agenesis by whole-exome sequencing. Oral Dis. 2019, 25, 523–534. [Google Scholar] [CrossRef]
- Wang, Y.; Groppe, J.C.; Wu, J.; Ogawa, T.; Mues, G.; D’Souza, R.N.; Kapadia, H. Pathogenic mechanisms of tooth agenesis linked to paired domain mutations in human PAX9. Hum. Mol. Genet. 2009, 18, 2863–2874. [Google Scholar] [CrossRef]
- Phillips, H.M.; Stothard, C.A.; Shaikh Qureshi, W.M.; Kousa, A.I.; Briones-Leon, J.A.; Khasawneh, R.R.; O’Loughlin, C.; Sanders, R.; Mazzotta, S.; Dodds, R.; et al. Pax9 is required for cardiovascular development and interacts with Tbx1 in the pharyngeal endoderm to control 4th pharyngeal arch artery morphogenesis. Development 2019, 146, dev177618. [Google Scholar] [CrossRef]
- Mensah, J.K.; Ogawa, T.; Kapadia, H.; Cavender, A.C.; D’Souza, R.N. Functional analysis of a mutation in PAX9 associated with familial tooth agenesis in humans. J. Biol. Chem. 2004, 279, 5924–5933. [Google Scholar] [CrossRef]
- Stockton, D.W.; Das, P.; Goldenberg, M.; D’Souza, R.N.; Patel, P.I. Mutation of PAX9 is associated with oligodontia. Nat. Genet. 2000, 24, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.L.; Nieminen, P.; Lammi, L.; Niebuhr, E.; Kreiborg, S. Novel mutation of the initiation codon of PAX9 causes oligodontia. J. Dent. Res. 2005, 84, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, S.; Keski-Filppula, R.; Alapulli, H.; Nieminen, P.; Anttonen, V. Familial oligodontia and regional odontodysplasia associated with a PAX9 initiation codon mutation. Clin. Oral Investig. 2019, 23, 4107–4111. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Stockton, D.W.; Bauer, C.; Shaffer, L.G.; D’Souza, R.N.; Wright, T.; Patel, P.I. Haploinsufficiency of PAX9 is associated with autosomal dominant hypodontia. Hum. Genet. 2002, 110, 371–376. [Google Scholar] [CrossRef]
- Sarkar, T.; Bansal, R.; Das, P. A novel G to A transition at initiation codon and exon-intron boundary of PAX9 identified in association with familial isolated oligodontia. Gene 2017, 635, 69–76. [Google Scholar] [CrossRef]
- Liang, J.; Qin, C.; Yue, H.; He, H.; Bian, Z. A novel initiation codon mutation of PAX9 in a family with oligodontia. Arch. Oral Biol. 2016, 61, 144–148. [Google Scholar] [CrossRef]
- Murakami, A.; Yasuhira, S.; Mayama, H.; Miura, H.; Maesawa, C.; Satoh, K. Characterization of PAX9 variant P20L identified in a Japanese family with tooth agenesis. PLoS ONE 2017, 12, e0186260. [Google Scholar] [CrossRef]
- Kapadia, H.; Frazier-Bowers, S.; Ogawa, T.; D’Souza, R.N. Molecular characterization of a novel PAX9 missense mutation causing posterior tooth agenesis. Eur. J. Hum. Genet. 2006, 14, 403–409. [Google Scholar] [CrossRef]
- Kist, R.; Watson, M.; Wang, X.; Cairns, P.; Miles, C.; Reid, D.J.; Peters, H. Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum. Mol. Genet. 2005, 14, 3605–3617. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, Q.; Chen, Y.; Luo, S.; Bao, L.; Xu, Y. A novel missense mutation in the paired domain of human PAX9 causes oligodontia. Am. J. Med. Genet. A 2007, 143, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Dou, J.; Huang, W.; Wang, F.; Wu, Y. Expanding the genetic spectrum of tooth agenesis using whole-exome sequencing. Clin. Genet. 2022, 102, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Bowers, S.A.; Guo, D.C.; Cavender, A.; Xue, L.; Evans, B.; King, T.; Milewicz, D.; D’Souza, R.N. A novel mutation in human PAX9 causes molar oligodontia. J. Dent. Res. 2002, 81, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-H.; Peng, H.-H.; Chou, H.-Y.E.; Hou, H.-H.; Liu, W.-W.; Kuo, M.Y.-P.; Lee, A.Y.-L.; Kao, H.-F.; Lin, H.-Y.; Chang, Y.-C.; et al. Downregulation of PAX1 in OSCC Enhances Stemness and Immunosuppression via IFIT1 and PD-L1 Pathways. Oral Dis. 2025, 31, 2071–2083. [Google Scholar] [CrossRef]
- Cheng, S.-J.; Chang, C.-F.; Ko, H.-H.; Liu, Y.-C.; Peng, H.-H.; Wang, H.-J.; Lin, H.-S.; Chiang, C.-P. Hypermethylated ZNF582 and PAX1 genes in oral scrapings collected from cancer-adjacent normal oral mucosal sites are associated with aggressive progression and poor prognosis of oral cancer. Oral Oncol. 2017, 75, 169–177. [Google Scholar] [CrossRef]
- Gokden, N.; Gokden, M.; Phan, D.C.; McKenney, J.K. The utility of PAX-2 in distinguishing metastatic clear cell renal cell carcinoma from its morphologic mimics: An immunohistochemical study with comparison to renal cell carcinoma marker. Am. J. Surg. Pathol. 2008, 32, 1462–1467. [Google Scholar] [CrossRef]
- Ozcan, A.; Liles, N.; Coffey, D.; Shen, S.S.; Truong, L.D. PAX2 and PAX8 Expression in Primary and Metastatic Müllerian Epithelial Tumors: A Comprehensive Comparison. Am. J. Surg. Pathol. 2011, 35, 1837–1847. [Google Scholar] [CrossRef]
- Ozcan, A.; de la Roza, G.; Ro, J.Y.; Shen, S.S.; Truong, L.D. PAX2 and PAX8 Expression in Primary and Metastatic Renal Tumors: A Comprehensive Comparison. Arch. Pathol. Lab. Med. 2012, 136, 1541–1551. [Google Scholar] [CrossRef]
- Ozcan, A.; Zhai, J.; Hamilton, C.; Shen, S.S.; Ro, J.Y.; Krishnan, B.; Truong, L.D. PAX-2 in the Diagnosis of Primary Renal Tumors: Immunohistochemical Comparison With Renal Cell Carcinoma Marker Antigen and Kidney-Specific Cadherin. Am. J. Clin. Pathol. 2009, 131, 393–404. [Google Scholar] [CrossRef]
- Lucas, E.; Niu, S.; Aguilar, M.; Molberg, K.; Carrick, K.; Rivera-Colon, G.; Gwin, K.; Wang, Y.; Zheng, W.; Castrillon, D.H.; et al. Utility of a PAX2, PTEN, and β-catenin Panel in the Diagnosis of Atypical Hyperplasia/Endometrioid Intraepithelial Neoplasia in Endometrial Polyps. Am. J. Surg. Pathol. 2023, 47, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma—The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet. Muscle 2012, 2, 25. [Google Scholar] [CrossRef]
- Mercado, G.E.; Barr, F.G. Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: Recent advances. Curr. Mol. Med. 2007, 7, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Mariño-Enríquez, A.; Fletcher, C.D.M.; Hornick, J.L. Expression of PAX3 Distinguishes Biphenotypic Sinonasal Sarcoma From Histologic Mimics. Am. J. Surg. Pathol. 2018, 42, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Fritchie, K.J.; Jin, L.; Wang, X.; Graham, R.P.; Torbenson, M.S.; Lewis, J.E.; Rivera, M.; Garcia, J.J.; Schembri-Wismayer, D.J.; Westendorf, J.J.; et al. Fusion gene profile of biphenotypic sinonasal sarcoma: An analysis of 44 cases. Histopathology 2016, 69, 930–936. [Google Scholar] [CrossRef]
- Wang, X.; Bledsoe, K.L.; Graham, R.P.; Asmann, Y.W.; Viswanatha, D.S.; Lewis, J.E.; Lewis, J.T.; Chou, M.M.; Yaszemski, M.J.; Jen, J.; et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat. Genet. 2014, 46, 666–668. [Google Scholar] [CrossRef]
- Huang, S.C.; Ghossein, R.A.; Bishop, J.A.; Zhang, L.; Chen, T.C.; Huang, H.Y.; Antonescu, C.R. Novel PAX3-NCOA1 Fusions in Biphenotypic Sinonasal Sarcoma With Focal Rhabdomyoblastic Differentiation. Am. J. Surg. Pathol. 2016, 40, 51–59. [Google Scholar] [CrossRef]
- Wong, W.J.; Lauria, A.; Hornick, J.L.; Xiao, S.; Fletcher, J.A.; Marino-Enriquez, A. Alternate PAX3-FOXO1 oncogenic fusion in biphenotypic sinonasal sarcoma. Genes Chromosomes Cancer 2016, 55, 25–29. [Google Scholar] [CrossRef]
- Le Loarer, F.; Laffont, S.; Lesluyes, T.; Tirode, F.; Antonescu, C.; Baglin, A.C.; Delespaul, L.; Soubeyran, I.; Hostein, I.; Pérot, G.; et al. Clinicopathologic and Molecular Features of a Series of 41 Biphenotypic Sinonasal Sarcomas Expanding Their Molecular Spectrum. Am. J. Surg. Pathol. 2019, 43, 747–754. [Google Scholar] [CrossRef]
- Jia, Z.; Gu, Z. PAX5 alterations in B-cell acute lymphoblastic leukemia. Front. Oncol. 2022, 12, 1023606. [Google Scholar] [CrossRef]
- Denk, D.; Bradtke, J.; König, M.; Strehl, S. PAX5 fusion genes in t(7;9)(q11.2;p13) leukemia: A case report and review of the literature. Mol. Cytogenet. 2014, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Schinnerl, D.; Fortschegger, K.; Kauer, M.; Marchante, J.R.; Kofler, R.; Den Boer, M.L.; Strehl, S. The role of the Janus-faced transcription factor PAX5-JAK2 in acute lymphoblastic leukemia. Blood 2015, 125, 1282–1291. [Google Scholar] [CrossRef]
- Jurado, S.; Fedl, A.S.; Jaritz, M.; Kostanova-Poliakova, D.; Malin, S.G.; Mullighan, C.G.; Strehl, S.; Fischer, M.; Busslinger, M. The PAX5-JAK2 translocation acts as dual-hit mutation that promotes aggressive B-cell leukemia via nuclear STAT5 activation. Embo J. 2022, 41, e108397. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Wu, X.; Tan, F.; Shi, Y.X.; Glass, T.; Liu, T.J.; Wathen, K.; Hess, K.R.; Gumin, J.; Lang, F.; et al. PAX6 suppresses growth of human glioblastoma cells. J. Neurooncol 2005, 71, 223–229. [Google Scholar] [CrossRef]
- Hegge, B.; Sjøttem, E.; Mikkola, I. Generation of a PAX6 knockout glioblastoma cell line with changes in cell cycle distribution and sensitivity to oxidative stress. BMC Cancer 2018, 18, 496. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, X.; Wang, J.; Liang, T.; Gu, Y.; Yang, D. Down-regulation of PAX6 by promoter methylation is associated with poor prognosis in non small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11452–11457. [Google Scholar]
- Li, Y.; Li, Y.; Liu, Y.; Xie, P.; Li, F.; Li, G. PAX6, a novel target of microRNA-7, promotes cellular proliferation and invasion in human colorectal cancer cells. Dig. Dis. Sci. 2014, 59, 598–606. [Google Scholar] [CrossRef]
- Mascarenhas, J.B.; Young, K.P.; Littlejohn, E.L.; Yoo, B.K.; Salgia, R.; Lang, D. PAX6 is expressed in pancreatic cancer and actively participates in cancer progression through activation of the MET tyrosine kinase receptor gene. J. Biol. Chem. 2009, 284, 27524–27532. [Google Scholar] [CrossRef]
- Lutz, F.; Hornburg, S.M.; Möller, K.; Viehweger, F.; Schlichter, R.; Menz, A.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; et al. PAX6 is a useful marker for pancreatic origin of neuroendocrine neoplasms: A tissue microarray study evaluating more than 19,000 tumors from 150 different tumor types. Hum. Pathol. 2024, 154, 105695. [Google Scholar] [CrossRef] [PubMed]
- Gorbokon, N.; Baltruschat, S.; Lennartz, M.; Luebke, A.M.; Höflmayer, D.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; Fraune, C.; Lebok, P.; et al. PAX8 expression in cancerous and non-neoplastic tissue: A tissue microarray study on more than 17,000 tumors from 149 different tumor entities. Virchows Arch. 2024, 485, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Bleu, M.; Gaulis, S.; Lopes, R.; Sprouffske, K.; Apfel, V.; Holwerda, S.; Pregnolato, M.; Yildiz, U.; Cordoʹ, V.; Dost, A.F.M.; et al. PAX8 activates metabolic genes via enhancer elements in Renal Cell Carcinoma. Nat. Commun. 2019, 10, 3739. [Google Scholar] [CrossRef]
- Corona, R.I.; Seo, J.-H.; Lin, X.; Hazelett, D.J.; Reddy, J.; Fonseca, M.A.S.; Abassi, F.; Lin, Y.G.; Mhawech-Fauceglia, P.Y.; Shah, S.P.; et al. Non-coding somatic mutations converge on the PAX8 pathway in ovarian cancer. Nat. Commun. 2020, 11, 2020. [Google Scholar] [CrossRef]
- Sereti, K.; Russo, A.E.; Raisner, R.; Ma, T.P.; Gascoigne, K.E. PAX8 Interacts with the SWI/SNF Complex at Enhancers to Drive Proliferation in Ovarian Cancer. Mol. Cancer Res. 2025, 23, 416–425. [Google Scholar] [CrossRef]
- Bleu, M.; Mermet-Meillon, F.; Apfel, V.; Barys, L.; Holzer, L.; Bachmann Salvy, M.; Lopes, R.; Amorim Monteiro Barbosa, I.; Delmas, C.; Hinniger, A.; et al. PAX8 and MECOM are interaction partners driving ovarian cancer. Nat. Commun. 2021, 12, 2442. [Google Scholar] [CrossRef]
- Ikari, N.; Aoyama, S.; Seshimo, A.; Suehiro, Y.; Motohashi, T.; Mitani, S.; Yoshina, S.; Tanji, E.; Serizawa, A.; Yamada, T.; et al. Somatic mutations and increased lymphangiogenesis observed in a rare case of intramucosal gastric carcinoma with lymph node metastasis. Oncotarget 2018, 9, 10808–10817. [Google Scholar] [CrossRef][Green Version]
- Tan, B.; Wang, J.; Song, Q.; Wang, N.; Jia, Y.; Wang, C.; Yao, B.; Liu, Z.; Zhang, X.; Cheng, Y. Prognostic value of PAX9 in patients with esophageal squamous cell carcinoma and its prediction value to radiation sensitivity. Mol. Med. Rep. 2017, 16, 806–816. [Google Scholar] [CrossRef]
- Xiong, Z.; Ren, S.; Chen, H.; Liu, Y.; Huang, C.; Zhang, Y.L.; Odera, J.O.; Chen, T.; Kist, R.; Peters, H.; et al. PAX9 regulates squamous cell differentiation and carcinogenesis in the oro-oesophageal epithelium. J. Pathol. 2018, 244, 164–175. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Paiboonrungruang, C.; Li, Y.; Peters, H.; Kist, R.; Xiong, Z. PAX9 in Cancer Development. Int. J. Mol. Sci. 2022, 23, 5589. [Google Scholar] [CrossRef]
- Hsu, D.S.; Acharya, C.R.; Balakumaran, B.S.; Riedel, R.F.; Kim, M.K.; Stevenson, M.; Tuchman, S.; Mukherjee, S.; Barry, W.; Dressman, H.K.; et al. Characterizing the developmental pathways TTF-1, NKX2-8, and PAX9 in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 5312–5317, Erratum in Proc. Natl. Acad. Sci. USA 2011, 108, 14705. [Google Scholar] [CrossRef]
- Kendall, J.; Liu, Q.; Bakleh, A.; Krasnitz, A.; Nguyen, K.C.; Lakshmi, B.; Gerald, W.L.; Powers, S.; Mu, D. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.S.; Ramanand, S.G.; Cuevas, I.C.; Gao, Y.; Lee, S.; Abbas, A.; Zhang, X.; Kumar, A.; Koduru, P.; Roy, S.; et al. A distinct mechanism of epigenetic reprogramming silences PAX2 and initiates endometrial carcinogenesis. J. Clin. Investig. 2025, 135, e190989. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, L.; Liu, H.; Qin, P.; Chen, R.; Liu, S.; Xiong, H.; Li, Y.; Yang, Z.; Xie, M.; et al. PAX2 is regulated by estrogen/progesterone through promoter methylation in endometrioid adenocarcinoma and has an important role in carcinogenesis via the AKT/mTOR signaling pathway. J. Pathol. 2024, 262, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Wang, J.; Li, Q.; Tao, X.; Chang, K.; Hua, K.; Yu, Y.; Wong, K.K.; Feng, W. DNA methylation promotes paired box 2 expression via myeloid zinc finger 1 in endometrial cancer. Oncotarget 2016, 7, 84785–84797. [Google Scholar] [CrossRef]
- Patrício, P.; Ramalho-Carvalho, J.; Costa-Pinheiro, P.; Almeida, M.; Barros-Silva, J.D.; Vieira, J.; Dias, P.C.; Lobo, F.; Oliveira, J.; Teixeira, M.R.; et al. Deregulation of PAX2 expression in renal cell tumours: Mechanisms and potential use in differential diagnosis. J. Cell. Mol. Med. 2013, 17, 1048–1058. [Google Scholar] [CrossRef]
- Palmisano, W.A.; Crume, K.P.; Grimes, M.J.; Winters, S.A.; Toyota, M.; Esteller, M.; Joste, N.; Baylin, S.B.; Belinsky, S.A. Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer Res. 2003, 63, 4620–4625. [Google Scholar]
- Zhao, L.; Li, S.; Gan, L.; Li, C.; Qiu, Z.; Feng, Y.; Li, J.; Li, L.; Li, C.; Peng, W.; et al. Paired box 5 is a frequently methylated lung cancer tumour suppressor gene interfering β-catenin signalling and GADD45G expression. J. Cell. Mol. Med. 2016, 20, 842–854. [Google Scholar] [CrossRef]
- Bhol, C.S.; Mishra, S.R.; Patil, S.; Sahu, S.K.; Kirtana, R.; Manna, S.; Shanmugam, M.K.; Sethi, G.; Patra, S.K.; Bhutia, S.K. PAX9 reactivation by inhibiting DNA methyltransferase triggers antitumor effect in oral squamous cell carcinoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2022, 1868, 166428. [Google Scholar] [CrossRef]
- Gouhier, A.; Dumoulin-Gagnon, J.; Lapointe-Roberge, V.; Harris, J.; Balsalobre, A.; Drouin, J. Pioneer factor Pax7 initiates two-step cell-cycle-dependent chromatin opening. Nat. Struct. Mol. Biol. 2024, 31, 92–101. [Google Scholar] [CrossRef]
- Singh, P.; Bhadada, S.K.; Arya, A.K.; Saikia, U.N.; Sachdeva, N.; Dahiya, D.; Kaur, J.; Brandi, M.L.; Rao, S.D. Aberrant Epigenetic Alteration of PAX1 Expression Contributes to Parathyroid Tumorigenesis. J. Clin. Endocrinol. Metab. 2022, 107, e783–e792. [Google Scholar] [CrossRef]
- Chen, Z.H.; Chen, Y.B.; Yue, H.R.; Zhou, X.J.; Ma, H.Y.; Wang, X.; Cao, X.C.; Yu, Y. PAX5-miR-142 feedback loop promotes breast cancer proliferation by regulating DNMT1 and ZEB1. Mol. Med. 2023, 29, 89. [Google Scholar] [CrossRef]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar] [CrossRef]
- Relaix, F.; Zoglio, V.; Chebouti, S.; de Lima, J.E.; Lafuste, P. Regulation and function of Pax3 in muscle stem cell heterogeneity and stress response. J. Muscle Res. Cell Motil. 2025, 46, 327–336. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.