
A Preliminary Approach to Oral Low-Dose Ketamine Self-Administration in Mice (Mus musculus)

 ,
,  ,
,  , and
, and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Animal Housing and Maintenance
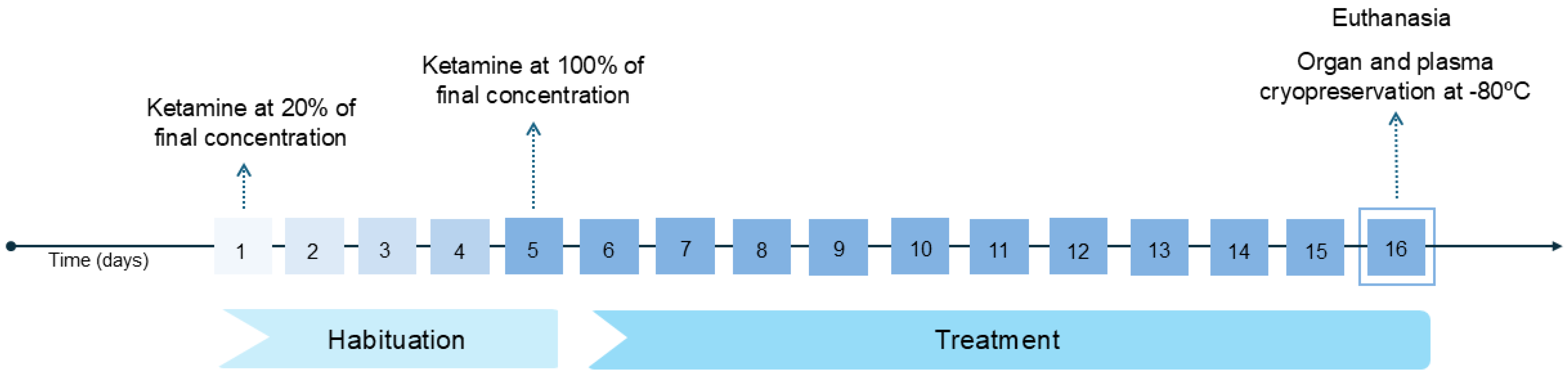
2.2. Experimental Design
2.3. Water Consumption and Ketamine Intake
2.4. Oxidative Stress Quantification in Brain, Liver, and Kidney Samples: Sample Preparation and PROTEIN Quantification
2.5. Analysis of Serum Biomarkers to Evaluate Liver and Kidney Function: Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST,) and Alkaline Phosphatase (ALP) Levels as Indicators of Liver Function, and Creatinine and Serum Urea Nitrogen (BUN) Levels as Indicators of Kidney Function
2.6. Evaluation of Additional Parameters in Kidney Samples
2.6.1. Caspase 3 and 9 Levels as Apoptosis INDUCTION Markers
2.6.2. NO as an Inflammation Marker
2.6.3. DNA Double-Strand Break Estimation as a DNA Damage Marker
2.7. Data Analysis
3. Results
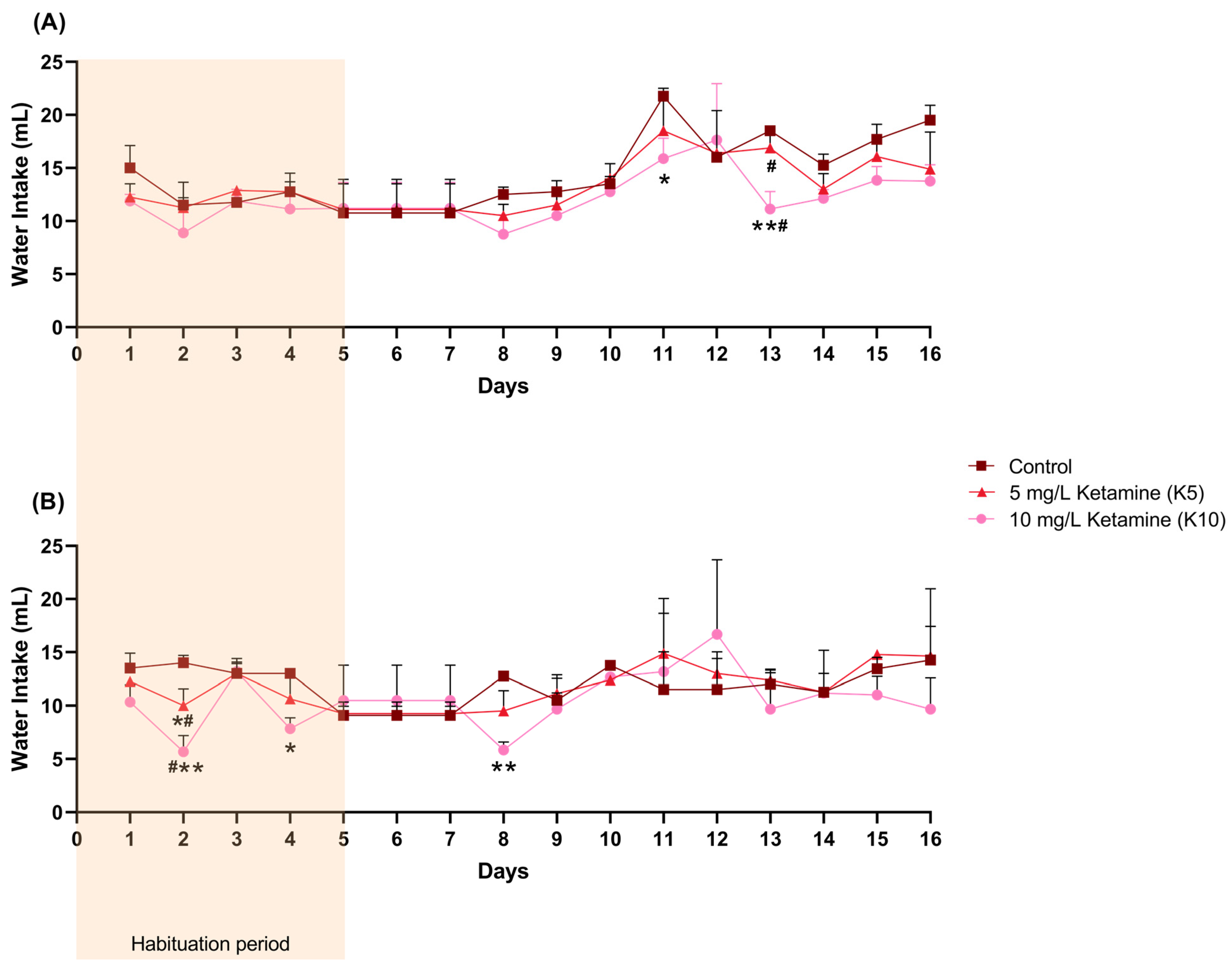
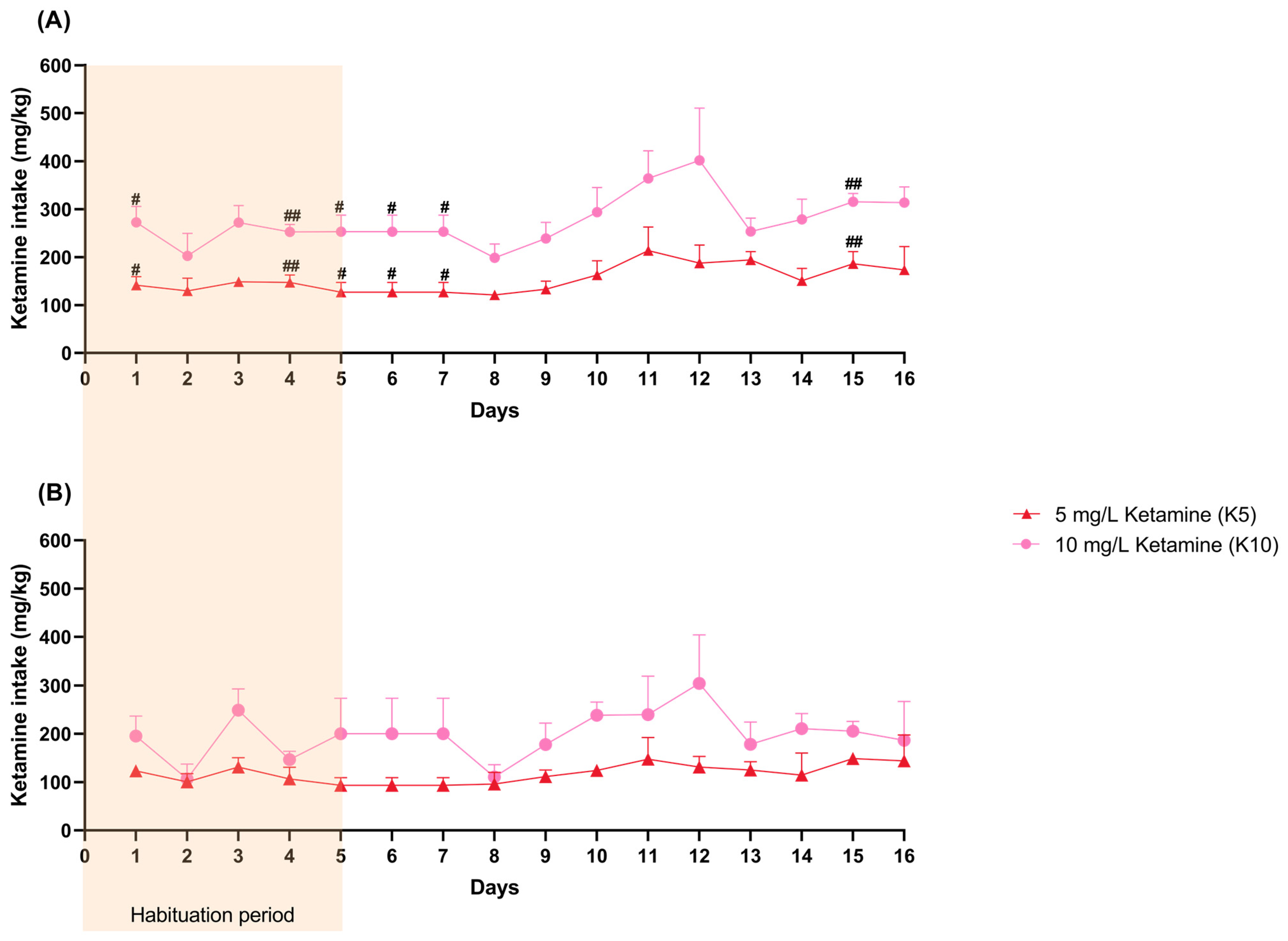
3.1. Water and Ketamine Intake
3.2. Oxidative Stress Evaluation
3.3. Liver and Kidney Function Assessment
3.4. Apoptosis, Inflammation, and DNA Damage in Kidney Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NMDA | N-methyl-D-aspartate |
| ROS | Reactive oxygen species |
| CI | Confidence interval |
| UTAD | University of Trás-os-Montes and Alto Douro |
| K5 | 5 mg/L of ketamine |
| K10 | 10 mg/L of ketamine |
| MDA | Malondialdehyde |
| PC | Protein carbonyls |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| GR | Glutathione reductase |
| GPx | Glutathione peroxidase |
| GST | Glutathione S-transferase |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| ALP | Alkaline phosphatase |
| BUN | Serum urea nitrogen |
| ANOVA | Two-way analysis of variance |
| SD | Standard deviation |
| IQR | Interquartile range |
References
- Félix, L.; Antunes, L.; Campos, S.; Venâncio, C.; Coimbra, A.M. Recreational Use of Ketamine and Its Interaction with NMDA Receptors. In Neuropathology of Drug Addictions and Substance Misuse; Academic Press: Cambridge, MA, USA, 2016; pp. 672–680. [Google Scholar]
- Dong, C.; Anand, K.J. Developmental neurotoxicity of ketamine in pediatric clinical use. Toxicol. Lett. 2013, 220, 53–60. [Google Scholar] [CrossRef]
- Lodge, D.; Mercier, M.S. Ketamine and phencyclidine: The good, the bad and the unexpected. Br. J. Pharmacol. 2015, 172, 4254–4276. [Google Scholar] [CrossRef]
- Kronenberg, R.H. Ketamine as an Analgesic. J. Pain Palliat. Care Pharmacother. 2002, 16, 27–35. [Google Scholar]
- Schoevers, R.A.; Chaves, T.V.; Balukova, S.M.; Rot, M.; Kortekaas, R. Oral ketamine for the treatment of pain and treatment-resistant depressiond. Br. J. Psychiatry 2016, 208, 108–113. [Google Scholar] [CrossRef]
- Strasburger, S.E.; Bhimani, P.M.; Kaabe, J.H.; Krysiak, J.T.; Nanchanatt, D.L.; Nguyen, T.N.; Pough, K.A.; Prince, T.A.; Ramsey, N.S.; Savsani, K.H.; et al. What is the mechanism of Ketamine’s rapid-onset antidepressant effect? A concise overview of the surprisingly large number of possibilities. J. Clin. Pharm. Ther. 2017, 42, 147–154. [Google Scholar] [CrossRef]
- Kreutzwiser, D.; Tawfic, Q.A. Expanding Role of NMDA Receptor Antagonists in the Management of Pain. CNS Drugs 2019, 33, 347–374. [Google Scholar] [CrossRef]
- Chang, L.; Zhang, K.; Pu, Y.; Qu, Y.; Wang, S.M.; Xiong, Z.; Ren, Q.; Dong, C.; Fujita, Y.; Hashimoto, K. Comparison of antidepressant and side effects in mice after intranasal administration of (R,S)-ketamine, (R)-ketamine, and (S)-ketamine. Pharmacol. Biochem. Behav. 2019, 181, 53–59. [Google Scholar] [CrossRef]
- Shimoyama, M.; Shimoyama, N.; Gorman, L.; Elliott, K.; Inturrisi, C.E. Oral ketamine is antinociceptive in the rat formalin test: Role of the metabolite, norketamine. Pain 1999, 81, 85–93. [Google Scholar] [CrossRef]
- Erdinc, M.; Uyar, E.; Kelle, I.; Akkoc, H. Anti-nociceptive effects of low dose ketamine in mice may be mediated by the serotonergic systems. Psychiat Clin. Psychy 2019, 29, 252–256. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, C.; Cao, Y.; Han, J. Repeated administration of low dose ketamine for the treatment of monoarthritic pain in the rat. Life Sci. 2000, 67, 261–267. [Google Scholar] [CrossRef]
- do Vale, E.M.; Xavier, C.C.; Nogueira, B.G.; Campos, B.C.; de Aquino, P.E.; da Costa, R.O.; Leal, L.K.; de Vasconcelos, S.M.; Neves, K.R.; de Barros Viana, G.S. Antinociceptive and Anti-Inflammatory Effects of Ketamine and the Relationship to Its Antidepressant Action and GSK3 Inhibition. Basic Clin. Pharmacol. Toxicol. 2016, 119, 562–573. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.; Aghajanian, G.; Duman, R.S. mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Venancio, C.; Antunes, L.; Felix, L.; Rodrigues, P.; Summavielle, T.; Peixoto, F. Chronic ketamine administration impairs mitochondrial complex I in the rat liver. Life Sci. 2013, 93, 464–470. [Google Scholar] [CrossRef]
- Fitzgibbon, E.J.; Hall, P.; Schroder, C.; Seely, J.; Viola, R. Low dose ketamine as an analgesic adjuvant in difficult pain syndromes: A strategy for conversion from parenteral to oral ketamine. J. Pain Symptom Manag. 2002, 23, 165–170. [Google Scholar] [CrossRef]
- Can, A.T.; Hermens, D.F.; Dutton, M.; Gallay, C.C.; Jensen, E.; Jones, M.; Scherman, J.; Beaudequin, D.A.; Yang, C.; Schwenn, P.E.; et al. Low dose oral ketamine treatment in chronic suicidality: An open-label pilot study. Transl. Psychiatry 2021, 11, 101. [Google Scholar] [CrossRef]
- Duman, R.S.; Li, N.; Liu, R.J.; Duric, V.; Aghajanian, G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012, 62, 35–41. [Google Scholar] [CrossRef]
- Corriger, A.; Pickering, G. Ketamine and depression: A narrative review. Drug Des. Devel Ther. 2019, 13, 3051–3067. [Google Scholar] [CrossRef]
- Blonk, M.I.; Koder, B.G.; van den Bemt, P.M.; Huygen, F.J. Use of oral ketamine in chronic pain management: A review. Eur. J. Pain 2010, 14, 466–472. [Google Scholar] [CrossRef]
- Andrade, C. Ketamine for Depression, 4: In What Dose, at What Rate, by What Route, for How Long, and at What Frequency? J. Clin. Psychiatry 2017, 78, e852–e857. [Google Scholar] [CrossRef]
- Aan Het Rot, M.; Zarate, C.A., Jr.; Charney, D.S.; Mathew, S.J. Ketamine for depression: Where do we go from here? Biol. Psychiatry 2012, 72, 537–547. [Google Scholar] [CrossRef]
- Holtman, J.R., Jr.; Crooks, P.A.; Johnson-Hardy, J.K.; Hojomat, M.; Kleven, M.; Wala, E.P. Effects of norketamine enantiomers in rodent models of persistent pain. Pharmacol. Biochem. Behav. 2008, 90, 676–685. [Google Scholar] [CrossRef]
- Kasikara, H.; Sungu, N.; Arslan, M.; Kucuk, A.; Ozturk, L.; Afandiyeva, N.; Kavutcu, M. Repeated Doses of Ketamine Affect the Infant Rat Urogenital System. Drug Des. Devel Ther. 2021, 15, 1157–1165. [Google Scholar] [CrossRef]
- Oliveira, L.; Spiazzi, C.M.; Bortolin, T.; Canever, L.; Petronilho, F.; Mina, F.G.; Dal-Pizzol, F.; Quevedo, J.; Zugno, A.I. Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1003–1008. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ganguly, S.; Srivastava, M.; Palit, G. Effect of ‘chronic’ versus ‘acute’ ketamine administration and its ‘withdrawal’ effect on behavioural alterations in mice: Implications for experimental psychosis. Behav. Brain Res. 2011, 216, 247–254. [Google Scholar] [CrossRef]
- Bedir, Z.; Erdem, K.T.O.; Ates, I.; Karakurt, T.C.O.; Gursul, C.; Onk, D.; Kurt, N.; Suleyman, Z.; Suleyman, H. Effects of ketamine, thiopental and their combination on the rat liver: A biochemical evaluation. Adv. Clin. Exp. Med. 2022, 31, 285–292. [Google Scholar] [CrossRef]
- Venâncio, C.; Félix, L.; Almeida, V.; Coutinho, J.; Antunes, L.; Peixoto, F.; Summavielle, T. Acute Ketamine Impairs Mitochondrial Function and Promotes Superoxide Dismutase Activity in the Rat Brain. Anesth. Analg. 2015, 120, 320–328. [Google Scholar] [CrossRef]
- Hughes, J.; Gobe, G. Identification and quantification of apoptosis in the kidney using morphology, biochemical and molecular markers. Nephrol. Carlton 2007, 12, 452–458. [Google Scholar] [CrossRef]
- Pantelias, K.; Grapsa, E. Drug abuse and kidney. Hippokratia 2001, 15, 4–8. [Google Scholar]
- Nebendahl, K.; Hauff, P. Drug Administration. In Small Animal Imaging; Kiessling, F., Pichler, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Rajandram, R.; Yap, N.; Ong, T.; Mun, K.; Wali, H.; Hasan, M.; Razack, A.; Mohd, M. Oral ketamine induced pathological changes of the urinary tract in a rat model. Malays. J. Pathol. 2017, 39, 47–53. [Google Scholar]
- Arantes-Rodrigues, R.; Henriques, A.; Pinto-Leite, R.; Faustino-Rocha, A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Seixas, F.; Gama, A.; Colaço, B.; Colaço, A.; et al. The effects of repeated oral gavage on the health of male CD-1 mice. Lab. Anim. 2012, 41, 129–134. [Google Scholar] [CrossRef]
- Onaolapo, O.J.; Ademakinwa, O.Q.; Olalekan, T.O.; Onaolapo, A.Y. Ketamine-induced behavioural and brain oxidative changes in mice: An assessment of possible beneficial effects of zinc as mono- or adjunct therapy. Psychopharmacol. Berl 2017, 234, 2707–2725. [Google Scholar] [CrossRef]
- Walker, M.K.; Boberg, J.R.; Walsh, M.T.; Wolf, V.; Trujillo, A.; Duke, M.S.; Palme, R.; Felton, L.A. A less stressful alternative to oral gavage for pharmacological and toxicological studies in mice. Toxicol. Appl. Pharmacol. 2012, 260, 65–69. [Google Scholar] [CrossRef]
- Ferguson, S.A.; Boctor, S.Y. Use of food wafers for multiple daily oral treatments in young rats. J. Am. Assoc. Lab. Anim. Sci. 2009, 48, 292–295. [Google Scholar]
- Azevedo, T.; Silva, J.; Peixoto, F.; Silvestre-Ferreira, A.C.; Gama, A.; Seixas, F.; Finimundy, T.C.; Barros, L.; Matos, M.; Oliveira, P.A.; et al. The role of natural compounds in rat mammary cancer: The beneficial effects of Santolina chamaecyparissus L. aqueous extract. Vet. Stanica 2023, 55, 45–61. [Google Scholar] [CrossRef]
- Li, X.; Li, S.; Zheng, W.; Pan, J.; Huang, K.; Chen, R.; Pan, T.; Liao, G.; Chen, Z.; Zhou, D.; et al. Environmental enrichment and abstinence attenuate ketamine-induced cardiac and renal toxicity. Sci. Rep. 2015, 5, 11611. [Google Scholar] [CrossRef]
- Deng, J.; Yu, L.; Liu, C.; Yu, K.; Shi, X.; Yeung, L.W.; Lam, P.K.; Wu, R.S.; Zhou, B. Hexabromocyclododecane-induced developmental toxicity and apoptosis in zebrafish embryos. Aquat. Toxicol. 2009, 93, 29–36. [Google Scholar] [CrossRef]
- Mesquita, C.S.; Oliveira, R.; Bento, F.; Geraldo, D.; Rodrigues, J.V.; Marcos, J.C. Simplified 2,4-dinitrophenylhydrazine spectrophotometric assay for quantification of carbonyls in oxidized proteins. Anal. Biochem. 2014, 458, 69–71. [Google Scholar] [CrossRef]
- Wallin, B.; Rosengren, B.; Shertzer, H.G.; Camejo, G. Lipoprotein oxidation and measurement of thiobarbituric acid reacting substances formation in a single microtiter plate: Its use for evaluation of antioxidants. Anal. Biochem. 1993, 208, 10–15. [Google Scholar] [CrossRef]
- Durak, I.; Yurtarslanl, Z.; Canbolat, O.; Akyol, O. A methodological approach to superoxide dismutase (SOD) activity assay based on inhibition of nitroblue tetrazolium (NBT) reduction. Clin. Chim. Acta 1993, 214, 103–104. [Google Scholar] [CrossRef]
- Claiborne, A. Catalase activity. In CRC Handbook of Methods for Oxygen Radical Research; CRC Press: Boca Raton, FL, USA, 1985; pp. 283–284. [Google Scholar]
- Massarsky, A.; Kozal, J.S.; Di Giulio, R.T. Glutathione and zebrafish: Old assays to address a current issue. Chemosphere 2017, 168, 707–715. [Google Scholar] [CrossRef]
- Habig, W.H.; Jakoby, W.B. Assays for differentiation of glutathione S-transferases. Methods Enzym. 1981, 77, 398–405. [Google Scholar]
- Alisik, M.; Neselioglu, S.; Erel, O. A colorimetric method to measure oxidized, reduced and total glutathione levels in erythrocytes. J. Lab. Med. 2019, 43, 269–277. [Google Scholar] [CrossRef]
- Floreani, M.; Petrone, M.; Debetto, P.; Palatini, P. A comparison between different methods for the determination of reduced and oxidized glutathione in mammalian tissues. Free Radic. Res 1997, 26, 449–455. [Google Scholar] [CrossRef]
- Singh, V.; Gera, R.; Purohit, M.P.; Patnaik, S.; Ghosh, D. Fluorometric Estimation of Glutathione in Cultured Microglial Cell Lysate. Bio-Protocol 2017, 7, e2304. [Google Scholar] [CrossRef]
- Brentnall, M.; Rodriguez-Menocal, L.; Ladron De Guevara, R.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef]
- Kuida, K. Caspase-9. Int. J. Biochem. Cell Biol. 2000, 32, 121–124. [Google Scholar] [CrossRef]
- Mincberg, M.; Gopas, J.; Tal, J. Minute virus of mice (MVMp) infection and NS1 expression induce p53 independent apoptosis in transformed rat fibroblast cells. Virology 2011, 412, 233–243. [Google Scholar] [CrossRef]
- Krishnan, M.; Kang, S.C. Vitexin inhibits acrylamide-induced neuroinflammation and improves behavioral changes in zebrafish larvae. Neurotoxicol. Teratol. 2019, 74, 106811. [Google Scholar] [CrossRef]
- Olive, P.L. DNA precipitation assay: A rapid and simple method for detecting DNA damage in mammalian cells. Environ. Mol. Mutagen. 1998, 11, 487–495. [Google Scholar] [CrossRef]
- Onaolapo, A.Y.; Ayeni, O.J.; Ogundeji, M.O.; Ajao, A.; Onaolapo, O.J.; Owolabi, A.R. Subchronic ketamine alters behaviour, metabolic indices and brain morphology in adolescent rats: Involvement of oxidative stress, glutamate toxicity and caspase-3-mediated apoptosis. J. Chem. Neuroanat. 2019, 96, 22–33. [Google Scholar] [CrossRef]
- Ganguly, S.; Panetta, J.C.; Roberts, J.K.; Schuetz, E.G. Ketamine Pharmacokinetics and Pharmacodynamics Are Altered by P-Glycoprotein and Breast Cancer Resistance Protein Efflux Transporters in Mice. Drug Metab. Dispos. 2018, 46, 1014–1022. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J. Metabolism and metabolomics of ketamine: A toxicological approach. Forensic Sci. Res. 2017, 2, 2–10. [Google Scholar] [CrossRef]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef]
- Domany, Y.; Bleich-Cohen, M.; Tarrasch, R.; Meidan, R.; Litvak-Lazar, O.; Stoppleman, N.; Schreiber, S.; Bloch, M.; Hendler, T.; Sharon, H. Repeated oral ketamine for out-patient treatment of resistant depression: Randomised, double-blind, placebo-controlled, proof-of-concept study. Br. J. Psychiatry 2019, 214, 20–26. [Google Scholar] [CrossRef]
- Ponton, E.; Turecki, G.; Nagy, C. Sex Differences in the Behavioral, Molecular, and Structural Effects of Ketamine Treatment in Depression. Int. J. Neuropsychopharmacol. 2022, 25, 75–84. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Q.; Zhang, R.; Wei, M.; Nie, Z.; Raza, F. Gender Differences in Adverse Events of Ketamine Drugs: A Real-World Study Based on FAERS. J. Clin. Pharm. Ther. 2024, 2024, 4898082. [Google Scholar] [CrossRef]
- Carrier, N.; Kabbaj, M. Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology 2013, 70, 27–34. [Google Scholar] [CrossRef]
- Morgan, C.J.; Perry, E.B.; Cho, H.S.; Krystal, J.H.; D’Souza, D.C. Greater vulnerability to the amnestic effects of ketamine in males. Psychopharmacology 2006, 187, 405–414. [Google Scholar] [CrossRef]
- Clements, J.A.; Nimmo, W.S.; Grant, I.S. Bioavailability, Pharmacokinetics, and Analgesic Activity of Ketamine in Humans. J. Pharm. Sci. 1982, 71, 539–542. [Google Scholar] [CrossRef]
- Lannes, B.; Micheletti, G.; Warter, J.M.; Kempf, E.; Di Scala, G. Behavioural, pharmacological, and biological effect of acute and chronic administration of ketamine in the rat. Neurosci. Lett. 1991, 128, 177–181. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Ozbek, E. Induction of oxidative stress in kidney. Int. J. Nephrol. 2012, 2012, 465897. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.E.; Youness, E.R.; Mohammed, N.A.; Omara, E.A.; Sleem, A.A. Effect of ketamine on oxidative stress following lipopolysaccharide administration. Comp. Clin. Pathol. 2013, 24, 53–63. [Google Scholar] [CrossRef]
- Breshears, M.A.; Confer, A.W. The Urinary System. In Pathologic Basis of Veterinary Disease; Zachary, J.F., Ed.; Elsevier: St. Louis, MO, USA, 2017; pp. 617–681. [Google Scholar]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative Stress in Chronic Kidney Disease. Iran. J. Kidney Dis. 2015, 9, 165–179. [Google Scholar]
- Adwas, A.A.; Elsayed, A.S.I.; Azab, A.E.; Quwaydir, F.A. Oxidative stress and antioxidant mechanisms in human body. J. Appl. Biotechnol. Bioeng. 2019, 6, 43–47. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2019, 54, 287–293. [Google Scholar] [CrossRef]
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J. Food Biochem. 2020, 44, 131–145. [Google Scholar] [CrossRef]
- Silva, F.C.C.; Cito, M.C.O.; Silva, M.I.G.; Moura, B.A.; Neto, M.R.A.; Feitosa, M.L.; Chaves, R.C.; Macedo, D.S.; Vasconcelos, S.M.M.; Fonteles, M.M.F.; et al. Behavioral alterations and pro-oxidant effect of a single ketamine administration to mice. Brain Res. Bull. 2010, 83, 9–15. [Google Scholar] [CrossRef]
- Liu, K.M.; Chuang, S.M.; Long, C.Y.; Lee, Y.L.; Wang, C.C.; Lu, M.C.; Lin, R.J.; Lu, J.H.; Jang, M.Y.; Wu, W.J.; et al. Ketamine-induced ulcerative cystitis and bladder apoptosis involve oxidative stress mediated by mitochondria and the endoplasmic reticulum. Am. J. Physiol. Ren. Physiol. 2015, 309, 318–331. [Google Scholar] [CrossRef]
- Rofael, H.Z. Effect of ketamine pretreatment on cocaine-mediated hepatotoxicity in rats. Toxicol. Lett. 2004, 152, 213–222. [Google Scholar] [CrossRef]
- Liang, J.; Wu, S.; Xie, W.; He, H. Ketamine ameliorates oxidative stress-induced apoptosis in experimental traumatic brain injury via the Nrf2 pathway. Drug Des. Devel Ther. 2018, 12, 845–853. [Google Scholar] [CrossRef]
- Tang, S.H.; Yu, J.G.; Li, J.J.; Sun, J.Y. Neuroprotective effect of ketamine on acute spinal cord injury in rats. Genet. Mol. Res. 2015, 14, 3551–3556. [Google Scholar] [CrossRef]
- Finco, D.R. Kidney Function. In Clinical Biochemistry of Domestic Animals, 5th ed.; Kaneko, J.J., Harvey, J.W., Bruss, M.L., Eds.; Academic Press: Cambridge, MA, USA, 1997; pp. 441–484. [Google Scholar]
- Uchino, S.; Bellomo, R.; Goldsmith, D. The meaning of the blood urea nitrogen/creatinine ratio in acute kidney injury. Clin. Kidney J. 2012, 5, 187–191. [Google Scholar] [CrossRef]
- Harrison, S.D.; Burdeshaw, J.A.; Crosby, R.G.; Cusic, A.M.; Denine, E.P. Hematology and Clinical Chemistry Reference Values for C57BL/6 x DBA/2 F1 Mice. Cancer Res. 1978, 38, 2636–2639. [Google Scholar]
- Serfilippi, L.M.; Pallman, D.R.; Russell, B. Serum Clinical Chemistry and Hematology Reference Values in Outbred Stocks of Albino Mice from Three Commonly Used Vendors and Two Inbred Strains of Albino Mice. Contemp. Top. Lab. Anim. Sci. 2003, 42, 46–52. [Google Scholar]
- Mendes, P.F.; Simon, K.A.; Hueza, I.M. Toxic and Immunotoxic Evaluation of Ketamine and/or Ethanol in Rats during 28 Days. Int. J. Pharm. Pharm. Sci. 2017, 9, 205. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Female | Male | |||||
|---|---|---|---|---|---|---|
| Control | K5 | K10 | Control | K5 | K10 | |
| ROS | 24.31 (23.75–28.71) | 23.42 (20.66–27.18) | 27.55 (25.10–29.05) | 27.58 (25.62–29.42) | 27.17 (24.91–28.57) | 27.73 (24.86–32.07) |
| MDA | 2.20 ± 0.27 | 2.03 ± 0.35 | 2.36 ± 0.50 | 2.24 ± 0.90 | 2.05 ± 0.55 | 2.36 ± 0.84 |
| PC | 7.63 (7.28–8.41) | 7.27 (7.05–7.29) | 7.26 (6.25–7.28) | 7.276 (7.25–7.30) | 7.277 (7.22–7.41) | 7.275 (7.19–7.30) |
| GSH | 0.15 ± 0.13 | 0.088 ± 0.08 | 0.091 ± 0.07 | 0.14 ± 0.05 | 0.13 ± 0.08 | 0.07 ± 0.08 |
| GSSG | 9.72 ± 4.40 | 13.82 ± 3.36 | 11.89 ± 3.05 | 12.00 ± 3.07 | 12.57 ± 3.17 | 13.54 ± 2.91 |
| SOD | 31.10 (29.25–32.81) | 35.12 (28.16–33.93) | 35.40 (30.69–40.83) | 36.87 ± 0.93 | 39.14 ± 4.76 | 41.54 ± 7.62 |
| CAT | 5.38 (5.19–6.23) | 8.30 (7.82–9.46) * | 8.32 (6.86–9.78) * | 8.85 ± 1.52 | 7.23 ± 1.74 | 10.70 ± 4.74 |
| GR | 8.49 ± 1.51 | 13.69 ± 5.59 | 12.28 ± 5.55 | 14.30 ± 1.84 | 9.11 ± 2.68 | 13.66 ± 11.53 |
| GPx | 20.15 ± 2.87 | 18.77 ± 6.36 | 21.82 ± 11.02 | 26.55 ± 5.22 | 27.83 ± 5.57 | 22.95 ± 8.74 |
| GST | 39.94 (38.55–51.77) | 53.16 (48.27–56.43) | 52.99 (45.48–71.52) | 58.89 ± 13.21 | 57.35 ± 9.59 | 69.10 ± 8.59 |
| Female | Male | |||||
|---|---|---|---|---|---|---|
| Control | K5 | K10 | Control | K5 | K10 | |
| ROS | 12.73 ± 2.92 | 12.05 ± 7.97 | 18.90 ± 5.05 | 19.10 ± 2.40 | 14.48 ± 6.36 | 17.34 ± 2.931 |
| MDA | 3.60 (2.06–3.69) | 1.93 (1.03–2.48) | 2.14 (1.48–2.78) | 2.77 ± 0.44 | 2.11 ± 1.14 | 1.78 ± 0.81 |
| PC | 1.82 ± 0.18 | 1.70 ± 0.57 | 1.37 ± 0.41 | 1.23 ± 0.62 | 1.61 ± 0.54 | 1.47 ± 0.50 |
| GSH | 348.4 (309.8–381.0) | 414.7 (317.4–524.1) | 393.0 (305.6–522.4) | 239.0 ± 129.4 | 449.5 ± 263.3 | 581.0 ± 194.6 |
| GSSG | 1511 (1371–1531) | 1835 (1583–2173) | 1695 (1355–2984) | 1713 ± 488.7 | 2198 ± 1305 | 2042 ± 727.1 |
| SOD | 373.2 (270.2–393.2) | 421.3 (363.0–483.9) | 347.8 (307.1–662.3) | 394.6 ± 77.02 | 401.8 ± 129.7 | 473.4 ± 111.9 |
| CAT | 4618 ± 1819 | 4190 ± 2089 | 6449 ± 3693 | 6959 ± 1571 | 5380 ± 2094 | 5886 ± 2328 |
| GR | 20.49 (19.72–21.36) | 15.42 (6.65–34.04) | 22.36 (12.66–60.10) | 22.16 (8.47–54.66) | 21.85 (16.15–61.75) | 33.44 (13.13–70.75) |
| GPx | 111.3 ± 70.07 | 96.38 ± 40.59 | 133.9 ± 30.07 | 149.8 ± 34.86 | 112.2 ± 55.68 | 109.3 ± 19.58 |
| GST | 58.50 (33.03–68.24) | 9.95 (4.21–75.26) | 81.71 (59.36–95.33) | 182.7 ± 80.27 | 152.6 ± 66.13 | 169.1 ± 56.58 |
| Female | Male | |||||
|---|---|---|---|---|---|---|
| Control | K5 | K10 | Control | K5 | K10 | |
| ROS | 11.71 ± 2.85 | 11.80 ± 4.99 | 12.03 ± 2.52 | 15.46 ± 5.39 | 17.33 ± 3.22 | 20.01 ± 4.82 |
| MDA | 2.27 (0.80–2.46) | 2.06 (1.77–4.50) | 2.06 (1.49–2.92) | 2.25 ± 0.79 | 2.60 ± 1.25 | 2.63 ± 0.91 |
| PC | 6.38 ± 1.47 | 5.78 ± 2.41 | 4.05 ± 1.02 | 6.48 ± 4.39 | 7.95 ± 5.29 | 5.58 ± 1.82 |
| GSH | 3.16 (1.69–7.23) | 3.39 (2.35–4.92) | 3.61 (3.17–4.19) | 4.39 ± 1.69 | 4.76 ± 2.14 | 3.72 ± 1.64 |
| GSSG | 26.76 ± 9.62 | 26.27 ± 10.52 | 28.27 ± 9.56 | 24.27 ± 7.02 | 31.53 ± 14.14 | 28.52 ± 11.64 |
| SOD | 171 (76.26–208.8) | 150.3 (116.0–208.3) | 178.2 (158.0–300.8) | 337.07 ± 62.87 | 271.63 ± 96.79 | 242.8 ± 77.09 |
| CAT | 2319 ± 779.03 | 2574 ± 932.25 | 1794 ± 932.79 | 2047 ± 1094 | 3194 ± 935.3 | 2943 ± 1156 |
| GR | 29.10 ± 2.91 | 39.92 ± 27.67 | 43.0 ± 25.20 | 34.45 ± 42.74 | 36.97 ± 24.60 | 40.59 ± 32.17 |
| GPx | 81. 70 ± 38.33 | 104.17 ± 55.78 | 123.59 ± 86.93 | 75.20 ± 21.64 | 97.22 ± 14.52 | 66.15 ± 29.53 |
| GST | 64.02 ± 31.23 | 63.28 ± 36.22 | 79.81 ± 32.69 | 60. 58 ± 35.71 | 47.76 ± 17.07 | 55.89 ± 27.49 |
| Female | Male | ||||||
|---|---|---|---|---|---|---|---|
| Control | K5 | K10 | Control | K5 | K10 | ||
| Liver Function | AST | 81.70 (58.30–105.0) | 235.7 (105.8–246.2) | 113.5 (48.80–265.3) | 144.1 ± 64.36 | 115.3 ± 56.04 | 87.16 ± 60.76 |
| ALT | 5.70 (5.20–6.10) | 2.20 (1.73–3.28) | 3.95 (2.60–6.18) | 3.10 (2.60–3.50) | 4.40 (3.50–7.0) | 2.65 (1.80–3.50) | |
| ALP | 57.80 (42.90–95.70) | 116.4 (86.25–142.8) | 86.60 (61.05–115.1) | 38.0 (31.40–57.80) | 50.35 (33.45–53.68) | 53.65 (39.18–83.33) | |
| Kidney Function | Creatinine | 1.0 ± 0.13 | 1.64 ± 1.28 | 1.33 ± 1.04 | 1.07 (0.65–1.93) | 0.80 (0.73–1.13) | 0.47 (0.37–1.23) |
| BUN | 31.90 ± 9.93 | 25.28 ± 5.78 | 31.86 ± 10.45 | 24.13 (12.28–37.24) | 31.21 (22.68–40.36) | 25.80 (21.63–36.82) | |
| Female | Male | ||||||
|---|---|---|---|---|---|---|---|
| Control | K5 | K10 | Control | K5 | K10 | ||
| Apoptosis | Caspase 3 | 75.82 (54.69–225.8) | 68.96 (38.47–82.54) | 69.12 (54.94–87.59) | 77.22 ± 28.14 | 77.35 ± 26.49 | 72.32 ± 16.62 |
| Caspase 9 | 148.2 ± 98.12 | 99.94 ± 20.39 | 102.6 ± 19.45 | 114.2 ± 21.75 | 105.3 ± 19.59 | 111.4 ± 16.24 | |
| Inflammation | NO | 0.82 (0.77–1.01) | 0.57 (0.48–0.75) | 0.75 (0.60–0.88) | 0.71 (0.57–1.00) | 0.93 (0.72–1.14) | 0.71 (0.66–1.24) |
| DNA damage | dsDNA breaks | 169.8 (92.81–217.0) | 181.5 (142.2–243.2) | 271.1 (130.5–460.5) | 337.0 ± 146.3 | 191.7 ± 91.28 | 304.3 ± 304.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, C.A.; Sampaio, L.; Félix, L.M.; Monteiro, S.M.; Antunes, L.; Venâncio, C. A Preliminary Approach to Oral Low-Dose Ketamine Self-Administration in Mice (Mus musculus). Curr. Issues Mol. Biol. 2025, 47, 592. https://doi.org/10.3390/cimb47080592
Rocha CA, Sampaio L, Félix LM, Monteiro SM, Antunes L, Venâncio C. A Preliminary Approach to Oral Low-Dose Ketamine Self-Administration in Mice (Mus musculus). Current Issues in Molecular Biology. 2025; 47(8):592. https://doi.org/10.3390/cimb47080592
Chicago/Turabian StyleRocha, Cláudia A., Luís Sampaio, Luís M. Félix, Sandra M. Monteiro, Luís Antunes, and Carlos Venâncio. 2025. "A Preliminary Approach to Oral Low-Dose Ketamine Self-Administration in Mice (Mus musculus)" Current Issues in Molecular Biology 47, no. 8: 592. https://doi.org/10.3390/cimb47080592
APA StyleRocha, C. A., Sampaio, L., Félix, L. M., Monteiro, S. M., Antunes, L., & Venâncio, C. (2025). A Preliminary Approach to Oral Low-Dose Ketamine Self-Administration in Mice (Mus musculus). Current Issues in Molecular Biology, 47(8), 592. https://doi.org/10.3390/cimb47080592