
Neuroinflammation: Mechanisms, Dual Roles, and Therapeutic Strategies in Neurological Disorders
Abstract
1. Introduction
2. Mechanisms of Neuroinflammation
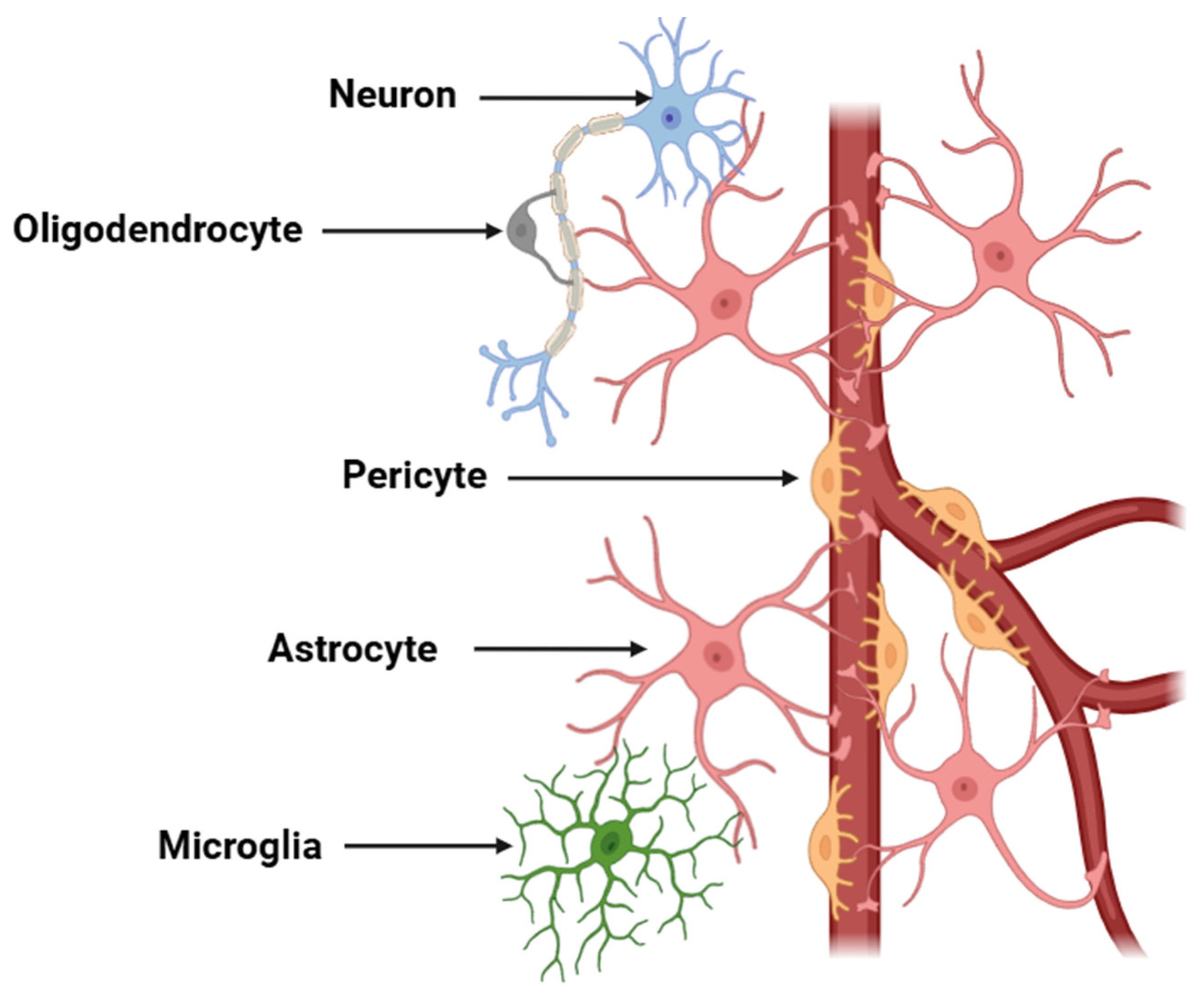
2.1. Cellular Mediators
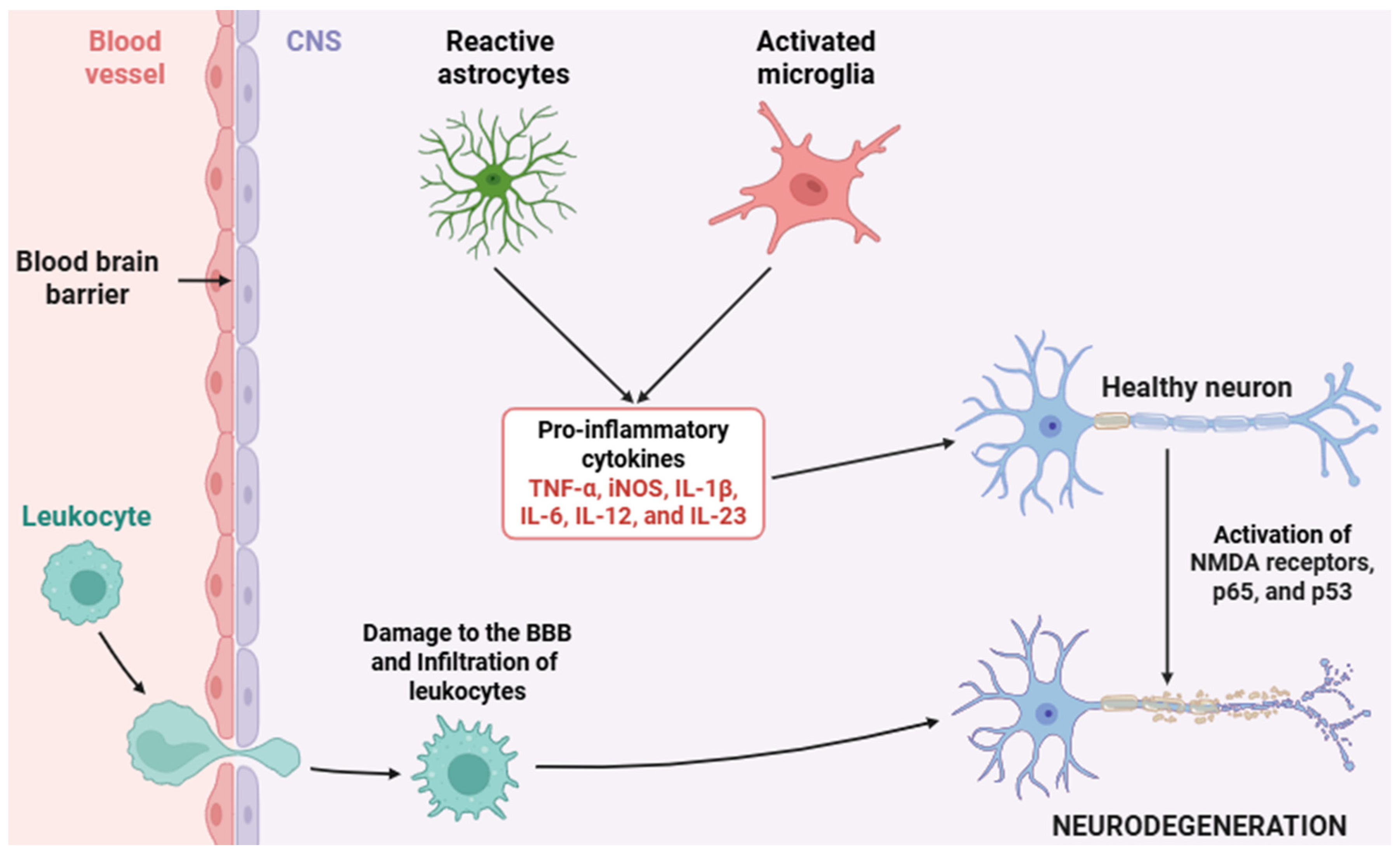
2.2. Pro-Inflammatory Mediators and Their Signaling Pathways
3. The Dual Role of Neuroinflammation
4. Therapeutic Strategies Targeting Neuroinflammation
4.1. Established Therapies
4.1.1. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
4.1.2. Corticosteroids
4.2. Experimental Approaches
4.2.1. Monoclonal Antibodies
4.2.2. Other Innovative Therapeutic Strategies
5. Future Directions and Research Gaps
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 15-LOX | 15-lipoxygenase |
| A1 | A1 phenotype astroglia |
| A2 | A2 phenotype astroglia |
| AAV | Anti-neutrophil cytoplasmic antibody-associated vasculitis |
| AD | Alzheimer’s disease |
| ALX/FPR2 | ALX/formyl peptide receptor 2 |
| AMPK | AMP-activated protein kinase |
| AP-1 | Activator protein 1 |
| Arg-1 | Arginase 1 |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| APC | Antigen-presenting cell |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid beta |
| BAFF | B-cell activating factor |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| BIM | Bcl-2 interacting mediator of cell death |
| C1q | Complement component 1q |
| C3 | Complement component 3 |
| C3a | Complement component 3a |
| C3aR | Complement component 3a receptor |
| C3b | Complement component 3b |
| C5 | Complement component 5 |
| C5a | Complement component 5a |
| C5aR1 | Complement component 5a receptor 1 |
| cAMP | Cyclic adenosine monophosphate |
| CARD | Caspase activation and recruitment domain |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| CD11b | Cluster of differentiation 11b |
| CD18 | Cluster of differentiation 18 |
| CD19 | Cluster of differentiation 19 |
| CD20 | Cluster of differentiation 20 |
| CD52 | Cluster of differentiation 52 |
| CD200 | Cluster of differentiation 200 |
| ChemR23 | Chemokine-like receptor 1 |
| CI | Confidence interval |
| c-Jun | c-Jun proto-oncogene |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| COX-1 | Cyclooxygenase 1 |
| COX-2 | Cyclooxygenase 2 |
| cPLA2 | Cytosolic phospholipase A2 |
| CR3 | Complement receptor 3 |
| CREB | cAMP response element-binding protein |
| CSF1R | Colony-stimulating factor 1 receptor |
| CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CX3CR1 | C-X3-C motif chemokine receptor 1 |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| DAMP | Damage-associated molecular pattern |
| DHA | Docosahexaenoic acid |
| DNA | Deoxyribonucleic acid |
| EP | E-prostanoid receptor (PGE2 receptor) |
| EP1 | E-type prostanoid receptor 1 |
| EP2 | E-type prostanoid receptor 2 |
| EP3 | E-type prostanoid receptor 3 |
| EP4 | E-type prostanoid receptor 4 |
| EPA | Eicosapentaenoic acid |
| ERK | Extracellular signal-regulated kinase |
| FASL | Fas ligand |
| GPCR | G protein-coupled receptor |
| GPR32 | G protein-coupled receptor 32 |
| GR | Glucocorticoid receptor |
| GRE | Glucocorticoid response element |
| HMGB1 | High mobility group box 1 |
| Hsp | Heat shock protein |
| HSP70 | Heat shock protein 70 |
| HSP90 | Heat shock protein 90 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IFN-β | Interferon beta |
| IFNβ-1a | Interferon beta-1a |
| IGF-1 | Insulin-like growth factor 1 |
| IKK | IκB kinase |
| IKKε | IκB kinase epsilon |
| IL-10 | Interleukin 10 |
| IL-12 | Interleukin 12 |
| IL-17 | Interleukin 17 |
| IL-17A | Interleukin 17A |
| IL-1R | Interleukin 1 receptor |
| IL-1α | Interleukin 1 alpha |
| IL-1β | Interleukin 1 beta |
| IL-23 | Interleukin 23 |
| IL-6 | Interleukin 6 |
| IL-6R | Interleukin 6 receptor |
| iNOS | Inducible nitric oxide synthase |
| IRAK1 | Interleukin 1 receptor-associated kinase 1 |
| IRAK4 | Interleukin 1 receptor-associated kinase 4 |
| IRF | Interferon regulatory factor |
| IRF3 | Interferon regulatory factor 3 |
| IκBα | Inhibitor of κB alpha |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LD | Lafora disease |
| LOX | Lipoxygenase |
| LXA4 | Lipoxin A4 |
| M1 | M1 phenotype microglia |
| M2 | M2 phenotype microglia |
| mAb | Monoclonal antibody |
| MaR1 | Maresin 1 |
| MAC | Membrane attack complex |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| MMP-2 | Matrix metalloproteinase 2 |
| MMP-9 | Matrix metalloproteinase 9 |
| MRI | Magnetic resonance imaging |
| MS | Multiple sclerosis |
| MyD88 | Myeloid differentiation primary response 88 |
| NCLs | Neuronal ceroid lipofuscinoses |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NLR | NOD-like receptor |
| NLRP3 | NLR family pyrin domain containing 3 |
| NMDA | N-methyl-D-aspartate |
| NMOSD | Neuromyelitis optica spectrum disorder |
| NO | Nitric oxide |
| NOX2 | NADPH oxidase 2 |
| NPSLE | Neuropsychiatric systemic lupus erythematosus |
| Nrf-2 | Nuclear factor erythroid 2-related factor 2 |
| NSAID | Non-steroidal anti-inflammatory drug |
| OPC | Oligodendrocyte precursor cell |
| OR | Odds ratio |
| P2X7 | Purinergic receptor P2X, ligand-gated ion channel 7 |
| P2Y12 | Purinergic receptor P2Y, G-protein coupled, 12 |
| p300 | E1A binding protein p300 |
| p38 | p38 mitogen-activated protein kinase |
| p50 | NF-κB p50 subunit |
| p53 | Tumor protein 53 |
| p65 | NF-κB p65 (RelA) subunit |
| PAMP | Pathogen-associated molecular pattern |
| PD | Parkinson’s disease |
| PD1 | Protectin D1 |
| PGE2 | Prostaglandin E2 |
| PGH2 | Prostaglandin H2 |
| PI3K/AKT | Phosphoinositide 3-kinase/Protein kinase B pathway |
| PPAR | Peroxisome proliferator-activated receptor |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| pro-IL-18 | Pro-interleukin 18 |
| pro-IL-1β | Pro-interleukin 1 beta |
| PRR | Pattern recognition receptor |
| PYD | Pyrin domain |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RLR | RIG-I-like receptor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RR | Relative risk |
| RRMS | Relapsing–remitting multiple sclerosis |
| RvD1 | Resolvin D1 |
| RvE1 | Resolvin E1 |
| S100B | S100 calcium-binding protein B |
| SPM | Specialized pro-resolving mediators |
| SRC-1 | Steroid receptor coactivator-1 |
| STAT | Signal transducer and activator of transcription |
| STAT1 | Signal transducer and activator of transcription 1 |
| TBI | Traumatic brain injury |
| TBK1 | TANK-binding kinase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TLR4 | Toll-like receptor 4 |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TNFR2 | Tumor necrosis factor receptor 2 |
| TNF-α | Tumor necrosis factor alpha |
| TRAF6 | TNF receptor-associated factor 6 |
| TRIF | TIR-domain-containing adapter-inducing interferon beta |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
References
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, A.G.; Zgavc, T.; Kooijman, R.; Hachimi-Idrissi, S.; Sarre, S.; Michotte, Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J. Neuroinflamm. 2010, 7, 74. [Google Scholar] [CrossRef]
- Kim, M.E.; Lee, J.S. Mechanisms and Emerging Regulators of Neuroinflammation: Exploring New Therapeutic Strategies for Neurological Disorders. Curr. Issues Mol. Biol. 2024, 47, 8. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Di Vito, A.; Donato, G.; Tomassoni, D. Molecular and Cellular Mechanisms of Neuroinflammation. Biomed. Res. Int. 2017, 2017, 8417183. [Google Scholar] [CrossRef]
- Afridi, R.; Bhusal, A.; Tsuda, M.; Ryu, H.; Suk, K. Function of Glial Cells in Neuroinflammatory and Neuroimmunological Responses II. Cells 2023, 12, 1750. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Figuera-Losada, M.; Rojas, C.; Slusher, B.S. Inhibition of microglia activation as a phenotypic assay in early drug discovery. J. Biomol. Screen 2014, 19, 17–31. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Liy, P.M.; Puzi, N.N.A.; Jose, S.; Vidyadaran, S. Nitric oxide modulation in neuroinflammation and the role of mesenchymal stem cells. Exp. Biol. Med. 2021, 246, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor κB (NF-κB)-Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Suter, M.R. p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 2007, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, X.; Escames, G.; Lei, W.; Zhang, X.; Li, M.; Jing, T.; Yao, Y.; Qiu, Z.; Wang, Z.; et al. The NLRP3 inflammasome: Contributions to inflammation-related diseases. Cell. Mol. Biol. Lett. 2023, 28, 51. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Gradisnik, L.; Velnar, T. Astrocytes in the central nervous system and their functions in health and disease: A review. World J. Clin. Cases 2023, 11, 3385–3394. [Google Scholar] [CrossRef]
- Nutma, E.; van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells 2020, 9, 600. [Google Scholar] [CrossRef]
- Bouvier, D.S.; Fixemer, S.; Heurtaux, T.; Jeannelle, F.; Frauenknecht, K.B.M.; Mittelbronn, M. The Multifaceted Neurotoxicity of Astrocytes in Ageing and Age-Related Neurodegenerative Diseases: A Translational Perspective. Front. Physiol. 2022, 13, 814889. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood-Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol Sci. 2023, 24, 17146. [Google Scholar] [CrossRef] [PubMed]
- Manich, G.; Recasens, M.; Valente, T.; Almolda, B.; González, B.; Castellano, B. Role of the CD200-CD200R Axis During Homeostasis and Neuroinflammation. Neuroscience 2019, 405, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Hippensteel, R.; Shimizu, S.; Nicolai, J.; Fatatis, A.; Meucci, O. Interactions between chemokines: Regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J. Biol. Chem. 2010, 285, 10563–10571. [Google Scholar] [CrossRef]
- Ledonne, A.; Mercuri, N.B. On the Modulatory Roles of Neuregulins/ErbB Signaling on Synaptic Plasticity. Int. J. Mol. Sci. 2019, 21, 275. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S.; Müller, V. From Homeostasis to Neuroinflammation: Insights into Cellular and Molecular Interactions and Network Dynamics. Cells 2025, 14, 54. [Google Scholar] [CrossRef]
- Mehta, S.L.; Arruri, V.; Vemuganti, R. Role of transcription factors, noncoding RNAs, epitranscriptomics, and epigenetics in post-ischemic neuroinflammation. J. Neurochem. 2024, 168, 3430–3448. [Google Scholar] [CrossRef]
- Lee, S.H.; Suk, K. Emerging roles of protein kinases in microglia-mediated neuroinflammation. Biochem. Pharmacol. 2017, 146, 1–9. [Google Scholar] [CrossRef]
- David, S.; López-Vales, R. Bioactive Lipid Mediators in the Initiation and Resolution of Inflammation after Spinal Cord Injury. Neuroscience 2021, 466, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.V.; Bastos, L.F.; Limborço-Filho, M.; Fiebich, B.L.; de Oliveira, A.C. Role of prostaglandins in neuroinflammatory and neurodegenerative diseases. Mediat. Inflamm. 2012, 2012, 946813. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2020, 87, 1–10. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- Pajares, M.; I Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Chan, A.; Ouyang, J.; Nguyen, K.; Jones, A.; Basso, S.; Karasik, R. Traumatic brain injuries: A neuropsychological review. Front. Behav. Neurosci. 2024, 18, 1326115. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef]
- Takahashi, K.; Nelvagal, H.R.; Lange, J.; Cooper, J.D. Glial Dysfunction and Its Contribution to the Pathogenesis of the Neuronal Ceroid Lipofuscinoses. Front. Neurol. 2022, 13, 886567. [Google Scholar] [CrossRef]
- Della Vecchia, S.; Marchese, M.; Santorelli, F.M. Glial Contributions to Lafora Disease: A Systematic Review. Biomedicines 2022, 10, 3103. [Google Scholar] [CrossRef] [PubMed]
- Baud, O.; Saint-Faust, M. Neuroinflammation in the Developing Brain: Risk Factors, Involvement of Microglial Cells, and Implication for Early Anesthesia. Anesth. Analg. 2019, 128, 718–725. [Google Scholar] [CrossRef]
- Bairamian, D.; Sha, S.; Rolhion, N.; Sokol, H.; Dorothée, G.; Lemere, C.A.; Krantic, S. Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 19. [Google Scholar] [CrossRef]
- Sun, Y.; Koyama, Y.; Shimada, S. Inflammation from Peripheral Organs to the Brain: How Does Systemic Inflammation Cause Neuroinflammation? Front. Aging Neurosci. 2022, 14, 903455. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Prakash, R.; Kumari, N.; Ali Khan, M.; Quaiyoom Khan, A.; Uddin, S.; Verma, S.; Ab Robertson, A.; Boltze, J.; Shadab Raza, S. MCC950 reduces autophagy and improves cognitive function by inhibiting NLRP3-dependent neuroinflammation in a rat model of Alzheimer’s disease. Brain Behav. Immun. 2024, 116, 70–84. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhu, J.; Wu, L.; Xu, G.; Dai, J.; Liu, X. Tetracycline inhibits local inflammation induced by cerebral ischemia via modulating autophagy. PLoS ONE 2012, 7, e48672. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Gulyaeva, N.V. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines 2022, 10, 1139. [Google Scholar] [CrossRef]
- Mallick, R.; Basak, S.; Chowdhury, P.; Bhowmik, P.; Das, R.K.; Banerjee, A.; Paul, S.; Pathak, S.; Duttaroy, A.K. Targeting Cytokine-Mediated Inflammation in Brain Disorders: Developing New Treatment Strategies. Pharmaceuticals 2025, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.; Ulu, A.; Hanson, C.; Cameron-Smith, E.; Bertoni, J.; Wuebker, J.; Fisher, A.; Siu, K.C.; Marmelat, V.; Adamec, J.; et al. Role of Specialized Pro-resolving Mediators in Reducing Neuroinflammation in Neurodegenerative Disorders. Front. Aging Neurosci. 2022, 14, 780811. [Google Scholar] [CrossRef]
- Valente, M.; Dentoni, M.; Bellizzi, F.; Kuris, F.; Gigli, G.L. Specialized Pro-Resolving Mediators in Neuroinflammation: Overview of Studies and Perspectives of Clinical Applications. Molecules 2022, 27, 4836. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, X.; Liu, S.; Shen, D.; Zhu, J.; Liu, K. Role of Resolvins in the Inflammatory Resolution of Neurological Diseases. Front. Pharmacol. 2020, 11, 612. [Google Scholar] [CrossRef]
- Sammons, M.; Popescu, M.C.; Chi, J.; Liberles, S.D.; Gogolla, N.; Rolls, A. Brain-body physiology: Local, reflex, and central communication. Cell 2024, 187, 5877–5890. [Google Scholar] [CrossRef]
- Florio, T.M. Emergent Aspects of the Integration of Sensory and Motor Functions. Brain Sci. 2025, 15, 162. [Google Scholar] [CrossRef]
- Powers, R.M.; Hevner, R.F.; Halpain, S. The Neuron Navigators: Structure, function, and evolutionary history. Front. Mol. Neurosci. 2023, 15, 1099554. [Google Scholar] [CrossRef] [PubMed]
- Demmings, M.D.; da Silva Chagas, L.; Traetta, M.E.; Rodrigues, R.S.; Acutain, M.F.; Barykin, E.; Datusalia, A.K.; German-Castelan, L.; Mattera, V.S.; Mazengenya, P.; et al. (Re)building the nervous system: A review of neuron-glia interactions from development to disease. J. Neurochem. 2025, 169, e16258. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells as Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Xu, L.; Wang, J.; Ding, Y.; Wang, L.; Zhu, Y.J. Current Knowledge of Microglia in Traumatic Spinal Cord Injury. Front. Neurol. 2022, 12, 796704. [Google Scholar] [CrossRef]
- Waisman, A.; Ginhoux, F.; Greter, M.; Bruttger, J. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015, 36, 625–636. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Liu, D.; Hsueh, S.C.; Tweedie, D.; Price, N.; Glotfelty, E.; Lecca, D.; Telljohann, R.; deCabo, R.; Hoffer, B.J.; Greig, N.H. Chronic inflammation with microglia senescence at basal forebrain: Impact on cholinergic deficit in Alzheimer’s brain haemodynamics. Brain Commun. 2024, 6, fcae204. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.L.; Singh, S.K.; Campos-Campos, J.; Colmena, C.; Campo-Palacio, I.; Alvarez-Gamez, K.; Caballero, O.; Jorda, A. Functions of Astrocytes under Normal Conditions and after a Brain Disease. Int. J. Mol. Sci. 2023, 24, 8434. [Google Scholar] [CrossRef] [PubMed]
- Rupareliya, V.P.; Singh, A.A.; Butt, A.M.; Hariharan, A.; Kumar, H. The “molecular soldiers” of the CNS: Astrocytes, a comprehensive review on their roles and molecular signatures. Eur. J. Pharmacol. 2023, 959, 176048. [Google Scholar] [CrossRef]
- Hösli, L.; Zuend, M.; Bredell, G.; Zanker, H.S.; Porto de Oliveira, C.E.; Saab, A.S.; Weber, B. Direct vascular contact is a hallmark of cerebral astrocytes. Cell Rep. 2022, 39, 110599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Cao, Y.; Yang, J. Astrocyte-Mediated Neuroinflammation in Neurological Conditions. Biomolecules 2024, 14, 1204. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Ye, Q.; Jo, J.; Wang, C.Y.; Oh, H.; Zhan, J.; Choy, T.J.; Kim, K.I.; D’Alessandro, A.; Reshetnyak, Y.K.; Jung, S.Y.; et al. Astrocytic Slc4a4 regulates blood-brain barrier integrity in healthy and stroke brains via a CCL2-CCR2 pathway and NO dysregulation. Cell Rep. 2024, 43, 114193. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Moulson, A.J.; Squair, J.W.; Franklin, R.J.M.; Tetzlaff, W.; Assinck, P. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Front. Cell. Neurosci. 2021, 15, 703810. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Wang, F.; Huang, N.X.; Xiao, L.; Mei, F. Oligodendrocytes and myelin: Active players in neurodegenerative brains? Dev. Neurobiol. 2022, 82, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Pukos, N.; Goodus, M.T.; Sahinkaya, F.R.; McTigue, D.M. Myelin status and oligodendrocyte lineage cells over time after spinal cord injury: What do we know and what still needs to be unwrapped? Glia 2019, 67, 2178–2202. [Google Scholar] [CrossRef]
- Zveik, O.; Rechtman, A.; Brill, L.; Vaknin-Dembinsky, A. Anti- and pro-inflammatory milieu differentially regulate differentiation and immune functions of oligodendrocyte progenitor cells. Immunology 2024, 171, 618–633. [Google Scholar] [CrossRef] [PubMed]
- López-Muguruza, E.; Matute, C. Alterations of Oligodendrocyte and Myelin Energy Metabolism in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 12912. [Google Scholar] [CrossRef]
- Boccazzi, M.; Raffaele, S.; Fumagalli, M. Not only myelination: The immune-inflammatory functions of oligodendrocytes. Neural Regen. Res. 2022, 17, 2661–2663. [Google Scholar]
- Chen, J.Q.A.; Wever, D.D.; McNamara, N.B.; Bourik, M.; Smolders, J.; Hamann, J.; Huitinga, I. Inflammatory microglia correlate with impaired oligodendrocyte maturation in multiple sclerosis. Front. Immunol. 2025, 15, 1522381. [Google Scholar] [CrossRef]
- Gualerzi, A.; Lombardi, M.; Verderio, C. Microglia-oligodendrocyte intercellular communication: Role of extracellular vesicle lipids in functional signalling. Neural Regen. Res. 2021, 16, 1194–1195. [Google Scholar]
- Zelic, M.; Blazier, A.; Pontarelli, F.; LaMorte, M.; Huang, J.; Tasdemir-Yilmaz, O.E.; Ren, Y.; Ryan, S.K.; Shapiro, C.; Morel, C.; et al. Single-cell transcriptomic and functional studies identify glial state changes and a role for inflammatory RIPK1 signaling in ALS pathogenesis. Immunity 2025, 58, 961–979.e8. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Maliar, N.L.; Talbot, E.J.; Edwards, A.R.; Khoronenkova, S.V. Microglial inflammation in genome instability: A neurodegenerative perspective. DNA Repair 2024, 135, 103634. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.; Li, H.; Wang, F.; Yao, S. Toll-like receptor 4: A potential therapeutic target for multiple human diseases. Biomed. Pharmacother. 2023, 166, 115338. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.; Lee, J.H.; Hwangbo, C. Toll-like receptor 4 (TLR4): New insight immune and aging. Immun. Ageing 2023, 20, 67. [Google Scholar] [CrossRef]
- Laird, M.H.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef]
- Shalaby, K.H.; Al Heialy, S.; Tsuchiya, K.; Farahnak, S.; McGovern, T.K.; Risse, P.A.; Suh, W.K.; Qureshi, S.T.; Martin, J.G. The TLR4-TRIF pathway can protect against the development of experimental allergic asthma. Immunology 2017, 152, 138–149. [Google Scholar] [CrossRef]
- Zheng, C.; Chen, J.; Chu, F.; Zhu, J.; Jin, T. Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Front. Mol. Neurosci. 2020, 12, 314. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Xu, J.; Xiao, K.; Xu, Y.; Guo, T.; Zhang, L.; Wang, J.; Zheng, H. β-TrCP Restricts Lipopolysaccharide (LPS)-Induced Activation of TRAF6-IKK Pathway Upstream of IκBα Signaling. Front. Immunol. 2018, 9, 2930. [Google Scholar] [CrossRef]
- Dhillon, B.; Aleithan, F.; Abdul-Sater, Z.; Abdul-Sater, A.A. The Evolving Role of TRAFs in Mediating Inflammatory Responses. Front. Immunol. 2019, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Mota, M.; Pizzi, M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim. Biophys. Acta 2016, 1862, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Almouhanna, F.; Chausse, B. Interferon γ: A master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 2022, 45, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu. Rev. Immunol. 2023, 41, 301–316. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Hanamsagar, R.; Torres, V.; Kielian, T. Inflammasome activation and IL-1β/IL-18 processing are influenced by distinct pathways in microglia. J. Neurochem. 2011, 119, 736–748. [Google Scholar] [CrossRef]
- Calovi, S.; Mut-Arbona, P.; Sperlágh, B. Microglia and the Purinergic Signaling System. Neuroscience 2019, 405, 137–147. [Google Scholar] [CrossRef]
- Tewari, M.; Michalski, S.; Egan, T.M. Modulation of Microglial Function by ATP-Gated P2X7 Receptors: Studies in Rat, Mice and Human. Cells 2024, 13, 161. [Google Scholar] [CrossRef]
- Irino, Y.; Nakamura, Y.; Inoue, K.; Kohsaka, S.; Ohsawa, K. Akt activation is involved in P2Y12 receptor-mediated chemotaxis of microglia. J. Neurosci. Res. 2008, 86, 1511–1519. [Google Scholar] [CrossRef]
- Zhang, H.J.; Chen, Y.T.; Hu, X.L.; Cai, W.T.; Wang, X.Y.; Ni, W.F.; Zhou, K.L. Functions and mechanisms of cytosolic phospholipase A2 in central nervous system trauma. Neural Regen. Res. 2023, 18, 258–266. [Google Scholar] [PubMed]
- Akundi, R.S.; Candelario-Jalil, E.; Hess, S.; Hüll, M.; Lieb, K.; Gebicke-Haerter, P.J.; Fiebich, B.L. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia 2005, 51, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, Q. Prostaglandin EP2 receptor: Novel therapeutic target for human cancers (Review). Int. J. Mol. Med. 2018, 42, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, Y.; Hou, R.; Hao, J.; Jiang, J. Inhibiting the PGE2 Receptor EP2 Mitigates Excitotoxicity and Ischemic Injury. ACS Pharmacol. Transl. Sci. 2020, 3, 635–643. [Google Scholar] [CrossRef]
- Surace, M.J.; Block, M.L. Targeting microglia-mediated neurotoxicity: The potential of NOX2 inhibitors. Cell. Mol. Life Sci. 2012, 69, 2409–2427. [Google Scholar] [CrossRef]
- Hu, C.F.; Wu, S.P.; Lin, G.J.; Shieh, C.C.; Hsu, C.S.; Chen, J.W.; Chen, S.H.; Hong, J.S.; Chen, S.J. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 12, 638381. [Google Scholar] [CrossRef]
- Liu, P.W.; Chen, M.F.; Tsai, A.P.; Lee, T.J. STAT1 mediates oroxylin a inhibition of iNOS and pro-inflammatory cytokines expression in microglial BV-2 cells. PLoS ONE 2012, 7, e50363. [Google Scholar] [CrossRef]
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385. [Google Scholar] [CrossRef]
- Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. The Nitration of Proteins, Lipids and DNA by Peroxynitrite Derivatives-Chemistry Involved and Biological Relevance. Stresses 2022, 2, 53–64. [Google Scholar] [CrossRef]
- Chen, Y.; Chu, J.M.T.; Chang, R.C.C.; Wong, G.T.C. The Complement System in the Central Nervous System: From Neurodevelopment to Neurodegeneration. Biomolecules 2022, 12, 337. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Pei, H. C1q and central nervous system disorders. Front. Immunol. 2023, 14, 1145649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fan, L.; Xia, S.; Yang, Q.; Zhang, Z.; Chen, H.; Zeng, H.; Fu, X.; Peng, Y.; Xu, C.; et al. Role of complement C1q/C3-CR3 signaling in brain injury after experimental intracerebral hemorrhage and the effect of minocycline treatment. Front. Immunol. 2022, 13, 919444. [Google Scholar] [CrossRef] [PubMed]
- Ramaglia, V.; Hughes, T.R.; Donev, R.M.; Ruseva, M.M.; Wu, X.; Huitinga, I.; Baas, F.; Neal, J.W.; Morgan, B.P. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 965–970. [Google Scholar] [CrossRef]
- Guo, R.F.; Ward, P.A. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, T.; Bosco, D.B.; Xie, M.; Zheng, J.; Dheer, A.; Ying, Y.; Wu, Q.; Lennon, V.A.; Wu, L.J. The complement C3-C3aR pathway mediates microglia-astrocyte interaction following status epilepticus. Glia 2021, 69, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.; Schartz, N.D.; Balderrama-Gutierrez, G.; Liang, H.Y.; Chu, S.H.; Selvan, P.; Gomez-Arboledas, A.; Petrisko, T.J.; Fonseca, M.I.; Mortazavi, A.; et al. Modulation of C5a–C5aR1 signaling alters the dynamics of AD progression. J. Neuroinflamm. 2022, 19, 178. [Google Scholar] [CrossRef]
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef]
- Warwick, C.A.; Keyes, A.L.; Woodruff, T.M.; Usachev, Y.M. The complement cascade in the regulation of neuroinflammation, nociceptive sensitization, and pain. J. Biol. Chem. 2021, 297, 101085. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Acharya, M.M.; Tenner, A.J. The Role of Complement in Synaptic Pruning and Neurodegeneration. Immunotargets Ther. 2021, 10, 373–386. [Google Scholar] [CrossRef]
- Song, J.; Wu, C.; Korpos, E.; Zhang, X.; Agrawal, S.M.; Wang, Y.; Faber, C.; Schäfers, M.; Körner, H.; Opdenakker, G.; et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep. 2015, 10, 1040–1054. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood-brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Joffre, C.; Rey, C.; Layé, S. N-3 Polyunsaturated Fatty Acids and the Resolution of Neuroinflammation. Front. Pharmacol. 2019, 10, 1022. [Google Scholar] [CrossRef] [PubMed]
- Salsinha, A.S.; Socodato, R.; Rodrigues, A.; Vale-Silva, R.; Relvas, J.B.; Pintado, M.; Rodríguez-Alcalá, L.M. Potential of omega-3 and conjugated fatty acids to control microglia inflammatory imbalance elicited by obesogenic nutrients. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2023, 1868, 159331. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, M.; Chiurchiù, V. Specialized Pro-resolving Lipid Mediators and Glial Cells: Emerging Candidates for Brain Homeostasis and Repair. Front. Cell. Neurosci. 2021, 15, 673549. [Google Scholar] [CrossRef]
- Dokalis, N.; Prinz, M. Resolution of neuroinflammation: Mechanisms and potential therapeutic option. Semin. Immunopathol. 2019, 41, 699–709. [Google Scholar] [CrossRef]
- Ji, R.R. Specialized Pro-Resolving Mediators as Resolution Pharmacology for the Control of Pain and Itch. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 273–293. [Google Scholar] [CrossRef]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Neurath, M.F. Resolution of inflammation: From basic concepts to clinical application. Semin. Immunopathol. 2019, 41, 627–631. [Google Scholar] [CrossRef]
- Chen, Y.; Kou, Y.; Ni, Y.; Yang, H.; Xu, C.; Fan, H.; Liu, H. Microglia efferocytosis: An emerging mechanism for the resolution of neuroinflammation in Alzheimer’s disease. J. Neuroinflamm. 2025, 22, 96. [Google Scholar] [CrossRef]
- Wu, S.Y.; Pan, B.S.; Tsai, S.F.; Chiang, Y.T.; Huang, B.M.; Mo, F.E.; Kuo, Y.M. BDNF reverses aging-related microglial activation. J. Neuroinflamm. 2020, 17, 210. [Google Scholar] [CrossRef]
- Wan, Y.; Gao, W.; Zhou, K.; Liu, X.; Jiang, W.; Xue, R.; Wu, W. Role of IGF-1 in neuroinflammation and cognition deficits induced by sleep deprivation. Neurosci. Lett. 2022, 776, 136575. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.A.; Planel, E.; Terrando, N. Impaired Resolution of Inflammation in Alzheimer’s Disease: A Review. Front. Immunol. 2017, 8, 1464. [Google Scholar] [CrossRef] [PubMed]
- Lotz, S.K.; Blackhurst, B.M.; Reagin, K.L.; Funk, K.E. Microbial Infections Are a Risk Factor for Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 15, 691136. [Google Scholar] [CrossRef] [PubMed]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation After Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef]
- Alsbrook, D.L.; Di Napoli, M.; Bhatia, K.; Biller, J.; Andalib, S.; Hinduja, A.; Rodrigues, R.; Rodriguez, M.; Sabbagh, S.Y.; Selim, M.; et al. Neuroinflammation in Acute Ischemic and Hemorrhagic Stroke. Curr. Neurol. Neurosci. Rep. 2023, 23, 407–431. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia Crosstalk in Neuroinflammatory Diseases. Front. Cell. Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef]
- Oyarce, K.; Cepeda, M.Y.; Lagos, R.; Garrido, C.; Vega-Letter, A.M.; Garcia-Robles, M.; Luz-Crawford, P.; Elizondo-Vega, R. Neuroprotective and Neurotoxic Effects of Glial-Derived Exosomes. Front. Cell. Neurosci. 2022, 16, 920686. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Amanollahi, M.; Jameie, M.; Heidari, A.; Rezaei, N. The Dialogue Between Neuroinflammation and Adult Neurogenesis: Mechanisms Involved and Alterations in Neurological Diseases. Mol. Neurobiol. 2023, 60, 923–959. [Google Scholar] [CrossRef]
- Zhu, Y.; Webster, M.J.; Mendez Victoriano, G.; Middleton, F.A.; Massa, P.T.; Weickert, C.S. Molecular Evidence for Altered Angiogenesis in Neuroinflammation-Associated Schizophrenia and Bipolar Disorder Implicate an Abnormal Midbrain Blood-Brain Barrier. Schizophr. Bull. 2024, sbae184. [Google Scholar] [CrossRef]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front. Cell. Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal. Transduct. Target Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef]
- Mancuso, M.R.; Kuhnert, F.; Kuo, C.J. Developmental angiogenesis of the central nervous system. Lymphat. Res. Biol. 2008, 6, 173–180. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef]
- Johnson, H.J.; Koshy, A.A. Understanding neuroinflammation through central nervous system infections. Curr. Opin. Neurobiol. 2022, 76, 102619. [Google Scholar] [CrossRef]
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Viviani, B.; Boraso, M.; Marchetti, N.; Marinovich, M. Perspectives on neuroinflammation and excitotoxicity: A neurotoxic conspiracy? Neurotoxicology 2014, 43, 10–20. [Google Scholar] [CrossRef]
- Mottahedin, A.; Ardalan, M.; Chumak, T.; Riebe, I.; Ek, J.; Mallard, C. Effect of Neuroinflammation on Synaptic Organization and Function in the Developing Brain: Implications for Neurodevelopmental and Neurodegenerative Disorders. Front. Cell. Neurosci. 2017, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, M.; Foss, J.F.; Giordano, T. Neuroinflammation, its Role in Alzheimer’s Disease and Therapeutic Strategie. J. Prev. Alzheimers Dis. 2023, 10, 686–698. [Google Scholar] [CrossRef]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Bjelobaba, I.; Savic, D.; Lavrnja, I. Multiple Sclerosis and Neuroinflammation: The Overview of Current and Prospective Therapies. Curr. Pharm. Des. 2017, 23, 693–730. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Peng, W.; Xie, Y.; Liao, C.; Bai, Y.; Wang, H.; Li, C. Spatiotemporal patterns of gliosis and neuroinflammation in presenilin 1/2 conditional double knockout mice. Front. Aging Neurosci. 2022, 14, 966153. [Google Scholar] [CrossRef]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef]
- Bowirrat, A. Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis. J. Pers. Med. 2022, 12, 1817. [Google Scholar] [CrossRef]
- McCarthy, M.M. Sex differences in neuroimmunity as an inherent risk factor. Neuropsychopharmacology 2019, 44, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Jagodic, M.; Piehl, F.; Wallström, E. Genetics of autoimmune neuroinflammation. Curr. Opin. Immunol. 2006, 18, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Murtaj, V.; Belloli, S.; Di Grigoli, G.; Pannese, M.; Ballarini, E.; Rodriguez-Menendez, V.; Marmiroli, P.; Cappelli, A.; Masiello, V.; Monterisi, C.; et al. Age and Sex Influence the Neuro-inflammatory Response to a Peripheral Acute LPS Challenge. Front. Aging Neurosci. 2019, 11, 299. [Google Scholar] [CrossRef]
- Caldarelli, M.; Rio, P.; Marrone, A.; Ocarino, F.; Chiantore, M.; Candelli, M.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Gut-Brain Axis: Focus on Sex Differences in Neuroinflammation. Int. J. Mol. Sci. 2024, 25, 5377. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Miller, D.B. Neuroinflammation disorders exacerbated by environmental stressors. Metabolism 2019, 100S, 153951. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef]
- Stocchi, F.; Bravi, D.; Emmi, A.; Antonini, A. Parkinson disease therapy: Current strategies and future research priorities. Nat. Rev. Neurol. 2024, 20, 695–707. [Google Scholar] [CrossRef]
- Talanki Manjunatha, R.; Habib, S.; Sangaraju, S.L.; Yepez, D.; Grandes, X.A. Multiple Sclerosis: Therapeutic Strategies on the Horizon. Cureus 2022, 14, e24895. [Google Scholar] [CrossRef] [PubMed]
- Arfeen, M.; Srivastava, A.; Srivastava, N.; Khan, R.A.; Almahmoud, S.A.; Mohammed, H.A. Design, classification, and adverse effects of NSAIDs: A review on recent advancements. Bioorg. Med. Chem. 2024, 112, 117899. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best. Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Temel, S.G.; Kahveci, Z. Cyclooxygenase-2 expression in astrocytes and microglia in human oligodendroglioma and astrocytoma. J. Mol. Histol. 2009, 40, 369–377. [Google Scholar] [CrossRef]
- Hiskens, M.I.; Schneiders, A.G.; Fenning, A.S. Selective COX-2 Inhibitors as Neuroprotective Agents in Traumatic Brain Injury. Biomedicines 2024, 12, 1930. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Puhl, A.C.; Milton, F.A.; Cvoro, A.; Sieglaff, D.H.; Campos, J.C.; Bernardes, A.; Filgueira, C.S.; Lindemann, J.L.; Deng, T.; Neves, F.A.; et al. Mechanisms of peroxisome proliferator activated receptor γ regulation by non-steroidal anti-inflammatory drugs. Nucl. Recept. Signal. 2015, 13, e004. [Google Scholar] [CrossRef]
- Radi, Z.A.; Khan, N.K. Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp. Toxicol. Pathol. 2006, 58, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Drożdżal, S.; Lechowicz, K.; Szostak, B.; Rosik, J.; Kotfis, K.; Machoy-Mokrzyńska, A.; Białecka, M.; Ciechanowski, K.; Gawrońska-Szklarz, B. Kidney damage from nonsteroidal anti-inflammatory drugs-Myth or truth? Review of selected literature. Pharmacol. Res. Perspect. 2021, 9, e00817. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Sabzwari, S.R.A.; Vargova, V. Cardiovascular Risk of Nonsteroidal Anti-Inflammatory Drugs: An Under-Recognized Public Health Issue. Cureus 2017, 9, e1144. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Miyake, S.; Mizuno, M.; Oka, N.; Kusunoki, S.; Yamamura, T. Selective COX-2 inhibitor celecoxib prevents experimental autoimmune encephalomyelitis through COX-2-independent pathway. Brain 2006, 129, 1984–1992. [Google Scholar] [CrossRef]
- Irrera, N.; Bitto, A. Evidence for using a dual COX 1/2 and 5-LOX inhibitor in neurodegenerative diseases. Neural Regen. Res. 2017, 12, 1077–1078. [Google Scholar]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef]
- Gwee, K.A.; Goh, V.; Lima, G.; Setia, S. Coprescribing proton-pump inhibitors with nonsteroidal anti-inflammatory drugs: Risks versus benefits. J. Pain Res. 2018, 11, 361–374. [Google Scholar] [CrossRef]
- Tsau, S.; Emerson, M.R.; Lynch, S.G.; LeVine, S.M. Aspirin and multiple sclerosis. BMC Med. 2015, 13, 153. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Mather, A.E.; Peters, R.; Lawrence, C.B.; Brough, D. Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun. 2020, 2, fcaa109. [Google Scholar] [CrossRef]
- Ding, P.; Gorenflo, M.P.; Zhu, X.; Xu, R. Aspirin Use and Risk of Alzheimer’s Disease: A 2-Sample Mendelian Randomization Study. J. Alzheimers Dis. 2023, 92, 989–1000. [Google Scholar] [CrossRef]
- Gao, X.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011, 76, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Badawoud, A.M.; Ali, L.S.; Abdallah, M.S.; El Sabaa, R.M.; Bahaa, M.M.; Elmasry, T.A.; Wahsh, E.; Yasser, M.; Eltantawy, N.; Eldesoqui, M.; et al. The relation between Parkinson’s disease and non-steroidal anti-inflammatories; a systematic review and meta-analysis. Front. Pharmacol. 2024, 15, 1434512. [Google Scholar] [CrossRef]
- Khrieba, M.O.; Hegazy, S.K.; Mustafa, W.; El-Haggar, S.M. Repurposing celecoxib as adjuvant therapy in patients with Parkinsonian disease: A new therapeutic dawn: Randomized controlled pilot study. Inflammopharmacology 2024, 32, 3729–3738. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, D.; Hallan, D.R.; Staub, J.; Rizk, E.; Zacko, J.C. Early Celecoxib use in Patients with Traumatic Brain Injury. Neurocrit. Care 2024, 40, 886–897. [Google Scholar] [CrossRef]
- Tsai, C.P.; Lin, F.C.; Lee, J.K.; Lee, C.T. Aspirin use associated with amyotrophic lateral sclerosis: A total population-based case-control study. J. Epidemiol. 2015, 25, 172–177. [Google Scholar] [CrossRef]
- Salomon-Zimri, S.; Pushett, A.; Russek-Blum, N.; Van Eijk, R.P.A.; Birman, N.; Abramovich, B.; Eitan, E.; Elgrart, K.; Beaulieu, D.; Ennist, D.L.; et al. Combination of ciprofloxacin/celecoxib as a novel therapeutic strategy for ALS. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef]
- Stockner, T.; Sterk, H.; Kaptein, R.; Bonvin, A.M. Molecular dynamics studies of a molecular switch in the glucocorticoid receptor. J. Mol. Biol. 2003, 328, 325–334. [Google Scholar] [CrossRef]
- Meijer, O.C.; Kalkhoven, E.; van der Laan, S.; Steenbergen, P.J.; Houtman, S.H.; Dijkmans, T.F.; Pearce, D.; de Kloet, E.R. Steroid receptor coactivator-1 splice variants differentially affect corticosteroid receptor signaling. Endocrinology 2005, 146, 1438–1448. [Google Scholar] [CrossRef]
- Van Moortel, L.; Verhee, A.; Thommis, J.; Houtman, R.; Melchers, D.; Delhaye, L.; Van Leene, C.; Hellemans, M.; Gevaert, K.; Eyckerman, S.; et al. Selective Modulation of the Human Glucocorticoid Receptor Compromises GR Chromatin Occupancy and Recruitment of p300/CBP and the Mediator Complex. Mol. Cell. Proteom. 2024, 23, 100741. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.P.; Greulich, F.; Ansari, S.A.; Uhlenhaut, N.H. Anti-inflammatory glucocorticoid action: Genomic insights and emerging concepts. Curr. Opin. Pharmacol. 2020, 53, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- González, M.V.; Jiménez, B.; Berciano, M.T.; González-Sancho, J.M.; Caelles, C.; Lafarga, M.; Muñoz, A. Glucocorticoids antagonize AP-1 by inhibiting the Activation/phosphorylation of JNK without affecting its subcellular distribution. J. Cell. Biol. 2000, 150, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Kimmel, S.C.; Levin, R.I.; Martiniuk, F.; Weissmann, G. A mechanism for the antiinflammatory effects of corticosteroids: The glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1992, 89, 9991–9995. [Google Scholar] [CrossRef]
- Atsuta, J.; Plitt, J.; Bochner, B.S.; Schleimer, R.P. Inhibition of VCAM-1 expression in human bronchial epithelial cells by glucocorticoids. Am. J. Respir. Cell. Mol. Biol. 1999, 20, 643–650. [Google Scholar] [CrossRef]
- Fukuzuka, K.; Edwards, C.K., 3rd; Clare-Salzer, M.; Copeland, E.M., 3rd; Moldawer, L.L.; Mozingo, D.W. Glucocorticoid and Fas ligand induced mucosal lymphocyte apoptosis after burn injury. J. Trauma. 2000, 49, 710–716. [Google Scholar] [CrossRef]
- López-Royuela, N.; Balsas, P.; Galán-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Bim is the key mediator of glucocorticoid-induced apoptosis and of its potentiation by rapamycin in human myeloma cells. Biochim. Biophys. Acta 2010, 1803, 311–322. [Google Scholar] [CrossRef]
- Salvador, E.; Shityakov, S.; Förster, C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014, 355, 597–605. [Google Scholar] [CrossRef]
- Mustafa, S.S. Steroid-induced secondary immune deficiency. Ann. Allergy Asthma Immunol. 2023, 130, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Durham, H.; Glover, L.; Ather, O.; Phillips, V.; Nemes, S.; Cousens, L.; Blomgran, P.; Ambery, P. Metabolic adverse events associated with systemic corticosteroid therapy-a systematic review and meta-analysis. BMJ Open 2022, 12, e061476. [Google Scholar] [CrossRef]
- Briot, K.; Roux, C. Glucocorticoid-induced osteoporosis. RMD Open 2015, 1, e000014. [Google Scholar] [CrossRef]
- Nasereddin, L.; Alnajjar, O.; Bashar, H.; Abuarab, S.F.; Al-Adwan, R.; Chellappan, D.K.; Barakat, M. Corticosteroid-Induced Psychiatric Disorders: Mechanisms, Outcomes, and Clinical Implications. Diseases 2024, 12, 300. [Google Scholar] [CrossRef]
- Durelli, L.; Cocito, D.; Riccio, A.; Barile, C.; Bergamasco, B.; Baggio, G.F.; Perla, F.; Delsedime, M.; Gusmaroli, G.; Bergamini, L. High-dose intravenous methylprednisolone in the treatment of multiple sclerosis: Clinical-immunologic correlations. Neurology 1986, 36, 238–243. [Google Scholar] [CrossRef]
- Barkhof, F.; Tas, M.W.; Frequin, S.T.; Scheltens, P.; Hommes, O.R.; Nauta, J.J.; Valk, J. Limited duration of the effect of methylprednisolone on changes on MRI in multiple sclerosis. Neuroradiology 1994, 36, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Havrdova, E.; Zivadinov, R.; Krasensky, J.; Dwyer, M.G.; Novakova, I.; Dolezal, O.; Ticha, V.; Dusek, L.; Houzvickova, E.; Cox, J.L.; et al. Randomized study of interferon beta-1a, low-dose azathioprine, and low-dose corticosteroids in multiple sclerosis. Mult. Scler. 2009, 15, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.; Probasco, J.; Irani, S.R.; Ances, B.; Benavides, D.R.; Bradshaw, M.; Christo, P.P.; Dale, R.C.; Fernandez-Fournier, M.; Flanagan, E.P.; et al. Autoimmune Encephalitis Alliance Clinicians Network. Autoimmune encephalitis: Proposed recommendations for symptomatic and long-term management. J. Neurol. Neurosurg. Psychiatry 2021, 92, 897–907. [Google Scholar] [CrossRef]
- Abbatemarco, J.R.; Yan, C.; Kunchok, A.; Rae-Grant, A. Antibody-mediated autoimmune encephalitis: A practical approach. Cleve. Clin. J. Med. 2021, 88, 459–471. [Google Scholar] [CrossRef]
- Stingl, C.; Cardinale, K.; Van Mater, H. An Update on the Treatment of Pediatric Autoimmune Encephalitis. Curr. Treatm. Opt. Rheumatol. 2018, 4, 14–28. [Google Scholar] [CrossRef]
- Zhou, X.; Luo, X.; He, Z.; Tang, D.; Li, Y.; Li, P. Efficacy of dexamethasone combined with intravenous immunoglobulin for the treatment of pediatric autoimmune encephalitis. Front. Neurol. 2025, 16, 1512908. [Google Scholar] [CrossRef] [PubMed]
- Kothari, M.; Wanjari, A.; Acharya, S.; Karwa, V.; Chavhan, R.; Kumar, S.; Kadu, A.; Patil, R. A Comprehensive Review of Monoclonal Antibodies in Modern Medicine: Tracing the Evolution of a Revolutionary Therapeutic Approach. Cureus 2024, 16, e61983. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Coisne, C.; Mao, W.; Engelhardt, B. Cutting edge: Natalizumab blocks adhesion but not initial contact of human T cells to the blood-brain barrier in vivo in an animal model of multiple sclerosis. J. Immunol. 2009, 182, 5909–5913. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, K. Natalizumab for multiple sclerosis: The dilemma of NOVA. Lancet Neurol. 2022, 21, 579–581. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Liu, L.; Gong, X.; Liu, J.; Sun, L.; Liu, X.; Wu, L.; Chen, L.; Wang, L.; et al. Fine Comparison of the Efficacy and Safety Between GB242 and Infliximab in Patients with Rheumatoid Arthritis: A Phase III Study. Rheumatol. Ther. 2022, 9, 175–189. [Google Scholar] [CrossRef]
- Traczewski, P.; Rudnicka, L. Adalimumab in dermatology. Br. J. Clin. Pharmacol. 2008, 66, 618–625. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Strangfeld, A.; Listing, J. Infection and musculoskeletal conditions: Bacterial and opportunistic infections during anti-TNF therapy. Best Pract. Res. Clin. Rheumatol. 2006, 20, 1181–1195. [Google Scholar] [CrossRef]
- Kim, S.Y.; Solomon, D.H. Tumor necrosis factor blockade and the risk of viral infection. Nat. Rev. Rheumatol. 2010, 6, 165–174. [Google Scholar] [CrossRef]
- Li, Y.; Ye, R.; Dai, H.; Lin, J.; Cheng, Y.; Zhou, Y.; Lu, Y. Exploring TNFR1: From discovery to targeted therapy development. J. Transl Med. 2025, 23, 71. [Google Scholar] [CrossRef]
- Torene, R.; Nirmala, N.; Obici, L.; Cattalini, M.; Tormey, V.; Caorsi, R.; Starck-Schwertz, S.; Letzkus, M.; Hartmann, N.; Abrams, K.; et al. Canakinumab reverses overexpression of inflammatory response genes in tumour necrosis factor receptor-associated periodic syndrome. Ann. Rheum Dis. 2017, 76, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.L.; Nidorf, S.M. Anti-inflammatory therapy with canakinumab for atherosclerotic disease: Lessons from the CANTOS trial. J. Thorac. Dis. 2018, 10, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Weickert, T.W.; Jacomb, I.; Lenroot, R.; Lappin, J.; Weinberg, D.; Brooks, W.S.; Brown, D.; Pellen, D.; Kindler, J.; Mohan, A.; et al. Adjunctive canakinumab reduces peripheral inflammation markers and improves positive symptoms in people with schizophrenia and inflammation: A randomized control trial. Brain Behav. Immun. 2024, 115, 191–200. [Google Scholar] [CrossRef]
- Parisi, S.; Ditto, M.C.; Ghellere, F.; Panaro, S.; Piccione, F.; Borrelli, R.; Fusaro, E. Update on tocilizumab in rheumatoid arthritis: A narrative review. Front. Immunol. 2025, 16, 1470488. [Google Scholar] [CrossRef]
- Yu, Y.M.; Jin, G.H.; Zhong, C.; Qian, H.; Wang, L.; Zhan, F. Exploring the role of interleukin-6 receptor blockade in epilepsy and associated neuropsychiatric conditions through a mendelian randomization study. World J. Psychiatry 2024, 14, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Cadiz, M.P.; Gibson, K.A.; Todd, K.T.; Nascari, D.G.; Massa, N.; Lilley, M.T.; Olney, K.C.; Al-Amin, M.M.; Jiang, H.; Holtzman, D.M.; et al. Aducanumab anti-amyloid immunotherapy induces sustained microglial and immune alterations. J. Exp. Med. 2024, 221, e20231363. [Google Scholar] [CrossRef]
- Nisticò, R.; Borg, J.J. Aducanumab for Alzheimer’s disease: A regulatory perspective. Pharmacol. Res. 2021, 171, 105754. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Masaki, K.; Tanaka, E.; Matsushita, T.; Isobe, N. The Efficacy of Eculizumab in the Acute Phase of Neuromyelitis Optica Spectrum Disorder: A Case Series Study. Cureus 2024, 16, e73205. [Google Scholar] [CrossRef] [PubMed]
- Gurkan, S.; Fyfe, B.; Weiss, L.; Xiao, X.; Zhang, Y.; Smith, R.J. Eculizumab and recurrent C3 glomerulonephritis. Pediatr. Nephrol. 2013, 28, 1975–1981. [Google Scholar] [CrossRef]
- Malagoli, P.; Dapavo, P.; Amerio, P.; Atzori, L.; Balato, A.; Bardazzi, F.; Bianchi, L.; Cattaneo, A.; Chiricozzi, A.; Congedo, M.; et al. Secukinumab in the Treatment of Psoriasis: A Narrative Review on Early Treatment and Real-World Evidence. Dermatol. Ther. 2024, 14, 2739–2757. [Google Scholar] [CrossRef] [PubMed]
- Shelton, S.K.; Bai, S.R.; Jordan, J.K.; Sheehan, A.H. Ixekizumab: A Review of Its Use for the Management of Moderate to Severe Plaque Psoriasis. Ann. Pharmacother. 2019, 53, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Huang, L.; Flores, J.; Ding, Y.; Li, P.; Peng, J.; Zuo, G.; Zhang, J.H.; Lu, J.; Tang, J.P. Secukinumab attenuates reactive astrogliosis via IL-17RA/(C/EBPβ)/SIRT1 pathway in a rat model of germinal matrix hemorrhage. CNS Neurosci. Ther. 2019, 25, 1151–1161. [Google Scholar] [CrossRef]
- Liu, S.; Deng, S.; Ding, Y.; Flores, J.J.; Zhang, X.; Jia, X.; Hu, X.; Peng, J.; Zuo, G.; Zhang, J.H.; et al. Secukinumab attenuates neuroinflammation and neurobehavior defect via PKCβ/ERK/NF-κB pathway in a rat model of GMH. Exp. Neurol. 2023, 360, 114276. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Dong, M.; Bai, L.; Zhang, W.; Liu, W.; Wang, F.; Zhu, X. FDA-Approved Secukinumab Alleviates Glial Activation and Immune Cell Infiltration in MPTP-Induced Mouse Model of Parkinson’s Disease. Inflammation 2025, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Langrish, C.L.; McKenzie, B.; Joyce-Shaikh, B.; Stumhofer, J.S.; McClanahan, T.; Blumenschein, W.; Churakovsa, T.; Low, J.; Presta, L.; et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Investig. 2006, 116, 1317–1326. [Google Scholar] [CrossRef]
- Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals 2021, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Ruck, T.; Nimmerjahn, F.; Wiendl, H.; Lünemann, J.D. Next-generation antibody-based therapies in neurology. Brain 2022, 145, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20+ B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 835–841. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Thaler, F.S.; Zimmermann, L.; Kammermeier, S.; Strippel, C.; Ringelstein, M.; Kraft, A.; Sühs, K.W.; Wickel, J.; Geis, C.; Markewitz, R.; et al. Rituximab Treatment and Long-term Outcome of Patients with Autoimmune Encephalitis: Real-world Evidence from the GENERATE Registry. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1088. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, S.T.; Moon, J.; Sunwoo, J.S.; Byun, J.I.; Lim, J.A.; Kim, T.J.; Shin, Y.W.; Lee, K.J.; Jun, J.S.; et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics 2016, 13, 824–832. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Boess, F.G.; Taylor, K.I.; Ricci, B.; Mollenhauer, B.; Poewe, W.; Boulay, A.; Anzures-Cabrera, J.; Vogt, A.; Marchesi, M.; et al. A Phase II Study to Evaluate the Safety and Efficacy of Prasinezumab in Early Parkinson’s Disease (PASADENA): Rationale, Design, and Baseline Data. Front. Neurol. 2021, 12, 705407. [Google Scholar] [CrossRef]
- Hutchison, R.M.; Fraser, K.; Yang, M.; Fox, T.; Hirschhorn, E.; Njingti, E.; Scott, D.; Bedell, B.J.; Kistner, K.M.; Cedarbaum, J.M.; et al. Cinpanemab in Early Parkinson Disease: Evaluation of Biomarker Results From the Phase 2 SPARK Clinical Trial. Neurology 2024, 102, e209137. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Hu, B.; Duan, S.; Wang, Z.; Li, X.; Zhou, Y.; Zhang, X.; Zhang, Y.W.; Xu, H.; Zheng, H. Insights Into the Role of CSF1R in the Central Nervous System and Neurological Disorders. Front. Aging Neurosci. 2021, 13, 789834. [Google Scholar] [CrossRef]
- Hupp, S.; Iliev, A.I. CSF-1 receptor inhibition as a highly effective tool for depletion of microglia in mixed glial cultures. J. Neurosci. Methods 2020, 332, 108537. [Google Scholar] [CrossRef]
- Jovanovic, J.; Liu, X.; Kokona, D.; Zinkernagel, M.S.; Ebneter, A. Inhibition of inflammatory cells delays retinal degeneration in experimental retinal vein occlusion in mice. Glia 2020, 68, 574–588. [Google Scholar] [CrossRef]
- Lehmann, M.L.; Weigel, T.K.; Poffenberger, C.N.; Herkenham, M. The Behavioral Sequelae of Social Defeat Require Microglia and Are Driven by Oxidative Stress in Mice. J. Neurosci. 2019, 39, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, M.S.; Joly-Amado, A.; Bickford, P.C.; Nash, K.R. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol. Ther. 2022, 231, 107989. [Google Scholar] [CrossRef]
- Ridderstad Wollberg, A.; Ericsson-Dahlstrand, A.; Juréus, A.; Ekerot, P.; Simon, S.; Nilsson, M.; Wiklund, S.J.; Berg, A.L.; Ferm, M.; Sunnemark, D.; et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5409–5414. [Google Scholar] [CrossRef]
- Suresh, P.; Phasuk, S.; Liu, I.Y. Modulation of microglia activation and Alzheimer’s disease: CX3 chemokine ligand 1/CX3CR and P2X7R signaling. Tzu. Chi. Med. J. 2021, 33, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, Y.; Qi, M.; Zhang, X.; Wu, D. Pioglitazone Modulates Microglia M1/M2 Polarization Through PPAR-γ Pathway and Exerts Neuroprotective Effects in Experimental Subarachnoid Hemorrhage. Mol. Neurobiol. 2025, 62, 5930–5946. [Google Scholar] [CrossRef]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 8979. [Google Scholar] [CrossRef]
- Schwaid, A.G.; Spencer, K.B. Strategies for Targeting the NLRP3 Inflammasome in the Clinical and Preclinical Space. J. Med. Chem. 2021, 64, 101–122. [Google Scholar] [CrossRef]
- Imre, G. Pyroptosis in health and disease. Am. J. Physiol.-Cell Physiol. 2024, 326, C784–C794. [Google Scholar] [CrossRef]
- Xia, L.; Liu, L.; Cai, Y.; Zhang, Y.; Tong, F.; Wang, Q.; Ding, J.; Wang, X. Inhibition of Gasdermin D-Mediated Pyroptosis Attenuates the Severity of Seizures and Astroglial Damage in Kainic Acid-Induced Epileptic Mice. Front. Pharmacol. 2022, 12, 751644. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Angiulli, F.; Conti, E.; Zoia, C.P.; Da Re, F.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Blood-Based Biomarkers of Neuroinflammation in Alzheimer’s Disease: A Central Role for Periphery? Diagnostics 2021, 11, 1525. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, P.; Rauti, R.; Voulgaris, D.; Shlomy, I.; Maoz, B.M.; Herland, A. Recent progress in translational engineered in vitro models of the central nervous system. Brain 2020, 143, 3181–3213. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Ma, C.; Seong, H.; Li, X.; Yu, X.; Xu, S.; Li, Y. Human Brain Organoid: A Versatile Tool for Modeling Neurodegeneration Diseases and for Drug Screening. Stem Cells Int. 2022, 2022, 2150680. [Google Scholar] [CrossRef]
- Mofazzal Jahromi, M.A.; Abdoli, A.; Rahmanian, M.; Bardania, H.; Bayandori, M.; Moosavi Basri, S.M.; Kalbasi, A.; Aref, A.R.; Karimi, M.; Hamblin, M.R. Microfluidic Brain-on-a-Chip: Perspectives for Mimicking Neural System Disorders. Mol. Neurobiol. 2019, 56, 8489–8512. [Google Scholar] [CrossRef]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Velardo, D.; Podestà, M.A.; Ponticelli, C. Immunosuppression-related neurological disorders in kidney transplantation. J. Nephrol. 2021, 34, 539–555. [Google Scholar] [CrossRef]
- Clarke, G.J.B.; Follestad, T.; Skandsen, T.; Zetterberg, H.; Vik, A.; Blennow, K.; Olsen, A.; Håberg, A.K. Chronic immunosuppression across 12 months and high ability of acute and subacute CNS-injury biomarker concentrations to identify individuals with complicated mTBI on acute CT and MRI. J. Neuroinflamm. 2024, 21, 109. [Google Scholar] [CrossRef]
- Castro, I.; Ruiz, J.; Tasias, M.; Montero, M.; Salavert, M. Central nervous system infections in immunocompromised patients. Rev. Esp. Quimioter. 2018, 31 (Suppl. S1), 56–61. [Google Scholar] [PubMed]
- Lee, D.H.; Lee, J.Y.; Hong, D.Y.; Lee, E.C.; Park, S.W.; Lee, Y.K.; Oh, J.S. Pharmacological Treatment for Neuroinflammation in Stress-Related Disorder. Biomedicines 2022, 10, 2518. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, R.; Giaccone, G.; Calvert, H.; Lobbezoo, M.; Eisenhauer, E.A. Targeted agents: How to select the winners in preclinical and early clinical studies? Eur. J. Cancer 2012, 48, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, F.; Beltrán, E.; Gerdes, L.A.; Becher, B. Single-cell multiomics in neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102180. [Google Scholar] [CrossRef]
- Li, S.; Lu, C.; Zhao, Z.; Lu, D.; Zheng, G. Uncovering neuroinflammation-related modules and potential repurposing drugs for Alzheimer’s disease through multi-omics data integrative analysis. Front. Aging Neurosci. 2023, 15, 1161405. [Google Scholar] [CrossRef]
- Geng, Y.; Wang, R.Y.; Dong, M.Y.; Qian, Y.L.; Wang, X.H.; Xia, W.W.; Shen, Y.; Zhang, K.Z. Integrated Analysis of Single-Cell and Transcriptome Data Reveals the Role and Regulatory Mechanisms of Neuroinflammation in Parkinson’s Disease. Inflammation 2025, 1–20. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Disease | NSAID Employed | Effects | References |
|---|---|---|---|
| Multiple Sclerosis (MS) | Aspirin | Aspirin exhibits several potential therapeutic benefits, including anti-inflammatory effects, promotion of oligodendrocyte precursor cell proliferation, and improvement of symptoms such as fatigue. Nevertheless, its use requires caution due to potential adverse effects, notably an increased risk of hemorrhagic stroke and inhibition of mitochondrial complex I activity, both of which are particularly relevant in this pathology. | [188] |
| Alzheimer’s Disease (AD) | Diclofenac | Diclofenac use was found to be associated with a reduced incidence of AD and slower progression of cognitive decline, underscoring the necessity for further research into the therapeutic potential of diclofenac in AD. | [189] |
| Aspirin | The findings of this analysis suggest a genetically mediated protective effect of aspirin use against AD, potentially modulated by coronary heart disease, blood pressure, and lipid levels. | [190] | |
| Parkinson’s Disease (PD) | Ibuprofen | Over six years of follow-up, 291 incident PD cases were identified. Ibuprofen use was associated with a significantly lower PD risk (RR = 0.62; 95% CI 0.42–0.93; p = 0.02), with a dose–response relationship (p = 0.01). In another study, the ORs for PD occurrence in patients who took NSAIDs, ibuprofen, and non-aspirin NSAIDs were 0.88 [95% CI (0.80–0.97), p = 0.01], 0.73 [95% CI (0.53–1.00), p = 0.05], and 0.85 [95% CI (0.7–0.97), p = 0.01], respectively. | [191,192] |
| Celecoxib | The celecoxib group exhibited a significant reduction in the levels of TLR4 (p = 0.004), TNF-α (p = 0.042), and α-Syn (p = 0.004), as well as a notable increase in the levels of BDNF (p = 0.0005) and Nrf-2 (p = 0.004), as compared to the control group. Additionally, UPDRS scores were significantly lower in the celecoxib group (p < 0.05). | [193] | |
| Traumatic Brain Injury (TBI) | Celecoxib | Celecoxib use in patients with TBI was found to be associated with a significantly lower 1-year mortality rate (6.4% vs. 10.0%; OR 0.61; 95% CI 0.46–0.80; p = 0.0003) and higher survival rate (96.1% vs. 93.1%; p < 0.0001) compared to the control group. Celecoxib was also linked to reduced rates of gastrostomy tube dependence, seizure activity, and myocardial infarction. | [194] |
| Amyotrophic Lateral Sclerosis (ALS) | Aspirin | Multivariate analysis revealed that aspirin use was independently inversely associated with ALS risk after adjustment for diphenhydramine, mefenamic acid, and steroid use. This inverse association was predominantly observed in individuals over 55 years of age. | [195] |
| Celecoxib (plus ciprofloxacin) | Biomarker analyses revealed significant PrimeC (celecoxib combined with ciprofloxacin)-induced alterations in neuron-derived exosomal TDP-43 and LC3 levels, the latter being a key marker of autophagy. These results support the safety and tolerability of PrimeC in ALS and offer preliminary evidence of its biological activity. | [196] |
| Disease | Corticosteroid Employed | Effects | References |
|---|---|---|---|
| Multiple Sclerosis (MS) | Prednisone | Durelli et al. [215] conducted a study involving 23 patients with RRMS, who were treated over a 15-day period with either a placebo or methylprednisolone, initiated at a dose of 15 mg/kg/day and gradually tapered. Following this initial phase, all patients received oral prednisone at a dose of 100 mg/day, tapered over 120 days. The authors reported a statistically significant clinical benefit (p < 0.05) in favor of methylprednisolone treatment on days 5, 10, and 15. | [215] |
| Prednisolone | Methylprednisolone therapy in MS markedly reduces both the severity and duration of clinical relapses and the number of gadolinium-enhancing T1 lesions on MRI. In a cohort of 13 MS patients undergoing 31 treatment cycles over a mean of 50 weeks, 609 active lesions were identified across 195 MRI scans. Post-treatment imaging revealed a 78% immediate reduction in enhancing lesions, indicating transient restoration of blood–brain barrier integrity and inflammatory suppression. Re-enhancement of lesions was rare. The therapeutic effect persisted for approximately 10 weeks. | [216] | |
| Methylprednisolone | In 2009, a randomized study was published involving 181 patients with RRMS, all of whom were receiving treatment with IFNβ-1a. Participants were assigned to receive one of three adjunctive treatments over a two-year period: (1) placebo, (2) oral azathioprine (50 mg/day), or (3) oral azathioprine (50 mg/day) combined with oral prednisolone (10 mg every other day). The combination of prednisolone and azathioprine was associated with a 20% reduction in the annualized relapse rate. | [217] | |
| Autoimmune Encephalitis | Prednisone | Employed as a first-line therapy, a common approach involves initiating oral prednisone at a dose of 1–2 mg/kg/day immediately following the completion of acute treatment, followed by a gradual taper over several weeks to months. | [218] |
| Methylprednisolone | As a first-line therapy for autoimmune encephalitis, treatment typically consists of intravenous methylprednisolone at 1 g per day for 5 consecutive days, followed by oral prednisone at 1 mg/kg/day (up to a maximum of 60–80 mg/day), with a gradual tapering protocol extending over 3 to 6 months. | [219] | |
| Employed as a first-line treatment in infantile autoimmune encephalitis, intravenous methylprednisolone is administered at a dose of 30 mg/kg/day for 3 to 5 days. | [220] | ||
| Dexamethasone (plus immunoglobulins) | Therapy with dexamethasone and intravenous immunoglobulin resulted in significant clinical improvement during both acute and non-acute phases of the disease. At the final follow-up, 90.2% of patients exhibited a favorable clinical outcome, with 43.9% achieving full recovery and a relapse rate of only 2.4%. No severe adverse events were observed. | [221] |
| Disease | mAb Employed | Clinical Trial Phase | Effects | References |
|---|---|---|---|---|
| Multiple Sclerosis (MS) | Natalizumab (Anti-α4 integrin) | Phase III | Inhibited leukocyte migration Reduced relapses | [253] |
| Ocrelizumab (Anti-CD20) | Phase III | Caused B-cell depletion Effective in RRMS and PPMS | [254] | |
| Alemtuzumab (Anti-CD52) | Phase III | Produced long-lasting lymphocyte depletion | [255] | |
| Ofatumumab (Anti-CD20) | Phase III | Effective in reducing disease activity | [256] | |
| Neuromyelitis Optica Spectrum Disorder (NMOSD) | Eculizumab (Anti-C5) | Phase III | Inhibited the complement cascade Prevented attacks | [257] |
| Satralizumab (Anti-IL-6R) | Phase III | Reduced relapse risk | [258] | |
| Inebilizumab (Anti-CD19) | Phase II/III | Reduced relapse rate | [259] | |
| Autoimmune Encephalitis | Rituximab (Anti-CD20) | Phase II | Induced B-cell depletion Improved psychiatric and neurologic symptoms | [260] |
| Tocilizumab (Anti-IL-6R) | Phase II | Effective for refractory or relapsing cases | [261] | |
| Alzheimer’s Disease (AD) | Aducanumab (Anti-Aβ plaques) | Phase III/Approved | Reduced Aβ plaques | [240] |
| Lecanemab (Anti-Aβ plaques) | Phase III | Provided modest cognitive benefit | [262] | |
| Donanemab (Anti-Aβ plaques) | Phase III | Slowed progression in early AD | [263] | |
| Parkinson’s Disease (PD) | Prasinezumab (Anti-α-synuclein) | Phase II | Targeted misfolded protein aggregates | [264] |
| Cinpanemab (Anti-α-synuclein) | Phase II | Under investigation | [265] | |
| CNS Vasculitis | Rituximab (Anti-CD20) | Phase II | Served as an alternative to cyclophosphamide in AAV-related cases | [266] |
| Neuropsychiatric Systemic Lupus Erythematosus (NPSLE) | Belimumab (Anti-BAFF) | Phase III | Caused B-cell suppression Reduced neuropsychiatric flare frequency | [267] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Domínguez, M. Neuroinflammation: Mechanisms, Dual Roles, and Therapeutic Strategies in Neurological Disorders. Curr. Issues Mol. Biol. 2025, 47, 417. https://doi.org/10.3390/cimb47060417
García-Domínguez M. Neuroinflammation: Mechanisms, Dual Roles, and Therapeutic Strategies in Neurological Disorders. Current Issues in Molecular Biology. 2025; 47(6):417. https://doi.org/10.3390/cimb47060417
Chicago/Turabian StyleGarcía-Domínguez, Mario. 2025. "Neuroinflammation: Mechanisms, Dual Roles, and Therapeutic Strategies in Neurological Disorders" Current Issues in Molecular Biology 47, no. 6: 417. https://doi.org/10.3390/cimb47060417
APA StyleGarcía-Domínguez, M. (2025). Neuroinflammation: Mechanisms, Dual Roles, and Therapeutic Strategies in Neurological Disorders. Current Issues in Molecular Biology, 47(6), 417. https://doi.org/10.3390/cimb47060417