

In Silico Analysis of s-DAPK-1: From Structure to Function and Regulation

Abstract
1. Introduction
2. Materials and Methods
2.1. Prediction of microRNAs (miRs) Targeting s-DAPK-1 mRNA
2.2. Protein Sequence Retrieval
2.3. Modeling and Validation of s-DAPK-1’s 3D Structure
2.4. Prediction of Physical and Chemical Parameters of s-DAPK-1
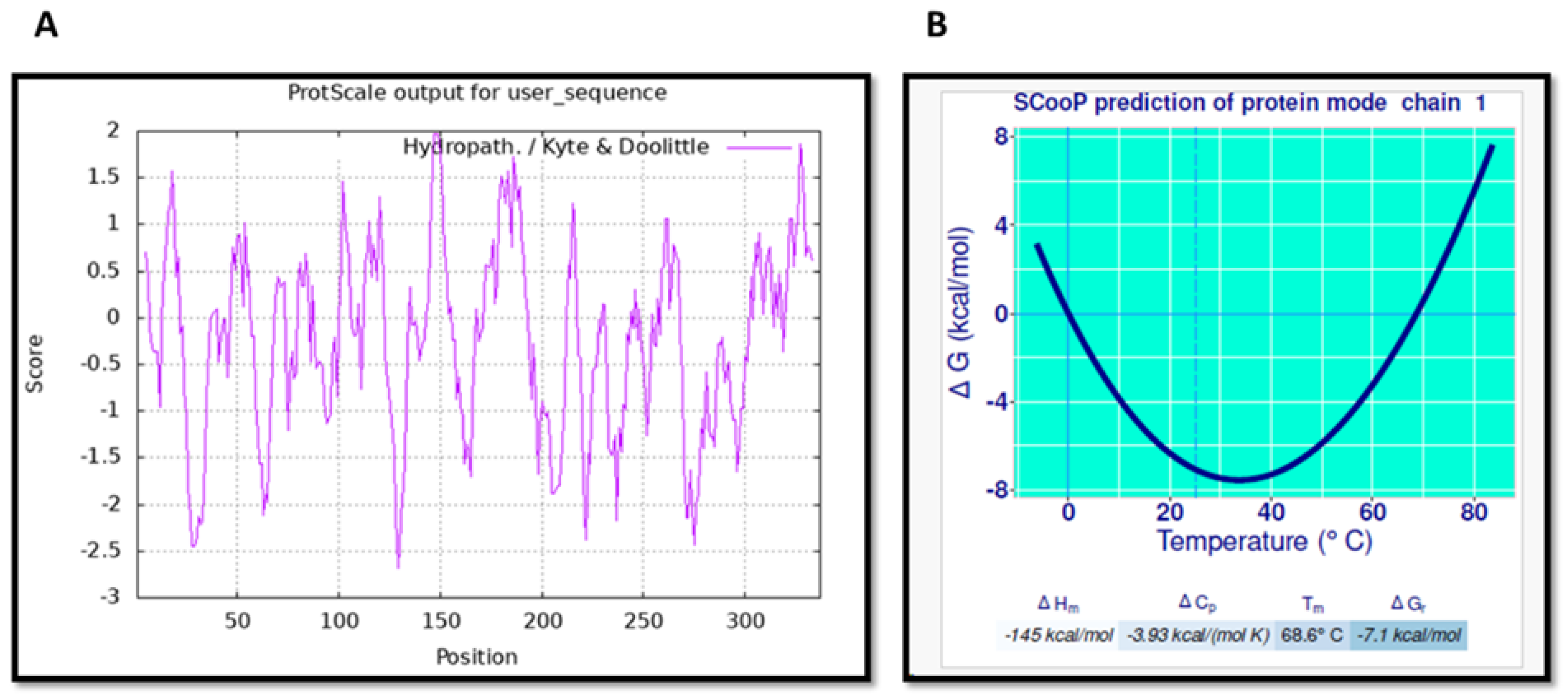
2.5. Prediction of s-DAPK-1’s Hydrophobicity and Thermodynamic Parameters
2.6. Prediction of Protein-Protein Interactions Involving s-DAPK-1
2.7. Protein–Protein Docking
2.8. Prediction of the Functions of the Modeled s-DAPK-1 Structure
3. Results
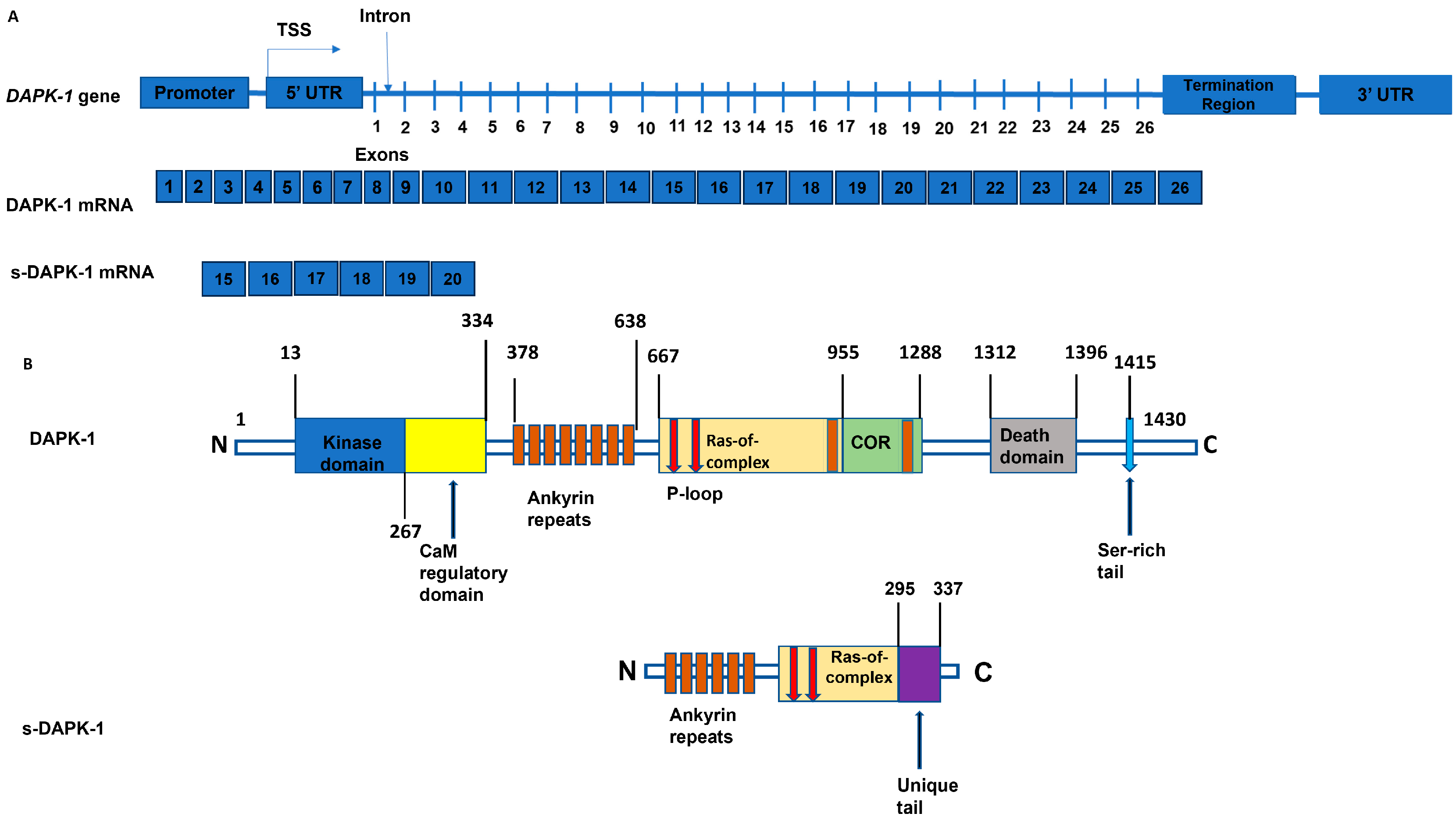
3.1. Proteins s-DAPK-1 and DAPK-1 Are Regulated by miRs
3.2. The 3D Structure of s-DAPK-1 Contains Ankyrin Repeats
3.3. Physicochemical Characterization of s-DAPK-1
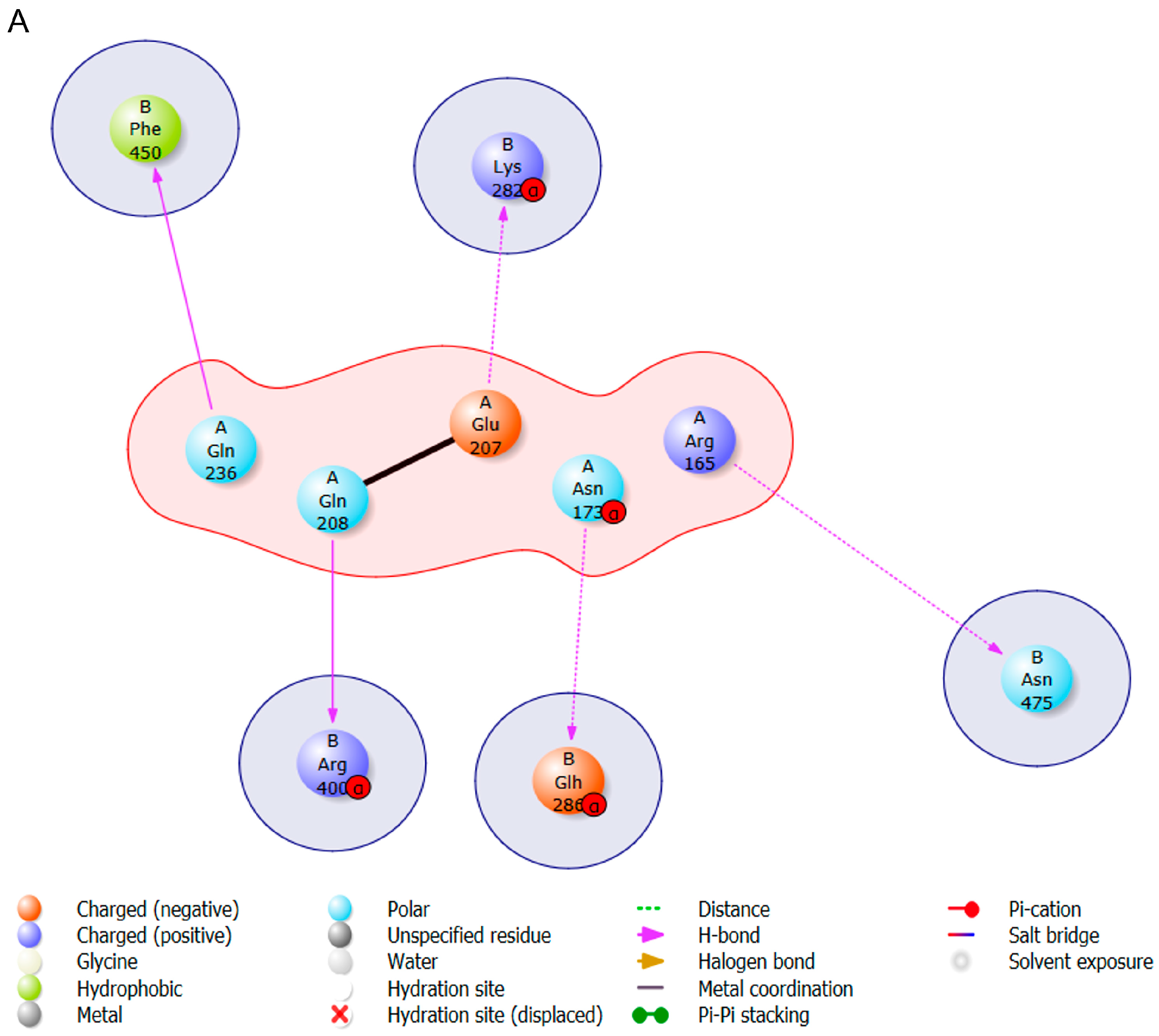
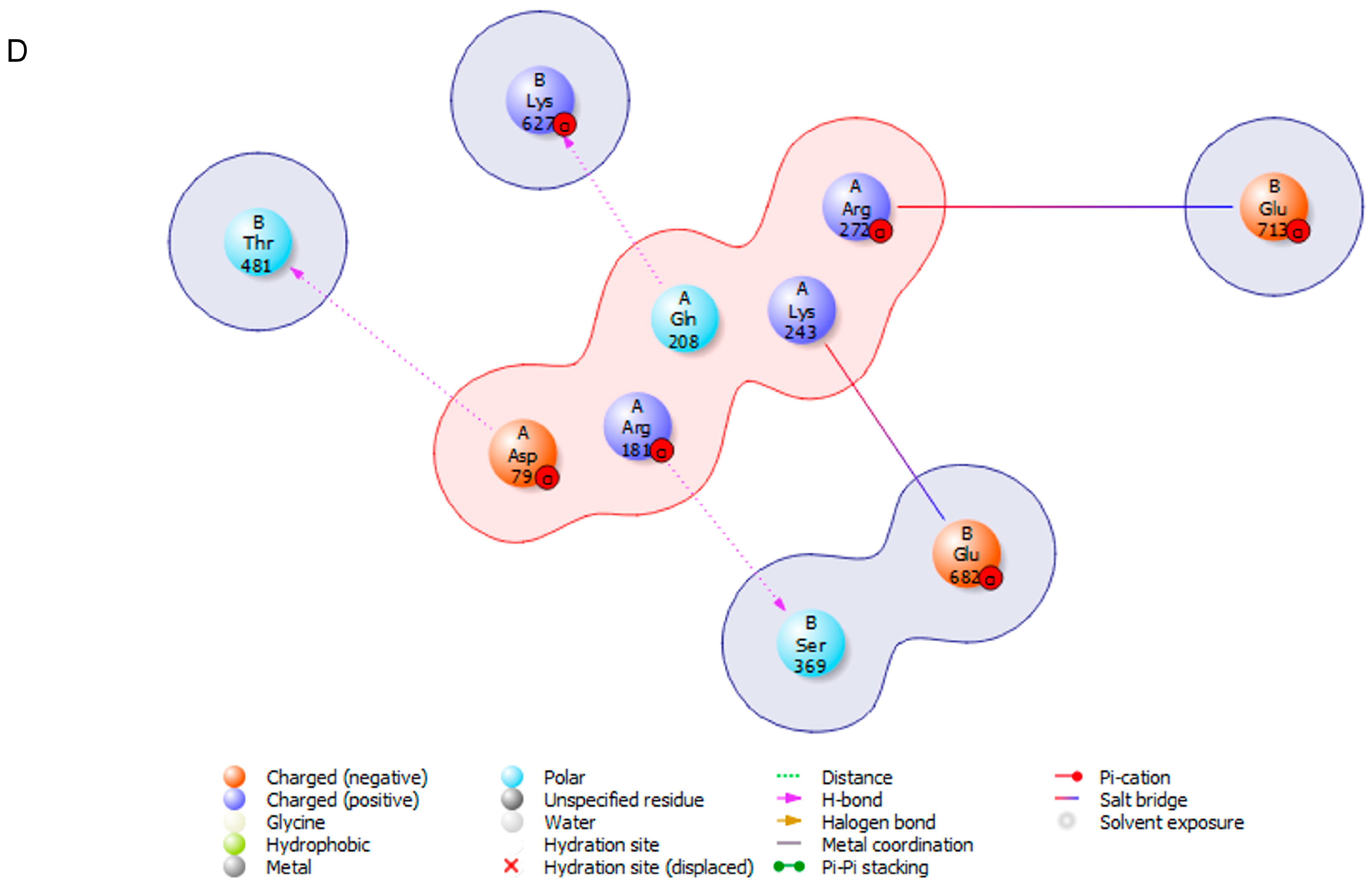
3.4. Protein s-DAPK-1 Interacts with Other Proteins
3.5. Protein s-DAPK-1 Performs Various Cellular Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gauthier, J.; Vincent, A.T.; Charette, S.J.; Derome, N. A brief history of bioinformatics. Brief. Bioinform. 2019, 20, 1981–1996. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, A.D.; Bateman, A. The Importance of Biological Databases in Biological Discovery. Curr. Protoc. Bioinform. 2015, 50, 1.1.1–1.1.8. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. Methods Mol. Biol. 2017, 1558, 3–39. [Google Scholar] [CrossRef]
- Gromiha, M.M. (Ed.) Chapter 6—Protein Stability. In Protein Bioinformatics; Academic Press: Singapore, 2010; pp. 209–245. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, J.; Lu, Y.; Ngo, F.Y.; Shuai, B.; Zhang, Z.-J.; Feng, Y.; Rong, J. In Silico Prediction of Quercetin Analogs for Targeting Death-Associated Protein Kinase 1 (DAPK1) Against Alzheimer’s Disease. Curr. Neuropharmacol. 2024, 22, 2353–2367. [Google Scholar] [CrossRef]
- Firoz, A.; Talwar, P. Role of death-associated protein kinase 1 (DAPK1) in retinal degenerative diseases: An in-silico approach towards therapeutic intervention. J. Biomol. Struct. Dyn. 2023, 42, 5686–5698. [Google Scholar] [CrossRef]
- Singh, P.; Talwar, P. Exploring putative inhibitors of Death Associated Protein Kinase 1 (DAPK1) via targeting Gly- Glu -Leu (G E L) and Pro- Glu -Asn (P E N) substrate recognition motifs. J. Mol. Graph. Model. 2017, 77, 153–167. [Google Scholar] [CrossRef]
- Tu, G.; Fu, T.; Yang, F.; Yao, L.; Xue, W.; Zhu, F. Prediction of GluN2B-CT1290–1310/DAPK1 Interaction by Protein−Peptide Docking and Molecular Dynamics Simulation. Molecules 2018, 23, 3018. [Google Scholar] [CrossRef] [PubMed]
- Makgoo, L.; Mosebi, S.; Mbita, Z. The Role of Death-Associated Protein Kinase-1 in Cell Homeostasis-Related Processes. Genes 2023, 14, 1274. [Google Scholar] [CrossRef]
- Lin, Y.; Stevens, C.; Hrstka, R.; Harrison, B.; Fourtouna, A.; Pathuri, S.; Vojtesek, B.; Hupp, T. An alternative transcript from the death-associated protein kinase 1 locus encoding a small protein selectively mediates membrane blebbing. FEBS J. 2008, 275, 2574–2584. [Google Scholar] [CrossRef]
- Zhang, T.; Kim, B.M.; Lee, T.H. Death-associated protein kinase 1 as a therapeutic target for Alzheimer’s disease. Transl. Neurodegener. 2024, 13, 4. [Google Scholar] [CrossRef]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Belal, C.; Wang, M.; Jia, N.; Zhang, W.; et al. DAPK1 Interaction with NMDA Receptor NR2B Subunits Mediates Brain Damage in Stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Chen, C.H.; Suizu, F.; Huang, P.; Schiene-Fischer, C.; Daum, S.; Zhang, Y.J.; Goate, A.; Chen, R.-H.; Zhou, X.Z.; et al. Death-Associated Protein Kinase 1 Phosphorylates Pin1 and Inhibits Its Prolyl Isomerase Activity and Cellular Function. Mol. Cell 2011, 42, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, C.; Li, K.; Ye, Y.; Shen, A.; Guo, L.; Chen, P.; Meng, C.; Wang, Q.; Yang, X.; et al. Death-associated protein kinase 1 suppresses hepatocellular carcinoma cell migration and invasion by upregulation of DEAD-box helicase 20. Cancer Sci. 2020, 111, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Tian, Y.; Qu, G.; Huang, K.; Hu, P.; Xue, Y.; Hu, B.; Luo, S. The targeting of DAPK1 with miR-190a-3p promotes autophagy in trophoblast cells. Mol. Reprod. Dev. 2024, 91, e23724. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, M.; Hong, Z.; Kang, J.; Pan, H.; Yan, C. MiR-130a-3p Has Protective Effects in Alzheimer’s Disease via Targeting DAPK1. Am. J. Alzheimer’s Dis. Other Dement. 2021, 36, 15333175211020572. [Google Scholar] [CrossRef]
- Zhai, C.; Tang, G.; Qian, G.; Hu, H.; Wang, S.; Yin, D.; Zhang, S. MicroRNA-98 attenuates cardiac ischemia-reperfusion injury through inhibiting DAPK1 expression. IUBMB Life 2018, 71, 166–176. [Google Scholar] [CrossRef]
- Rani, V.; Sengar, R.S. Biogenesis and mechanisms of microRNA-mediated gene regulation. Biotechnol. Bioeng. 2022, 119, 685–692. [Google Scholar] [CrossRef]
- Wang, L.; Shui, X.; Mei, Y.; Xia, Y.; Lan, G.; Hu, L.; Zhang, M.; Gan, C.-L.; Li, R.; Tian, Y.; et al. miR-143-3p Inhibits Aberrant Tau Phosphorylation and Amyloidogenic Processing of APP by Directly Targeting DAPK1 in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7992. [Google Scholar] [CrossRef]
- Tian, X.; Xu, L.; Wang, P. MiR-191 inhibits TNF-α induced apoptosis of ovarian endometriosis and endometrioid carcinoma cells by targeting DAPK1. Int. J. Clin. Exp. Pathol. 2015, 8, 4933–4942. [Google Scholar]
- Tian, Y.; Yan, M.; Zheng, J.; Li, R.; Lin, J.; Xu, A.; Liang, Y.; Zheng, R.; Yuan, Y. miR-483-5p decreases the radiosensitivity of nasopharyngeal carcinoma cells by targeting DAPK1. Lab. Investig. 2019, 99, 602–611. [Google Scholar] [CrossRef]
- Li, D.; Xu, D.; Xu, Y.; Chen, L.; Li, C.; Dai, X.; Zhang, L.; Zheng, L. MicroRNA-141-3p targets DAPK1 and inhibits apoptosis in rat ovarian granulosa cells. Cell Biochem. Funct. 2017, 35, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, L. Alternative splicing: Human disease and quantitative analysis from high-throughput sequencing. Comput. Struct. Biotechnol. J. 2020, 19, 183–195. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Wu, C. Alternative Splicing and Isoforms: From Mechanisms to Diseases. Genes 2022, 13, 401. [Google Scholar] [CrossRef]
- Cieply, B.; Carstens, R.P. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 311–326. [Google Scholar] [CrossRef]
- Wang, Q.; Weng, S.; Zhong, W.; Lin, Y.; Yu, Y.; Huang, Y.; Ge, L.; Zhang, X.; Xue, F.; Assaraf, Y.G.; et al. Modulation of DAPK1 expression by its alternative splice variant DAPK1-215 in cancer. J. Transl. Med. 2025, 23, 85. [Google Scholar] [CrossRef]
- Skoufos, G.; Kakoulidis, P.; Tastsoglou, S.; Zacharopoulou, E.; Kotsira, V.; Miliotis, M.; Mavromati, G.; Grigoriadis, D.; Zioga, M.; Velli, A.; et al. TarBase-v9.0 extends experimentally supported miRNA–Gene interactions to cell-types and virally encoded miRNAs. Nucleic Acids Res. 2023, 52, D304–D310. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinform. 2020, 69, e90. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5.8.1–5.8.15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Pucci, F.; Kwasigroch, J.M.; Rooman, M. SCooP: An accurate and fast predictor of protein stability curves as a function of temperature. Bioinformatics 2017, 33, 3415–3422. [Google Scholar] [CrossRef] [PubMed]
- Canzler, S.; Fischer, M.; Ulbricht, D.; Ristic, N.; Hildebrand, P.W.; Staritzbichler, R. ProteinPrompt: A webserver for predicting protein-protein interactions. Bioinform. Adv. 2022, 2, vbac059. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Cozzetto, D.; Minneci, F.; Currant, H.; Jones, D.T. FFPred 3: Feature-based function prediction for all Gene Ontology domains. Sci. Rep. 2016, 6, 31865. [Google Scholar] [CrossRef]
- Wu, K.; Mu, X.Y.; Jiang, J.; Tan, M.; Wang, R.; Zhou, W.; Wang, X.; He, Y.; Li, M.; Liu, Z. miRNA 26a 5p and miR 26b 5p inhibit the proliferation of bladder cancer cells by regulating PDCD10. Oncol. Rep. 2018, 40, 3523–3532. [Google Scholar] [CrossRef]
- Liang, L.; Zeng, J.-H.; Wang, J.-Y.; He, R.-Q.; Ma, J.; Chen, G.; Cai, X.-Y.; Hu, X.-H. Down-regulation of miR-26a-5p in hepatocellular carcinoma: A qRT-PCR and bioinformatics study. Pathol. Res. Pract. 2017, 213, 1494–1509. [Google Scholar] [CrossRef]
- Gasparini, P.; Cascione, L.; Landi, L.; Carasi, S.; Lovat, F.; Tibaldi, C.; Alì, G.; D’incecco, A.; Minuti, G.; Chella, A.; et al. microRNA classifiers are powerful diagnostic/prognostic tools in ALK-, EGFR-, and KRAS-driven lung cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Xie, M.; Jing, X.; Jiang, H.; Wu, X.; Wang, X.; Shu, Y. Loss of miR-26b-5p promotes gastric cancer progression via miR-26b-5p-PDE4B/CDK8-STAT3 feedback loop. J. Transl. Med. 2023, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Heise, R.; Skazik, C.; Marquardt, Y.; Czaja, K.; Sebastian, K.; Kurschat, P.; Gan, L.; Denecke, B.; Ekanayake-Bohlig, S.; Wilhelm, K.-P.; et al. Dexpanthenol modulates gene expression in skin wound healing in vivo. Ski. Pharmacol. Physiol. 2012, 25, 241–248. [Google Scholar] [CrossRef]
- Shimomura, Y.; Ito, M. Human hair keratin-associated proteins. J. Investig. Dermatol. Symp. Proc. 2005, 10, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Moiana, M.; Aranda, F.; de Larrañaga, G. A focus on the roles of histones in health and diseases. Clin. Biochem. 2021, 94, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pusarla, R.H.; Bhargava, P. Histones in functional diversification. Core histone variants. FEBS J. 2005, 272, 5149–5168. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. [Google Scholar] [CrossRef]
- Martin, T.A.; Li, A.X.; Sanders, A.J.; Ye, L.; Frewer, K.; Hargest, R.; Jiang, W.G. NUPR1 and its potential role in cancer and pathological conditions (Review). Int. J. Oncol. 2021, 58, 21. [Google Scholar] [CrossRef]
- Vadlamudi, R.K.; Bagheri-Yarmand, R.; Yang, Z.; Balasenthil, S.; Nguyen, D.; Sahin, A.A.; Hollander, P.D.; Kumar, R. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004, 5, 575–585, Erratum in Cancer Cell. 2004, 6, 101; Erratum in Cancer Cell. 2013, 23, 421–422. [Google Scholar] [CrossRef]
- Puthalakath, H.; Huang, D.C.; O’Reilly, L.A.; King, S.M.; Strasser, A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 1999, 3, 287–296. [Google Scholar] [CrossRef]
- Kang, H.M.; Noh, K.H.; Chang, T.K.; Park, D.; Cho, H.-S.; Lim, J.H.; Jung, C.-R. Ubiquitination of MAP1LC3B by pVHL is associated with autophagy and cell death in renal cell carcinoma. Cell Death Dis. 2019, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.-P.; Cao, J.; Lee, S.-J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M.K. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zheng, Q.; Erramilli, S.K.; Pan, M.; Park, S.; Xie, Y.; Li, J.; Fei, J.; Kossiakoff, A.A.; Liu, L.; et al. K29-linked ubiquitin signaling regulates proteotoxic stress response and cell cycle. Nat. Chem. Biol. 2021, 17, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Kedves, A.T.; Gleim, S.; Liang, X.; Bonal, D.M.; Sigoillot, F.; Harbinski, F.; Sanghavi, S.; Benander, C.; George, E.; Gokhale, P.C.; et al. Recurrent ubiquitin B silencing in gynecological cancers establishes dependence on ubiquitin C. J. Clin. Investig. 2017, 127, 4554–4568. [Google Scholar] [CrossRef]
- Compton, S.L.; Behrend, E.N. PRAF1: A Golgi complex transmembrane protein that interacts with viruses. Biochem. Cell Biol. 2006, 84, 940–948. [Google Scholar] [CrossRef]
- Gougeon, P.Y.; Prosser, D.C.; Da-Silva, L.F.; Ngsee, J.K. Disruption of Golgi morphology and trafficking in cells expressing mutant prenylated rab acceptor-1. J. Biol. Chem. 2002, 277, 36408–36414. [Google Scholar] [CrossRef]
- Zheng, W.; Wuyun, Q.; Li, Y.; Zhang, C.; Freddolino, P.L.; Zhang, Y. Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data. Nat. Methods 2024, 21, 279–289. [Google Scholar] [CrossRef]
- Kustatscher, G.; Collins, T.; Gingras, A.-C.; Guo, T.; Hermjakob, H.; Ideker, T.; Lilley, K.S.; Lundberg, E.; Marcotte, E.M.; Ralser, M.; et al. Understudied proteins: Opportunities and challenges for functional proteomics. Nat. Methods 2022, 19, 774–779. [Google Scholar] [CrossRef]
- Forslund, K.; Sonnhammer, E.L. Predicting protein function from domain content. Bioinformatics 2008, 24, 1681–1687, Erratum in Bioinformatics 2009, 25, 1214. [Google Scholar] [CrossRef]
- Yoodee, S.; Thongboonkerd, V. Bioinformatics and computational analyses of kidney stone modulatory proteins lead to solid experimental evidence and therapeutic potential. Biomed. Pharmacother. 2023, 159, 114217. [Google Scholar] [CrossRef] [PubMed]
- Rahmatbakhsh, M.; Gagarinova, A.; Babu, M. Bioinformatic Analysis of Temporal and Spatial Proteome Alternations During Infections. Front. Genet. 2021, 12, 667936. [Google Scholar] [CrossRef]
- Calderón-González, K.G.; Hernández-Monge, J.; Herrera-Aguirre, M.E.; Luna-Arias, J.P. Bioinformatics Tools for Proteomics Data Interpretation. Adv. Exp. Med. Biol. 2016, 919, 281–341. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, W.; Yan, Z.; Zhao, B.; Zhao, J.; Feng, W.; Chen, X.; Li, C.; Liu, K.-X. MicroRNA-26b-5p Targets DAPK1 to Reduce Intestinal Ischemia/Reperfusion Injury via Inhibition of Intestinal Mucosal Cell Apoptosis. Dig. Dis. Sci. 2021, 67, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Gu, J.; Yu, D.; Wang, M.; Zhang, J.; Ji, R.; Jiang, P.; Zhang, X. Circ1811 suppresses gastric cancer progression by regulating the miR-632/DAPK1 axis. Gene 2024, 910, 148331. [Google Scholar] [CrossRef]
- Ner-Gaon, H.; Halachmi, R.; Savaldi-Goldstein, S.; Rubin, E.; Ophir, R.; Fluhr, R. Intron retention is a major phenomenon in alternative splicing inArabidopsis. Plant J. 2004, 39, 877–885. [Google Scholar] [CrossRef]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef]
- Tan, S.; Guo, J.; Huang, Q.; Chen, X.; Li-Ling, J.; Li, Q.; Ma, F. Retained introns increase putative microRNA targets within 3′ UTRs of human mRNA. FEBS Lett. 2007, 581, 1081–1086. [Google Scholar] [CrossRef]
- Schmitz, U.; Pinello, N.; Jia, F.; Alasmari, S.; Ritchie, W.; Keightley, M.-C.; Shini, S.; Lieschke, G.J.; Wong, J.J.; Rasko, J.E.J. Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol. 2017, 18, 216. [Google Scholar] [CrossRef]
- Navarro, E.; Mallén, A.; Hueso, M. Dynamic Variations of 3′UTR Length Reprogram the mRNA Regulatory Landscape. Biomedicines 2021, 9, 1560. [Google Scholar] [CrossRef]
- Chen, P.-A. The LncRNA TPT1-AS1 Promotes the Survival of Neuroendocrine Prostate Cancer Cells by Facilitating Autophagy. Am. J. Cancer Res. 2024, 14, 2103–2123. [Google Scholar] [CrossRef] [PubMed]
- Bibi, A.; Madè, A.; Greco, S.; Garcia-Manteiga, J.M.; Tascini, A.S.; Tastsoglou, S.; Zaccagnini, G.; Leszek, P.; Gaetano, C.; Martelli, F. Circular PVT1 Promotes Cardiac Fibroblast Activation Interacting with MiR-30a-5p and MiR-125b-5p. Cell Death Dis. 2025, 16, 325. [Google Scholar] [CrossRef] [PubMed]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25. [Google Scholar] [CrossRef]
- Gurzau, A.D.; Blewitt, M.E.; Czabotar, P.E.; Murphy, J.M.; Birkinshaw, R.W. Relating SMCHD1 structure to its function in epigenetic silencing. Biochem. Soc. Trans. 2020, 48, 1751–1763. [Google Scholar] [CrossRef]
- Berezovsky, I.N.; Guarnera, E.; Zheng, Z. Basic units of protein structure, folding, and function. Prog. Biophys. Mol. Biol. 2017, 128, 85–99. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Analyzing protein structure and function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.; Iyer, L. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef]
- Hao, Y.; Zong, X.; Ren, P.; Qian, Y.; Fu, A. Basic Helix-Loop-Helix (bHLH) Transcription Factors Regulate a Wide Range of Functions in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 7152. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Kumar, A.; Balbach, J. Folding and Stability of Ankyrin Repeats Control Biological Protein Function. Biomolecules 2021, 11, 840. [Google Scholar] [CrossRef]
- Kemege, K.E.; Hickey, J.M.; Lovell, S.; Battaile, K.P.; Zhang, Y.; Hefty, P.S. Ab initio structural modeling of and experimental validation for Chlamydia trachomatis protein CT296 reveal structural similarity to Fe(II) 2-oxoglutarate-dependent enzymes. J. Bacteriol. 2011, 193, 6517–6528. [Google Scholar] [CrossRef] [PubMed]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef]
- Li, J.; Mahajan, A.; Tsai, M.D. Ankyrin repeat: A unique motif mediating protein-protein interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef]
- Lin, Y.; Stevens, C.; Harrison, B.; Pathuri, S.; Amin, E.; Hupp, T.R. The alternative splice variant of DAPK-1, s-DAPK-1, induces proteasome-independent DAPK-1 destabilization. Mol. Cell Biochem. 2009, 328, 101–107. [Google Scholar] [CrossRef]
- Klose, J.; Wendt, N.; Kubald, S.; Krause, E.; Fechner, K.; Beyermann, M.; Bienert, M.; Rudolph, R.; Rothemund, S. Hexa-histidin tag position influences disulfide structure but not binding behavior of in vitro folded N-terminal domain of rat corticotropin-releasing factor receptor type 2a. Protein Sci. 2004, 13, 2470–2475. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.R.; Mrozkiewicz, M.K.; McGrath, W.J.; Listwan, P.; Kobe, B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003, 12, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein-protein interactions. Bioinformatics 2007, 23, 1909–1918. [Google Scholar] [CrossRef]
- Morris, R.; Black, K.A.; Stollar, E.J. Uncovering protein function: From classification to complexes. Essays Biochem. 2022, 66, 255–285. [Google Scholar] [CrossRef]
- Stevers, L.M.; Sijbesma, E.; Botta, M.; MacKintosh, C.; Obsil, T.; Landrieu, I.; Cau, Y.; Wilson, A.J.; Karawajczyk, A.; Eickhoff, J.; et al. Modulators of 14-3-3 Protein-Protein Interactions. J. Med. Chem. 2018, 61, 3755–3778. [Google Scholar] [CrossRef]
- Rudnizky, S.; Malik, O.; Bavly, A.; Pnueli, L.; Melamed, P.; Kaplan, A. Nucleosome mobility and the regulation of gene expression: Insights from single-molecule studies. Protein Sci. 2017, 26, 1266–1277. [Google Scholar] [CrossRef]
- Damiescu, R.; Efferth, T.; Dawood, M. Dysregulation of different modes of programmed cell death by epigenetic modifications and their role in cancer. Cancer Lett. 2024, 584, 216623. [Google Scholar] [CrossRef] [PubMed]
- Paramkusham, S.; Mamidala, E.; Madhukar, J. Structural and Functional Insights into Riboflavin-Binding Proteins in Whooping crane (Grus americana) and Other Avian Species: An In Silico Approach. J. Neonatal Surg. 2025, 14, 563–569. [Google Scholar]
- Jeba, S.H.; Hasan, M.A.; Faisal, A.; Khatun, M.U.S.; Chowdhury, A.H.; Islam, N.; Miah, M. In silico approach of exploring the structural and functional characteristics of a hypothetical protein from monkeypox virus: A potential insight for antiviral therapeutics. Microbes Infect. Dis./Microbes Infect. Dis. 2024, 29, 13596535241255199. [Google Scholar] [CrossRef]
- Saikia, A.; Palherkar, D.A.; Hiremath, L. In silico Analysis and Structural Prediction of a Hypothetical Protein from Leishmania Major. Biomed. Biotechnol. Res. J. (BBRJ) 2021, 5, 320–326. [Google Scholar] [CrossRef]
- Arshad, M.; Bhatti, A.; John, P. Identification and in Silico Analysis of Functional SNPs of Human TAGAP Protein: A Comprehensive Study. PLoS ONE 2018, 13, e0188143. [Google Scholar] [CrossRef]
- Khan, A.; Ahmed, H.; Jahan, N.; Ali, S.R.; Amin, A.; Morshed, M.N. An in silico Approach for Structural and Functional Annotation of Salmonella enterica serovar typhimurium Hypothetical Protein R_27. Dir. Open Access J. 2016, 20, 31–42. Available online: https://doaj.org/article/2368937bffb84fb0bc5c2843e7d2b2aa (accessed on 16 April 2025).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted microRNA | Function | Binding Site | References |
|---|---|---|---|
| hsa-miR-26a-5p | Tumor suppressor in hepatocellular carcinoma and lung cancer | 5′ UTR | [41,42,43] |
| hsa-miR-26b-5p | Tumor suppressor in gastric cancer | 5′ UTR | [44] |
| Parameters | Values |
|---|---|
| Number of amino acids | 337 |
| Molecular weight (Da) | 36,739.07 |
| Theoretical pI | 8.96 |
| Extinction coefficients | 27,680 a 26,930 b |
| Half-life | 30 h (mammalian reticulocytes, in vitro) >20 h (yeast, in vivo) >10 h (Escherichia coli, in vivo) |
| Instability index | 40.67 |
| Aliphatic index | 87.74 |
| Grand average of hydropathicity (GRAVY) | −0.221 |
| Protein Name | Uniprot ID | Score (1 = Binding) | Function |
|---|---|---|---|
| Keratin-associated protein 4–12 | Q9BQ66 | 0.8173 | Important for making hair strong, this happens when disulfide bonds connect cysteines of hair keratins [45,46] |
| Histone H2B type 2-E | Q16778 | 0.7173 | Responsible for wrapping and compacting DNA into chromatin, thus limiting DNA accessibility to cellular machinery that uses DNA as a template [47,48] |
| Prion protein | Q53YK7 | 0.7093 | Promotes tumor progression [49] |
| Nuclear protein 1 | O60356 | 0.7107 | Promotes cancer progression [50] |
| Dynein light chain 1, cytoplasmic | P63167 | 0.7200 | Regulates apoptosis by sequestering BCL2L11 in microtubules [51,52] |
| Microtubule-associated protein 1 light chain 3 beta, isoform CRA_c | Q658J6 | 0.7200 | Regulates autophagy [53] |
| Microtubule-associated proteins 1A/1B light chain 3B | Q9GZQ8 | 0.7200 | Involved in the formation of autophagosomes [54] |
| Polyubiquitin-B | P0CG47 | 0.7133 | Regulates protein ubiquitination [55,56] |
| Epididymis secretory protein Li 50 | Q5U5U6 | 0.7133 | Targets cellular proteins for degradation by the 26S proteosome [57] |
| Prenylated Rab acceptor protein 1 | Q9UI14 | 0.7133 | Necessary for the vesicle formation from the Golgi complex [58,59] |
| (a) | |||
| Biological Process Predictions | |||
| GO Term | Name | Probability Score | SVM Reliability |
| GO:1903506 | regulation of nucleic acid-templated transcription | 0.829 | H |
| GO:2001141 | regulation of RNA biosynthetic process | 0.813 | H |
| GO:0051252 | regulation of RNA metabolic process | 0.808 | H |
| GO:0051171 | regulation of nitrogen compound metabolic process | 0.769 | H |
| GO:0034645 | cellular macromolecule biosynthetic process | 0.767 | H |
| GO:0006355 | regulation of transcription, DNA-templated | 0.760 | H |
| GO:0009116 | nucleoside metabolic process | 0.719 | H |
| GO:0006810 | transport | 0.718 | H |
| GO:0019222 | regulation of metabolic process | 0.710 | H |
| GO:0006796 | phosphate-containing compound metabolic process | 0.704 | H |
| GO:0055086 | nucleobase-containing small molecule metabolic process | 0.684 | H |
| GO:0044281 | small molecule metabolic process | 0.675 | H |
| GO:0009059 | macromolecule biosynthetic process | 0.646 | H |
| GO:0010468 | regulation of gene expression | 0.638 | H |
| GO:0006163 | purine nucleotide metabolic process | 0.634 | H |
| GO:0045184 | establishment of protein localization | 0.608 | H |
| GO:0019637 | organophosphate metabolic process | 0.605 | H |
| GO:0045333 | cellular respiration | 0.577 | H |
| GO:0051641 | cellular localization | 0.571 | H |
| GO:0006091 | generation of precursor metabolites and energy | 0.551 | H |
| GO:0055114 | oxidation-reduction process | 0.547 | H |
| GO:0009259 | ribonucleotide metabolic process | 0.534 | H |
| GO:0051649 | establishment of localization in cell | 0.531 | H |
| GO:0006082 | organic acid metabolic process | 0.523 | H |
| GO:0006412 | translation | 0.510 | H |
| Molecular function predictions | |||
| GO:0008270 | zinc ion binding | 0.932 | H |
| GO:0003723 | RNA binding | 0.754 | H |
| GO:0003824 | catalytic activity | 0.715 | H |
| GO:0015631 | tubulin binding | 0.713 | H |
| GO:0003676 | nucleic acid binding | 0.708 | H |
| GO:0000166 | nucleotide binding | 0.687 | H |
| GO:0035639 | purine ribonucleoside triphosphate binding | 0.679 | H |
| GO:0017076 | purine nucleotide binding | 0.669 | H |
| GO:0001883 | purine nucleoside binding | 0.659 | H |
| GO:0008092 | cytoskeletal protein binding | 0.658 | H |
| GO:0016491 | oxidoreductase activity | 0.611 | H |
| GO:0001882 | nucleoside binding | 0.609 | H |
| GO:0032549 | ribonucleoside binding | 0.601 | H |
| GO:0030554 | adenyl nucleotide binding | 0.552 | H |
| GO:0003779 | actin binding | 0.541 | H |
| GO:0016740 | transferase activity | 0.533 | H |
| GO:0019901 | protein kinase binding | 0.530 | H |
| GO:0003677 | DNA binding | 0.520 | H |
| GO:0046872 | metal ion binding | 0.741 | L |
| GO:0005102 | receptor binding | 0.691 | L |
| GO:0036094 | small molecule binding | 0.666 | L |
| GO:0043169 | cation binding | 0.595 | L |
| GO:0032403 | protein complex binding | 0.554 | L |
| GO:0019904 | protein domain specific binding | 0.539 | L |
| (b) | |||
| Biological Process Predictions | |||
| GO Term | Name | Probability Score | SVM Reliability |
| GO:0006796 | phosphate-containing compound metabolic process | 0.813 | H |
| GO:0008380 | RNA splicing | 0.807 | H |
| GO:0006811 | ion transport | 0.753 | H |
| GO:0019222 | regulation of metabolic process | 0.735 | H |
| GO:0006810 | transport | 0.672 | H |
| GO:0016310 | phosphorylation | 0.648 | H |
| GO:0000398 | mRNA splicing, via spliceosome | 0.638 | H |
| GO:0044281 | small molecule metabolic process | 0.614 | H |
| GO:0051171 | regulation of nitrogen compound metabolic process | 0.598 | H |
| GO:0006396 | RNA processing | 0.566 | H |
| GO:0019637 | organophosphate metabolic process | 0.557 | H |
| GO:1903506 | regulation of nucleic acid-templated transcription | 0.552 | H |
| GO:2001141 | regulation of RNA biosynthetic process | 0.551 | H |
| GO:0009059 | macromolecule biosynthetic process | 0.546 | H |
| GO:0006397 | mRNA processing | 0.539 | H |
| GO:0071345 | cellular response to cytokine stimulus | 0.527 | H |
| GO:0006082 | organic acid metabolic process | 0.511 | H |
| GO:0055086 | nucleobase-containing small molecule metabolic process | 0.510 | H |
| GO:0051252 | regulation of RNA metabolic process | 0.510 | H |
| GO:0006355 | regulation of transcription, DNA-templated | 0.505 | H |
| GO:0055114 | oxidation-reduction process | 0.503 | H |
| GO:0008152 | metabolic process | 0.925 | L |
| GO:0050896 | response to stimulus | 0.869 | L |
| GO:0051716 | cellular response to stimulus | 0.836 | L |
| GO:0044237 | cellular metabolic process | 0.791 | L |
| Molecular Function Predictions | |||
| GO:0003824 | catalytic activity | 0.973 | H |
| GO:0035639 | purine ribonucleoside triphosphate binding | 0.939 | H |
| GO:0017076 | purine nucleotide binding | 0.938 | H |
| GO:0030554 | adenyl nucleotide binding | 0.925 | H |
| GO:0032549 | ribonucleoside binding | 0.921 | H |
| GO:0000166 | nucleotide binding | 0.914 | H |
| GO:0001883 | purine nucleoside binding | 0.911 | H |
| GO:0005524 | ATP binding | 0.899 | H |
| GO:0001882 | nucleoside binding | 0.844 | H |
| GO:0016817 | hydrolase activity, acting on acid anhydrides | 0.766 | H |
| GO:0016773 | phosphotransferase activity, alcohol group as acceptor | 0.759 | H |
| GO:0044822 | poly(A) RNA binding | 0.758 | H |
| GO:0016301 | kinase activity | 0.751 | H |
| GO:0017111 | nucleoside-triphosphatase activity | 0.740 | H |
| GO:0004386 | helicase activity | 0.666 | H |
| GO:0003676 | nucleic acid binding | 0.650 | H |
| GO:0016462 | pyrophosphatase activity | 0.636 | H |
| GO:0003723 | RNA binding | 0.633 | H |
| GO:0016818 | hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides | 0.596 | H |
| GO:0016740 | transferase activity | 0.593 | H |
| GO:0016491 | oxidoreductase activity | 0.523 | H |
| GO:0008092 | cytoskeletal protein binding | 0.517 | H |
| GO:0046872 | metal ion binding | 0.887 | L |
| GO:0097159 | organic cyclic compound binding | 0.869 | L |
| GO:0016772 | transferase activity, transferring phosphorus-containing groups | 0.862 | L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makgoo, L.; Mosebi, S.; Mbita, Z. In Silico Analysis of s-DAPK-1: From Structure to Function and Regulation. Curr. Issues Mol. Biol. 2025, 47, 416. https://doi.org/10.3390/cimb47060416
Makgoo L, Mosebi S, Mbita Z. In Silico Analysis of s-DAPK-1: From Structure to Function and Regulation. Current Issues in Molecular Biology. 2025; 47(6):416. https://doi.org/10.3390/cimb47060416
Chicago/Turabian StyleMakgoo, Lilian, Salerwe Mosebi, and Zukile Mbita. 2025. "In Silico Analysis of s-DAPK-1: From Structure to Function and Regulation" Current Issues in Molecular Biology 47, no. 6: 416. https://doi.org/10.3390/cimb47060416
APA StyleMakgoo, L., Mosebi, S., & Mbita, Z. (2025). In Silico Analysis of s-DAPK-1: From Structure to Function and Regulation. Current Issues in Molecular Biology, 47(6), 416. https://doi.org/10.3390/cimb47060416