
Plasma Sclerostin Levels in Rheumatoid Arthritis Women on TNF-α Inhibitor Therapy

 , and
, and 
Abstract
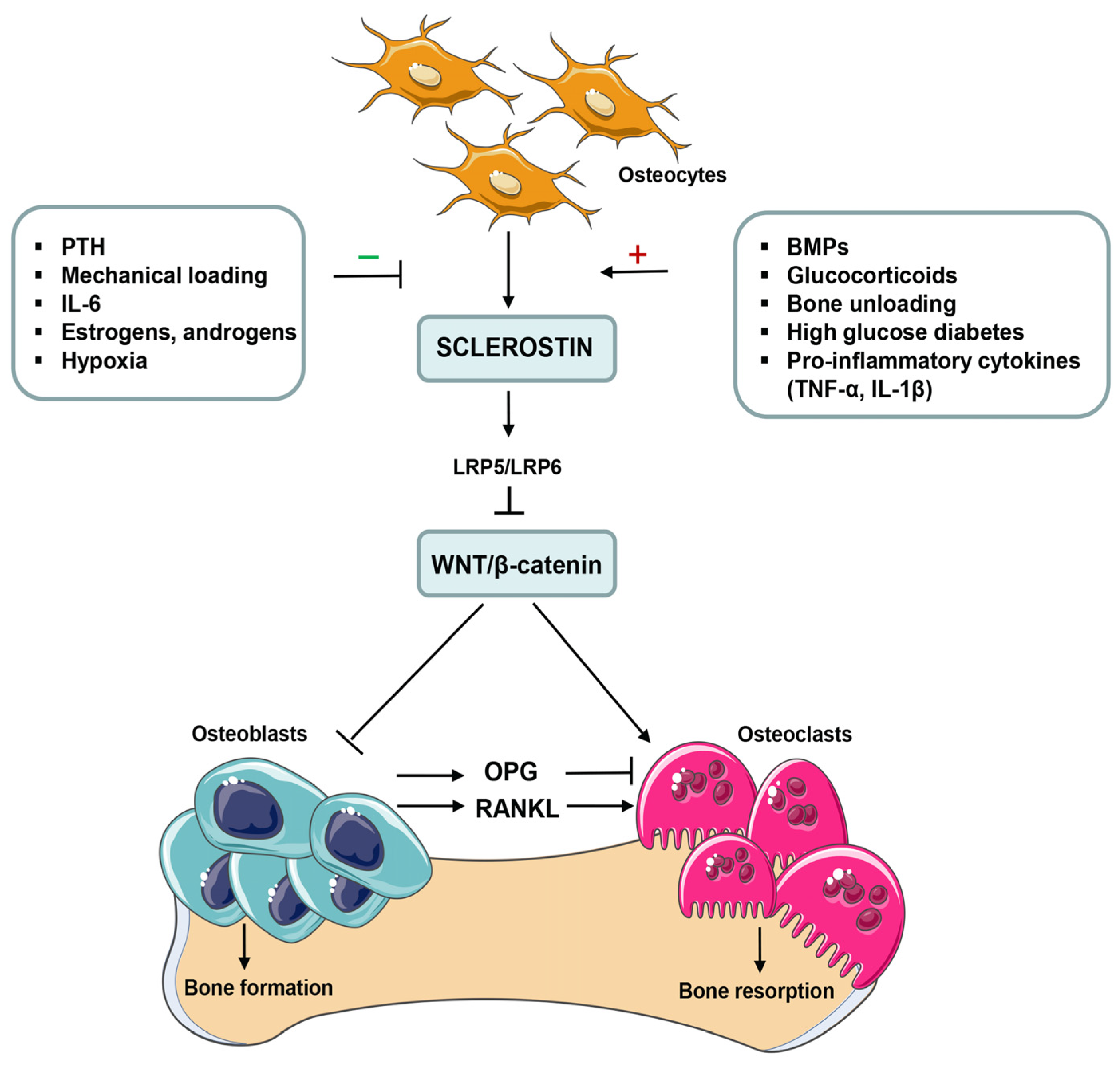
1. Introduction
2. Results
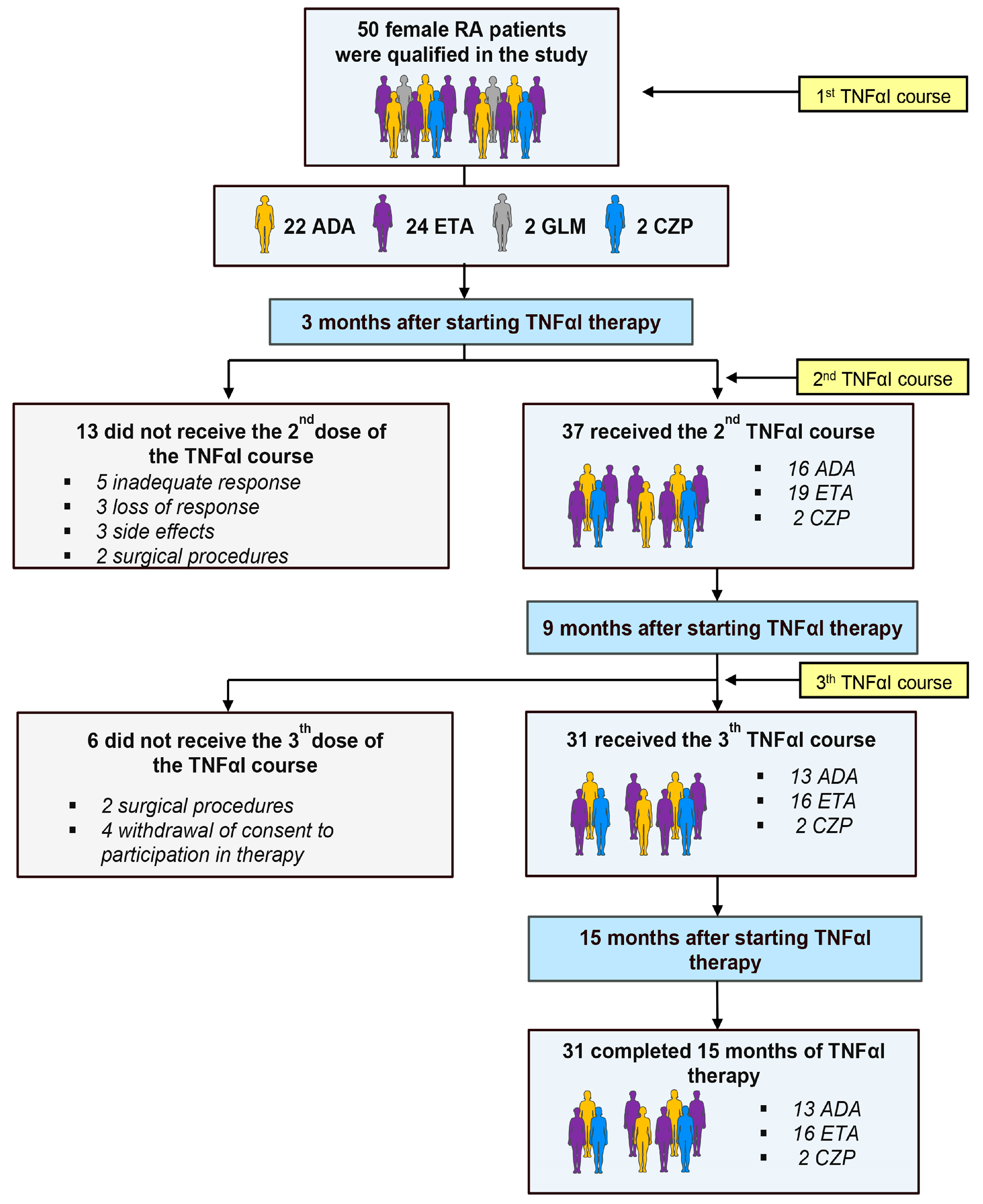
2.1. Clinical Response to TNFαI Therapy
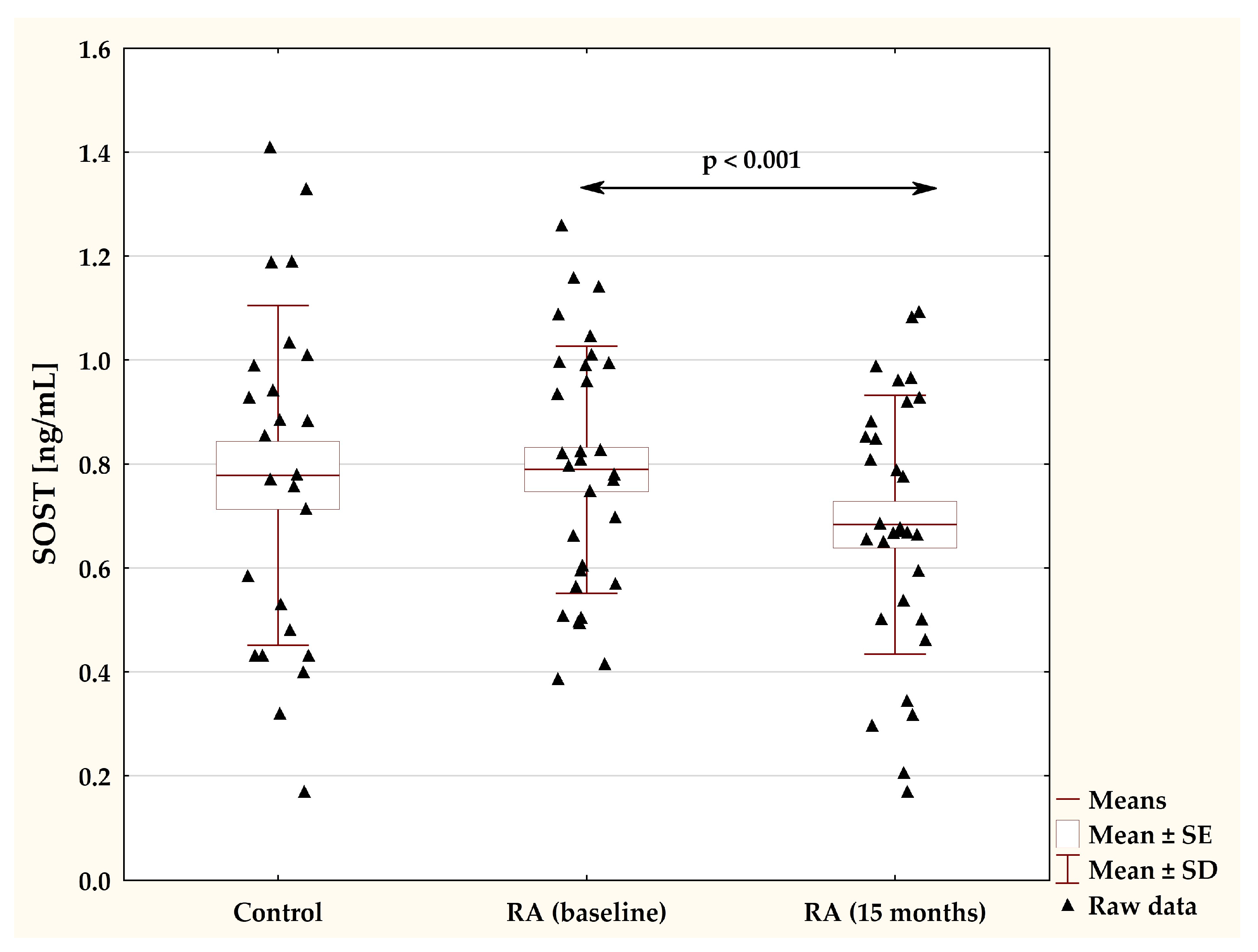
2.2. Effects of TNFαI Therapy on Circulating Sclerostin Levels
2.3. Plasma Sclerostin Levels in Pre- and Postmenopausal Women with RA
2.4. Correlations between Sclerostin and Demographic, Clinical and Laboratory Parameters in RA Patients under TNFαI Treatment
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Clinical and Laboratory Assessments
Immunoassay of Sclerostin
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akram, M.; Daniyal, M.; Sultana, S.; Owais, A.; Akhtar, N.; Zahid, R.; Said, F.; Bouyahya, A.; Ponomarev, E.; Ali Shariat, M.; et al. Traditional and modern management strategies for rheumatoid arthritis. Clin. Chim. Acta 2021, 512, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulas, T.; Nikas, S.N.; Trontzas, P.; Kitas, G.D. Biologic therapies and systemic bone loss in rheumatoid arthritis. Au-toimmun. Rev. 2013, 12, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Cici, D.; Corrado, A.; Rotondo, C.; Cantatore, F.P. Wnt signaling and biological therapy in rheumatoid arthritis and spon-dyloarthritis. Int. J. Mol. Sci. 2019, 20, 5552. [Google Scholar] [CrossRef]
- Marini, F.; Giusti, F.; Palmini, G.; Brandi, M.S. Role of Wnt signaling and sclerostin in bone and as therapeutic targets in skeletal disorders. Osteoporos. Int. 2023, 34, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, T.J.; Keen, J.A.; Wells, L.M.; Roberts, S.J. Novel insights on the effect of sclerostin on bone and other organs. J. Endocrinol. 2023, 257, e220209. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Böttcher, M.; Cromme, C.; König, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra35. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, C.; He, J.; Gu, W.; Yi, C.; Chen, B.; Wang, Q.; Tang, F.; Xu, J.; Yue, H.; et al. Sclerostin and its associations with bone metabolism markers and sex hormones in healthy community-dwelling elderly individuals and adolescents. Front. Cell Dev. Biol. 2020, 8, 57. [Google Scholar] [CrossRef]
- Kuipers, A.L.; Zhang, Y.; Yu, S.; Kammerer, C.M.; Nestlerode, C.S.; Chu, Y.; Bunker, C.H.; Patrick, A.L.; Wheeler, V.W.; Miljkovic, I.; et al. Relative influence of heritability, environment and genetics on serum sclerostin. Osteoporos. Int. 2014, 25, 905–912. [Google Scholar] [CrossRef]
- Ardawi, M.S.; Al-Kadi, H.A.; Rouzi, A.A.; Qari, M.H. Determinants of serum sclerostin in healthy pre- and postmenopausal women. J. Bone. Miner. Res. 2011, 26, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Jaśkiewicz, Ł.; Chmielewski, G.; Kuna, J.; Stompór, T.; Krajewska-Włodarczyk, M. The role of sclerostin in rheumatic diseases: A review. J. Clin. Med. 2023, 12, 6248. [Google Scholar] [CrossRef] [PubMed]
- Jura-Półtorak, A.; Szeremeta, A.; Olczyk, K.; Zoń-Giebel, A.; Komosińska-Vassev, K. Bone metabolism and RANKL/OPG ratio in rheumatoid arthritis women treated with TNF-α Inhibitors. J. Clin. Med. 2021, 10, 2905. [Google Scholar] [CrossRef] [PubMed]
- Vincent, C.; Findlay, D.M.; Welldon, K.J.; Wijenayaka, A.R.; Zheng, T.S.; Haynes, D.R.; Fazzalari, N.L.; Evdokiou, A.; Atkins, G.J. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mito-gen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J. Bone Miner. Res. 2009, 24, 1434–1449. [Google Scholar] [CrossRef] [PubMed]
- Matzelle, M.M.; Gallant, M.A.; Condon, K.W.; Walsh, N.C.; Manning, C.A.; Stein, G.S.; Lian, J.B.; Burr, D.B.; Gravallese, E.M. Resolution of inflammation induces osteoblast function and regulates the Wnt signaling pathway. Arthritis Rheum. 2012, 64, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Brabnikova-Maresova, K.; Jarosova, K.; Pavelka, K.; Stepan, J.J. Serum sclerostin in high-activity adult patients with juvenile idiopathic arthritis. Arthritis Res. Ther. 2014, 16, 460. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, Z.; Akgol, G.; Gulkesen, A.; Kaya, A.; Kaman, D.; Ulusoy, H. Clinical correlation and determination of Dkk-1 and sclerostin levels in patients with rheumatoid arthritis. Med. Sci. 2020, 9, 1053–1060. [Google Scholar] [CrossRef]
- Gharbia, O.; Hegazy, A.; Elhelaly, R.; ElGhaweet, A. Serum sclerostin in rheumatoid-induced osteoporosis. Egypt. Rheumatol. Rehabil. 2020, 47, 22. [Google Scholar] [CrossRef]
- Fayed, A.; Elgohary, R.; Fawzy, M. Evaluating the role of serum sclerostin as an indicator of activity and damage in Egyptian patients with rheumatoid arthritis: University hospital experience. Clin. Rheumatol. 2020, 39, 1121–1130. [Google Scholar] [CrossRef]
- Cauli, A.; Dessole, G.; Porru, G.; Piga, M.; Vacca, A.; Ibba, V.; Garau, P.; Mathieu, A. AB0114 Light (TNFSF14), cathepsin-K, DKK-1 and sclerostin in rheumatoid arthritis patients: Effect of anti TNF-alpha treatment in the WNT/beta-catenin network signaling. Ann. Rheum. Dis. 2013, 71, 644. [Google Scholar] [CrossRef]
- Mehaney, D.A.; Eissa, M.; Anwar, S.; Fakhr El-Din, S. Serum sclerostin level among egyptian rheumatoid arthritis patients: Relation to disease activity, bone mineral density and radiological grading. Acta Reumatol. Port. 2015, 40, 268–274. [Google Scholar]
- Świerkot, J.; Gruszecka, K.; Matuszewska, A.; Wiland, P. Assessment of the effect of methotrexate therapy on bone metabolism in patients with rheumatoid arthritis. Arch. Immunol. Ther. Exp. 2015, 63, 397–404. [Google Scholar] [CrossRef]
- Vargas-Muñoz, V.M.; Jimenez-Andrade, M.C.; Villarreal-Salcido, J.C.; Martinez-Martinez, A.; Acosta-Gonzalez, R.I.; Lu-go-Zamudio, G.E.; Conde-Mercado, J.M.; Barbosa-Cobos, R.E.; Matias-Morales, F.A.; Ramirez-Rosas, M.B.; et al. Association between sclerostin and bone mineral density in a Mexican sample of women with rheumatoid arthritis: A pilot study. J. Arthritis 2015, S1, 1–6. [Google Scholar]
- Lim, M.J.; Kwon, S.R.; Joo, K.; Son, M.J.; Park, S.G.; Park, W. Early effects of tumor necrosis factor inhibition on bone homeostasis after soluble tumor necrosis factor receptor use. Korean J. Intern. Med. 2014, 29, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Paccou, J.; Mentaverri, R.; Renard, C.; Liabeuf, S.; Fardellone, P.; Massy, Z.A.; Brazier, M.; Kamel, S. The relationships between serum sclerostin, bone mineral density, and vascular calcification in rheumatoid arthritis. J. Clin. Endocrinol. Metab. 2014, 99, 4740–4748. [Google Scholar] [CrossRef] [PubMed]
- Dhakad, U.; Sahoo, R.R.; Goel, A.P.; Pradhan, S.; Srivastava, R.; Das, S.K. Serum sclerostin levels in rheumatoid arthritis and correlation with disease activity and bone mineral density. Indian J. Rheumatol. 2019, 14, 28–31. [Google Scholar] [CrossRef]
- Pacifici, R. Role of T cells in ovariectomy induced bone loss-revisited. J. Bone Miner. Res. 2012, 27, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.E.; Abdelsamad, A.M.; Helmy, A.; Farouk, N. Serum sclerostin levels in rheumatoid arthritis. Indian J. Rheumatol. 2015, 10, 117–120. [Google Scholar] [CrossRef]
- El-Bakry, S.; Saber, N.; Zidan, H.; Samaha, D. Sclerostin as an innovative insight towards understanding rheumatoid arthritis. Egypt. Rheumatol. 2016, 38, 71–75. [Google Scholar] [CrossRef]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondy-litis. Arthritis Rheum. 2009, 60, 3257–3262. [Google Scholar] [CrossRef]
- Piec, I.; Washbourne, C.; Tang, J.; Fisher, E.; Greeves, J.; Jackson, S.; Fraser, W.D. How accurate is your sclerostin measurement? comparison between three commercially available sclerostin elisa kits. Calcif. Tissue. Int. 2016, 98, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Ying, H.; Du, J.; Shen, B. Serum sclerostin levels in patients with ankylosing spondylitis and rheumatoid arthritis: A systematic review and meta-analysis. BioMed Res. Int. 2017, 2017, 9295313. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Adami, G.; Giollo, A.; Viapiana, O.; Malavolta, N.; Saviola, G.; Bortolotti, R.; Idolazzi, L.; Bertoldo, F.; Rossini, M.; et al. Acute effects of glucocorticoid treatment, TNFα or IL-6R blockade on bone turnover markers and Wnt inhibitors in early rheumatoid arthritis: A pilot study. Calcif. Tissue Int. 2020, 106, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Orsolini, G.; Adami, S.; Viapiana, O.; Idolazzi, L.; Gatti, D.; Rossini, M. Effects of TNF inhibitors on parathyroid hormone and Wnt signaling antagonists in rheumatoid arthritis. Calcif. Tissue Int. 2016, 99, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Perpétuo, I.P.; Caetano-Lopes, J.; Rodrigues, A.M.; Campanilho-Marques, R.; Ponte, C.; Canhão, H.; Ainola, M.; Fonseca, J.E. Effect of tumor necrosis factor inhibitor therapy on osteoclasts precursors in rheumatoid arthritis. BioMed Res. Int. 2017, 2017, 2690402. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, K.; Horváth, Á.; Végh, E.; Pusztai, A.; Szentpétery, Á.; Pethö, Z.; Váncsa, A.; Bodnár, N.; Csomor, P.; Hamar, A.; et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin. Rheumatol. 2020, 39, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Pattacini, L.; Boiardi, L.; Casali, B.; Salvarani, C. Differential effects of anti-TNF-alpha drugs on fibroblast-like synoviocyte apoptosis. Rheumatology 2010, 49, 480–489. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S. The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef]
- Szeremeta, A.; Jura-Półtorak, A.; Zoń-Giebel, A.; Olczyk, K.; Komosińska-Vassev, K. Effects of etanercept and adalimumab on serum levels of cartilage remodeling markers in women with rheumatoid arthritis. J. Clin. Med. 2023, 12, 5185. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Woman with RA Which Responded to TNFαI Therapy (n = 31) | |
|---|---|---|
| Baseline (T0) | 15 Months after TNFαI Therapy (T3) | |
| Age (years) | 45.87 ± 12.28 | |
| Premenopausal females, n (%) | 17 (54.84) | |
| Postmenopausal females, n (%) | 14 (45.16) | |
| Disease duration (years) | 5 (3–11) | |
| Growth (cm) | 163.77 ± 6.63 | |
| Weight (kg) | 65.89 ± 14.60 | |
| BMI (kg/m2) | 24.62 ± 5.65 | |
| IgM-RF (+), n (%) | 28 (90.32) | |
| Anti-CCP (+), n (%) | 26 (83.87) | |
| ESR (mm/h) | 17.0 (10.0–34.0) | 13.0 (8.0–18.0) a |
| CRP (mg/L) | 6.3 (3.08–14.0) | 4.0 (1.5–5.1) a |
| Calcium C (mmol/L) | 2.30 ± 0.11 | 2.31 ± 0.11 |
| Phosphorus (mmol/L) | 1.36 ± 0.20 | 1.37 ± 0.21 |
| ALP (U/L) | 168.5 (152.5–202) | 165.5 (149.5–192) |
| SJC28, n | 7 (5–10) | 0 (0–0) a |
| TJC28, n | 12 (9–16) | 0 (0–0) a |
| VAS, (0–100 mm) | 80 (80–80) | 15 (5–20) a |
| DAS28-ESR | 5.78 (5.51–6.24) | 2.19 (1.75–2.51) a |
| Bone mineral density (BMD) | ||
| Lumbar L2-L4 BMD (g/cm2) | 0.89 (0.73–1.00) | 0.92 (0.79–1.03) |
| Neck femur BMD (g/cm2) | 0.83 (0.69–0.78) | 0.85 (0.77–0.85) |
| T-score | ||
| Lumbar L2-L4 T-score | −2.05 (−2.93–0.32) | −1.70 (−2.75–−0.65) |
| Neck femur T-score | −0.30 (−1.30–0.40) | −0.30 (−1.80–0.50) |
| Z-score | ||
| Lumbar L2-L4 Z-score | −1.43 (−2.38–0.23) | −1.15 (−2.00–0.15) |
| Neck femur Z-score | −0.10 (−0.90–0.10) | 0.00 (−0.70–0.10) |
| Patients which responded to TNFαI therapy, n (%) | ||
| ETA (Enbrel®) | 16 (51.62) | |
| ADA (Humira®) | 13 (41.93) | |
| CZP (Cimzia®) | 2 (6.45) | |
| Woman with RA Which Responded to TNFαI Therapy (n = 31) | ||||
|---|---|---|---|---|
| Parameter | Time of TNFαI Therapy (ADA or ETA or CZP) | |||
| Baseline (T0) | 3 mths. (T1) | 9 mths. (T2) | 15 mths. (T3) | |
| The inflammatory variables | ||||
| ESR (mm/h) | 17.0 (10.0–34.0) | 14.0 (9.0–23.0) | 13.0 (9.0–18.0) a | 13.0 (8.0–18.0) a |
| CRP (mg/L) | 6.3 (3.08–14.0) | 4.0 (2.0–9.0) | 4.0 (2.0–4.3) a | 4.0 (1.5–5.1) a |
| The clinical variables | ||||
| SJC28, n | 7 (5–10) | 2 (0–3) a,c | 0 (0–0) a,b | 0 (0–0) a,b |
| TJC28, n | 12 (9–16) | 4 (2–7) a,c | 1 (0–2) a,b | 0 (0–0) a,b,c |
| VAS, (0–100 mm) | 80 (80–80) | 40 (30–50) a,c | 20 (10–30) a,b | 15 (5–20) a,b |
| DAS28-ESR | 5.78 (5.51–6.24) | 3.92 (3.08–4.42) a,c | 2.75 (2.24–3.13) a,b | 2.19 (1.75–2.51) a,b,c |
| Disease activity, n (%) | ||||
| High (>5.1) | 31 (100) | 2 (6.45) | 0 (0) | 0 (0) |
| Moderate (>3.2 and ≤5.1) | 0 (0) | 20 (64.52) | 3 (9.68) | 0 (0) |
| Low (≤3.2 and >2.6) | 0 (0) | 4 (12.91) | 14 (45.16) | 5 (16.13) |
| Remission (≤2.6) | 0 (0) | 5 (16.13) | 14 (45.16) | 26 (83.87) |
| Parameter/ Stage of Menopause | SOST [ng/mL] | ||
|---|---|---|---|
| Baseline (T0) | 15 Months after TNFαI Therapy (T3) | p-Value | |
| Premenopausal Patients (n = 17) | 0.72 ± 0.24 | 0.61 ± 0.26 | 0.015 |
| Postmenopausal Patients (n = 14) | 0.88 ± 0.22 | 0.78 ± 0.20 | 0.027 |
| p-Value | 0.060 | 0.061 | |
| Parameter | Woman with RA Who Responded to TNFαI Therapy (n = 31) | |||
|---|---|---|---|---|
| SOST (ng/mL) | ||||
| Baseline (T0) | 15 Months after TNFαI Therapy (T3) | |||
| r | p | r | p | |
| Age (years) | 0.490 | 0.005 | ||
| Disease duration (years) | 0.342 | 0.059 | 0.426 | 0.017 |
| ESR (mm/h) | 0.428 | 0.016 | 0.220 | 0.235 |
| CRP (mg/L) | 0.249 | 0.177 | 0.078 | 0.678 |
| ALP (U/L) | 0.013 | 0.953 | −0.192 | 0.370 |
| SJC28, n | −0.235 | 0.203 | 0.085 | 0.648 |
| TJC28, n | −0.095 | 0.612 | 0.349 | 0.055 |
| VAS, (0–100 mm) | 0.115 | 0.538 | 0.222 | 0.230 |
| DAS28-ESR | 0.417 | 0.020 | 0.468 | 0.008 |
| Parameter | SOST in RA Patients (n = 31) at Baseline (T0) | |
|---|---|---|
| β | p-Value | |
| Age [years] | 0.008 | 0.028 |
| DAS28-ESR | 0.128 | 0.134 |
| Parameter | Healthy Controls | RA Patients at Baseline (T0) | p-Value |
|---|---|---|---|
| n = 26 | n = 31 | ||
| Age (years) | 46.12 ± 10.91 | 45.87 ± 12.28 | 0.938 |
| Premenopausal females, n (%) | 16 (61.54) | 17 (54.84) | – |
| Postmenopausal females, n (%) | 10 (38.46) | 14 (45.16) | – |
| Disease duration (years) | – | 5 (3–11) | – |
| Growth (cm) | 166.64 ± 6.23 | 163.77 ± 6.63 | 0.386 |
| Weight (kg) | 64.0 (62.0–67.0) | 64.0 (57.0–70.0) | 0.797 |
| BMI (kg/m2) | 23.65 (22.15–24.84) | 24.09 (21.01–25.91) | 0.382 |
| IgM-RF (+), n (%) | 0 (0) | 28 (90.32) | – |
| Anti-CCP (+), n (%) | 0 (0) | 26 (83.87) | – |
| RBCs (106/µL) | 4.28 ± 0.23 | 4.22 ± 0.24 | 0.380 |
| Hb (g/dL) | 13.08 ± 0.97 | 12.89 ± 1.20 | 0.562 |
| Ht (%) | 38.68 ± 3.10 | 38.31 ± 3.47 | 0.703 |
| Platelet (103/µL) | 263.88 ± 49.43 | 258.20 ± 69.70 | 0.748 |
| WBCs (103/μL) | 8.16 ± 1.63 | 7.79 ± 1.79 | 0.461 |
| ESR (mm/h) | 9.0 (8.0–12.0) | 17.0 (10.0–34.0) | <0.001 |
| CRP (mg/L) | 0.61 (0.40–2.81) | 6.3 (3.08–14.0) | <0.001 |
| Calcium C (mmol/L) | 2.28 ± 0.09 | 2.30 ± 0.11 | 0.324 |
| Phosphorus (mmol/L) | 1.34 ± 0.25 | 1.36 ± 0.20 | 0.698 |
| ALP (U/L) | 161.0 (142.0–172.0) | 168.5 (152.5–202.0) | 0.066 |
| ALT (U/L) | 20.50 (13.0–26.0) | 21.50 (13.50–32.0) | 0.405 |
| AST (U/L) | 19.0 (17.0–24.0) | 20.50 (18.0–26.50) | 0.209 |
| Creatinine (mg/dL) | 0.86 (0.81–0.92) | 0.70 (0.58–0.80) | <0.001 |
| SJC28, n | – | 7 (5–10) | – |
| TJC28, n | – | 12 (9–16) | – |
| VAS, (0–100 mm) | – | 80 (80–80) | – |
| DAS28-ESR | – | 5.78 (5.51–6.24) | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szeremeta, A.; Jura-Półtorak, A.; Zoń-Giebel, A.; Olczyk, K.; Komosińska-Vassev, K. Plasma Sclerostin Levels in Rheumatoid Arthritis Women on TNF-α Inhibitor Therapy. Pharmaceuticals 2024, 17, 666. https://doi.org/10.3390/ph17060666
Szeremeta A, Jura-Półtorak A, Zoń-Giebel A, Olczyk K, Komosińska-Vassev K. Plasma Sclerostin Levels in Rheumatoid Arthritis Women on TNF-α Inhibitor Therapy. Pharmaceuticals. 2024; 17(6):666. https://doi.org/10.3390/ph17060666
Chicago/Turabian StyleSzeremeta, Anna, Agnieszka Jura-Półtorak, Aleksandra Zoń-Giebel, Krystyna Olczyk, and Katarzyna Komosińska-Vassev. 2024. "Plasma Sclerostin Levels in Rheumatoid Arthritis Women on TNF-α Inhibitor Therapy" Pharmaceuticals 17, no. 6: 666. https://doi.org/10.3390/ph17060666
APA StyleSzeremeta, A., Jura-Półtorak, A., Zoń-Giebel, A., Olczyk, K., & Komosińska-Vassev, K. (2024). Plasma Sclerostin Levels in Rheumatoid Arthritis Women on TNF-α Inhibitor Therapy. Pharmaceuticals, 17(6), 666. https://doi.org/10.3390/ph17060666