
Solubility of Poorly Soluble Drugs in Phosphatidylcholine-Based Drug Delivery Systems: Comparison of the Loading Capacity in the Bulk Formulation and Its Dispersed State



Abstract
1. Introduction
2. Results and Discussion
2.1. Direct Loading of the Bulk Formulations
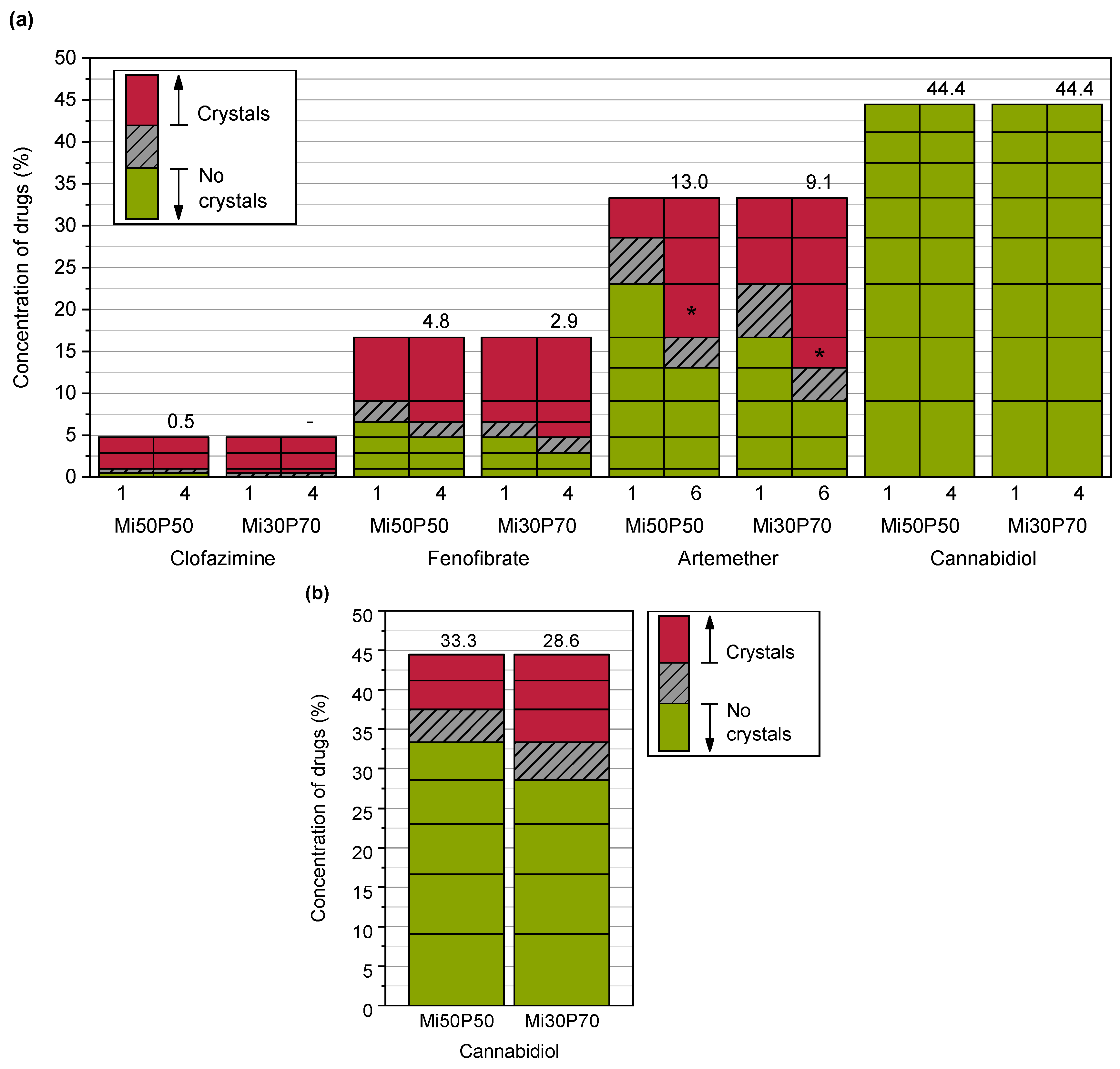
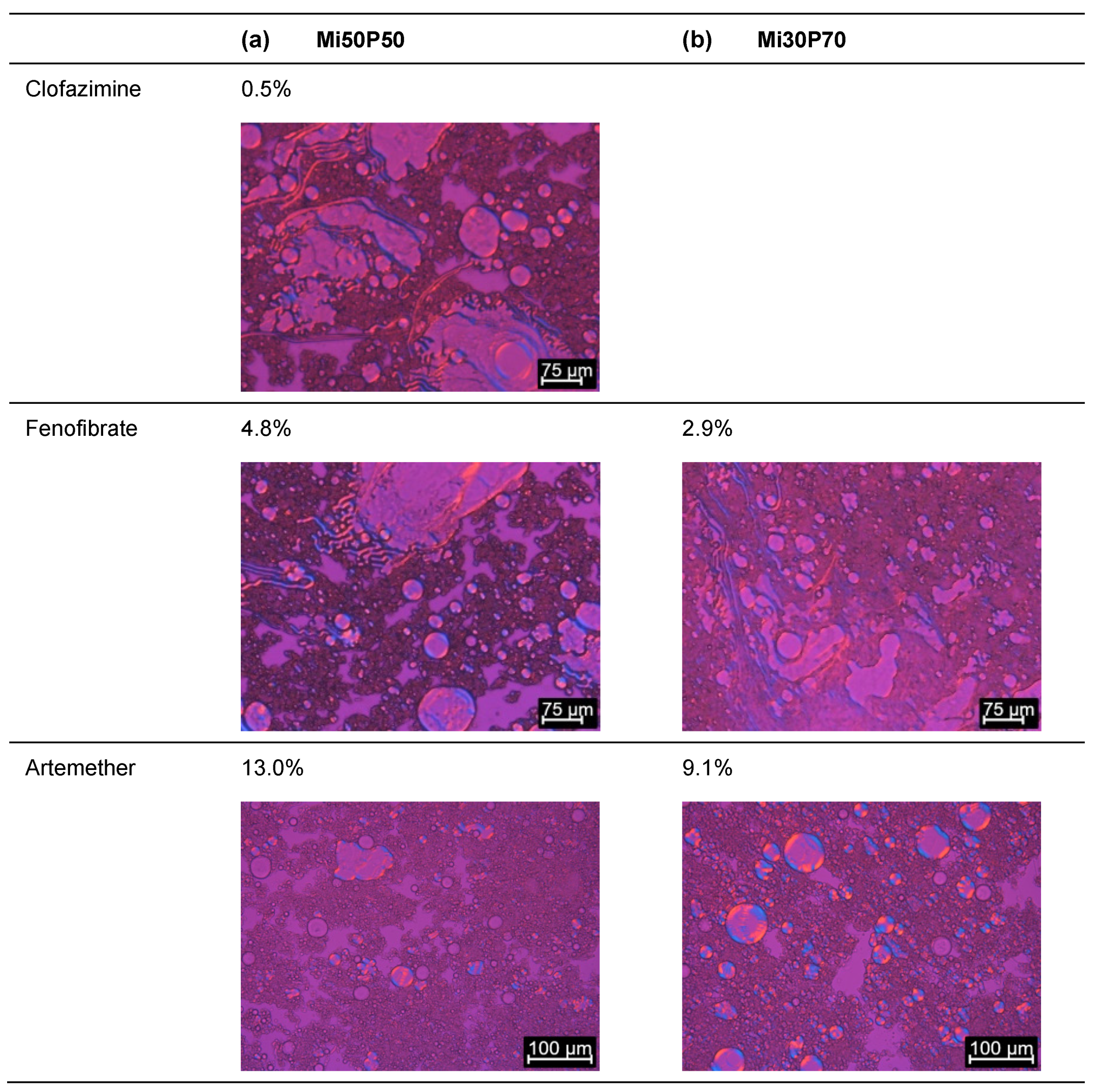
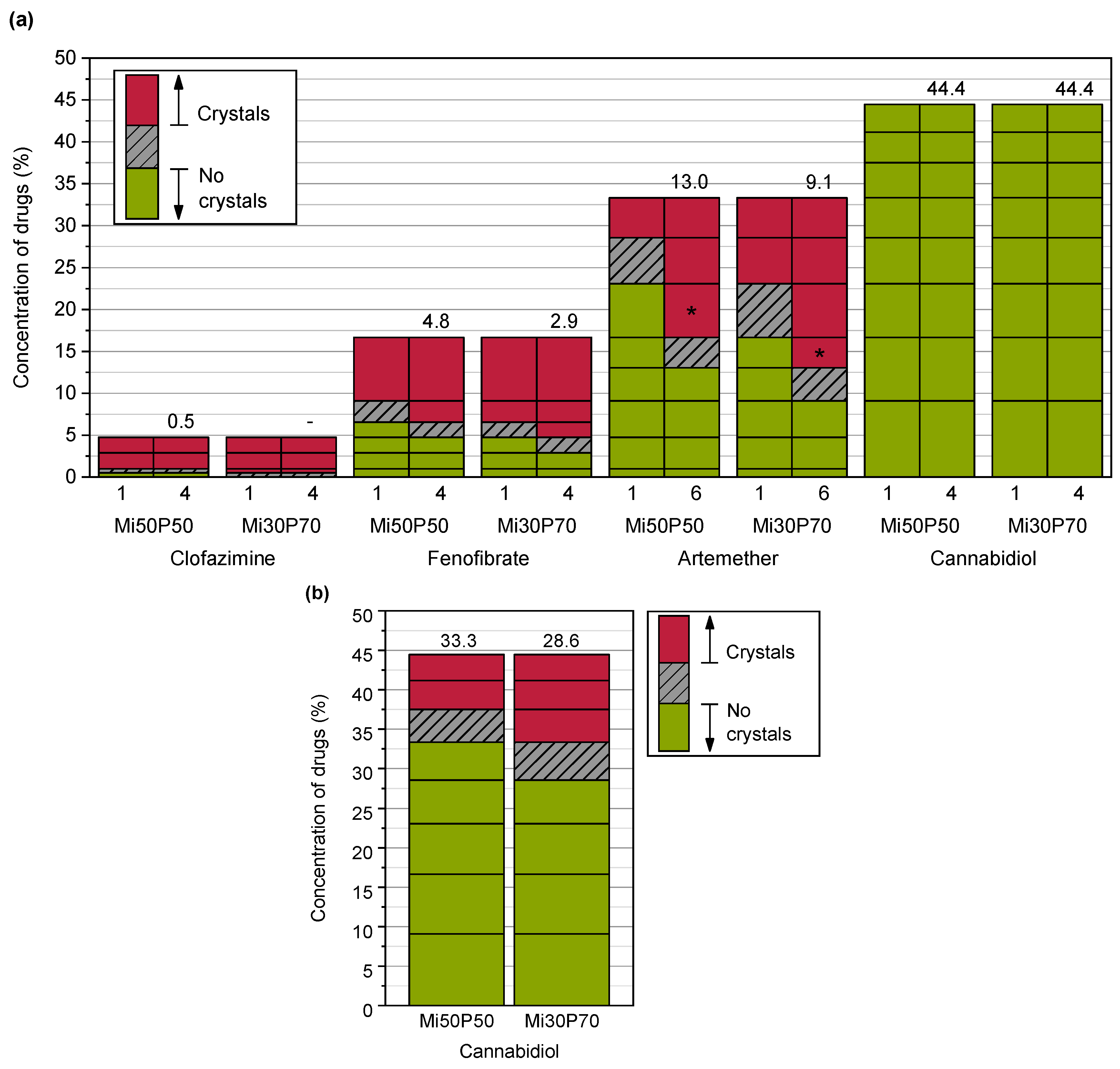
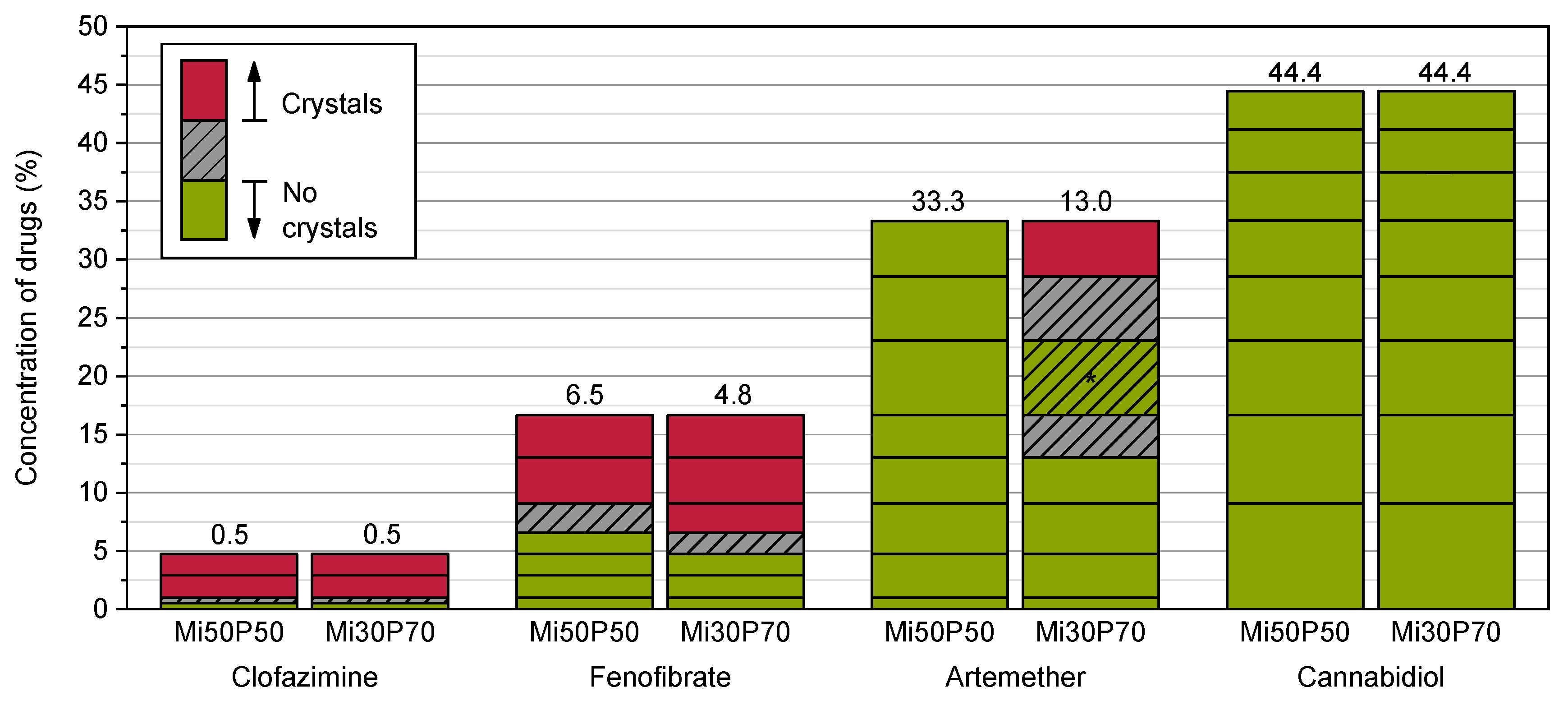
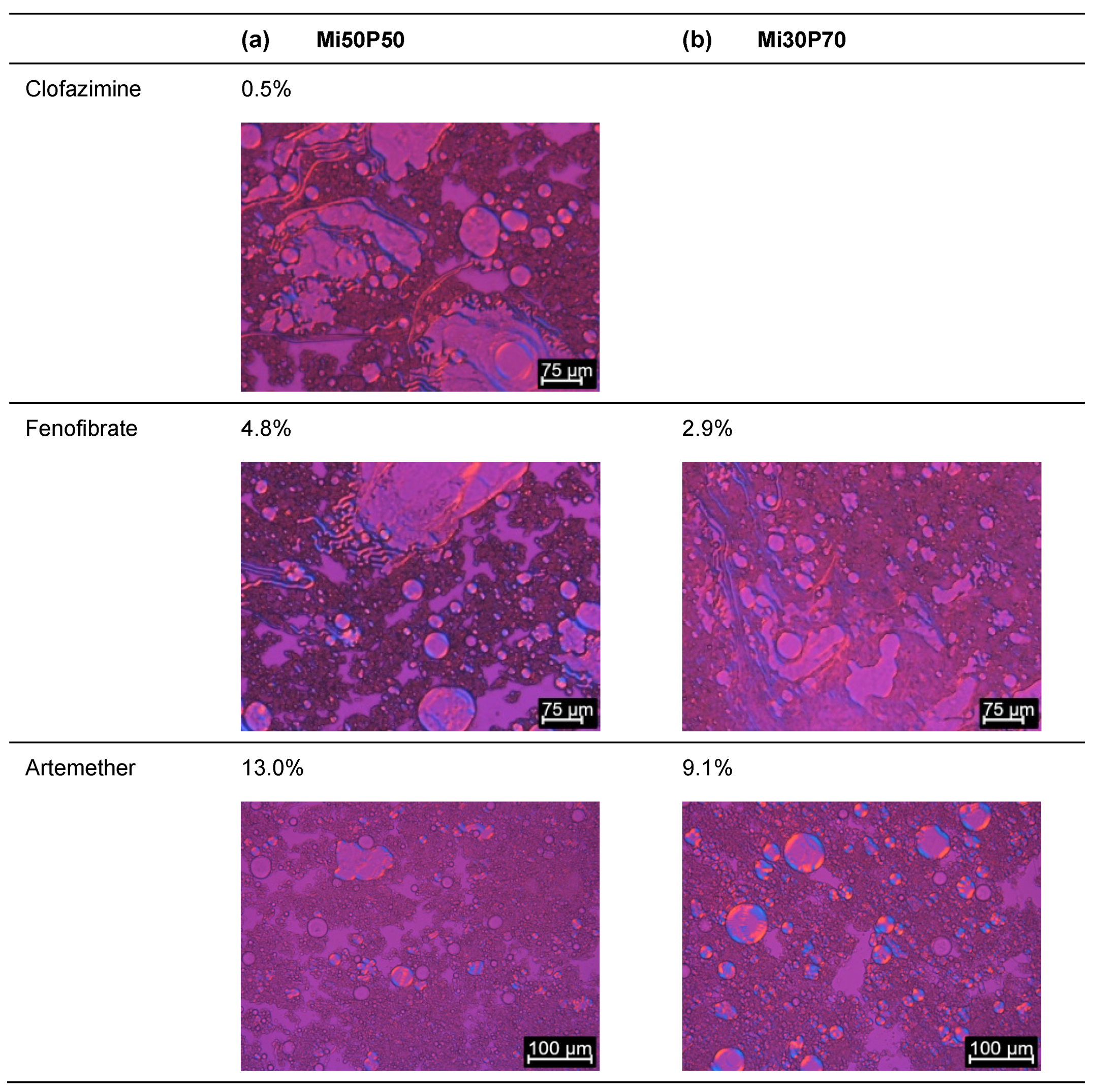
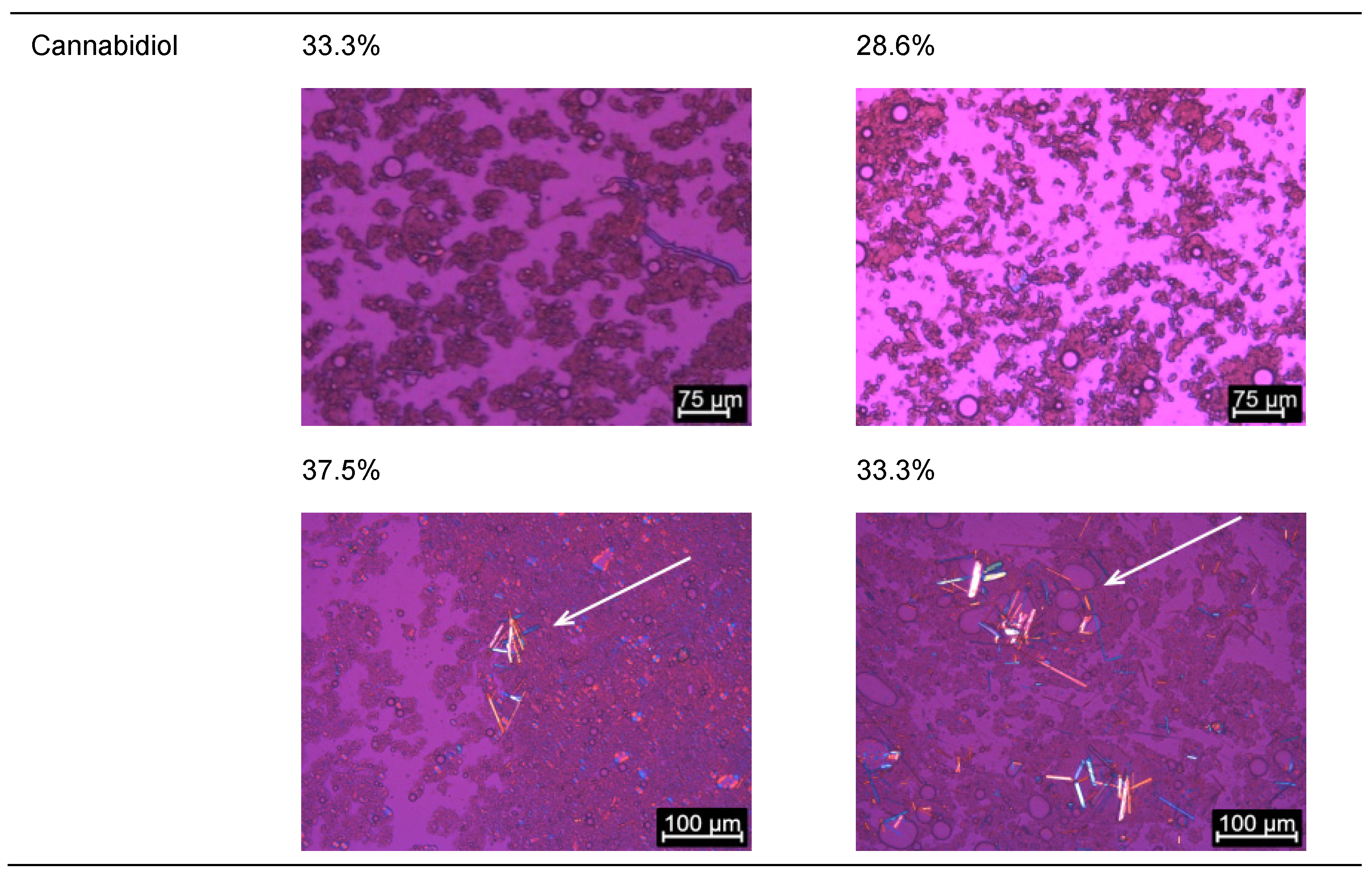
2.1.1. Determination of Solubility through the Detection of Drug Crystals in the Bulk Formulations
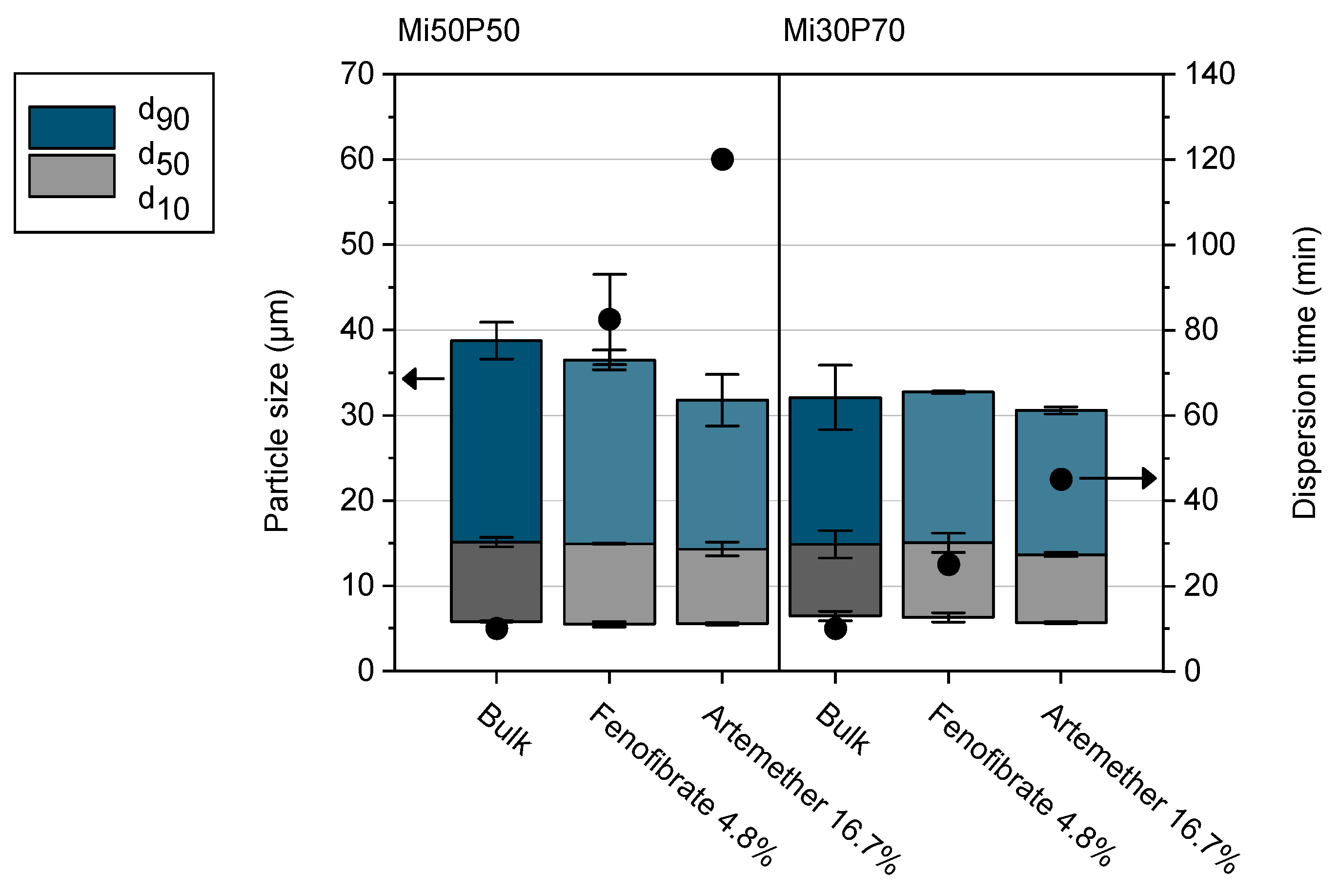
2.1.2. Dispersion of Loaded Bulk Formulations
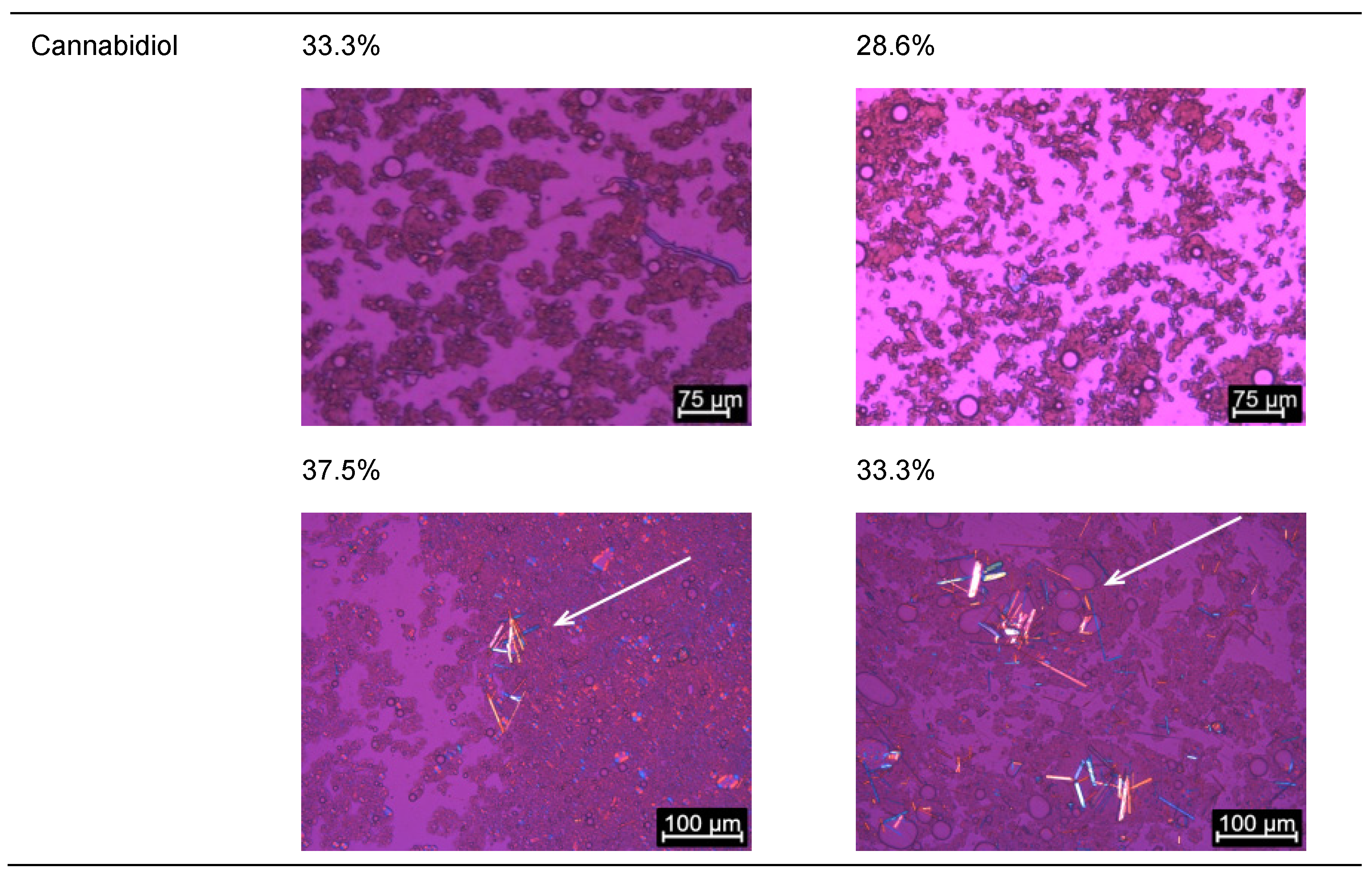
2.1.3. Evaluation of Loading Capacities
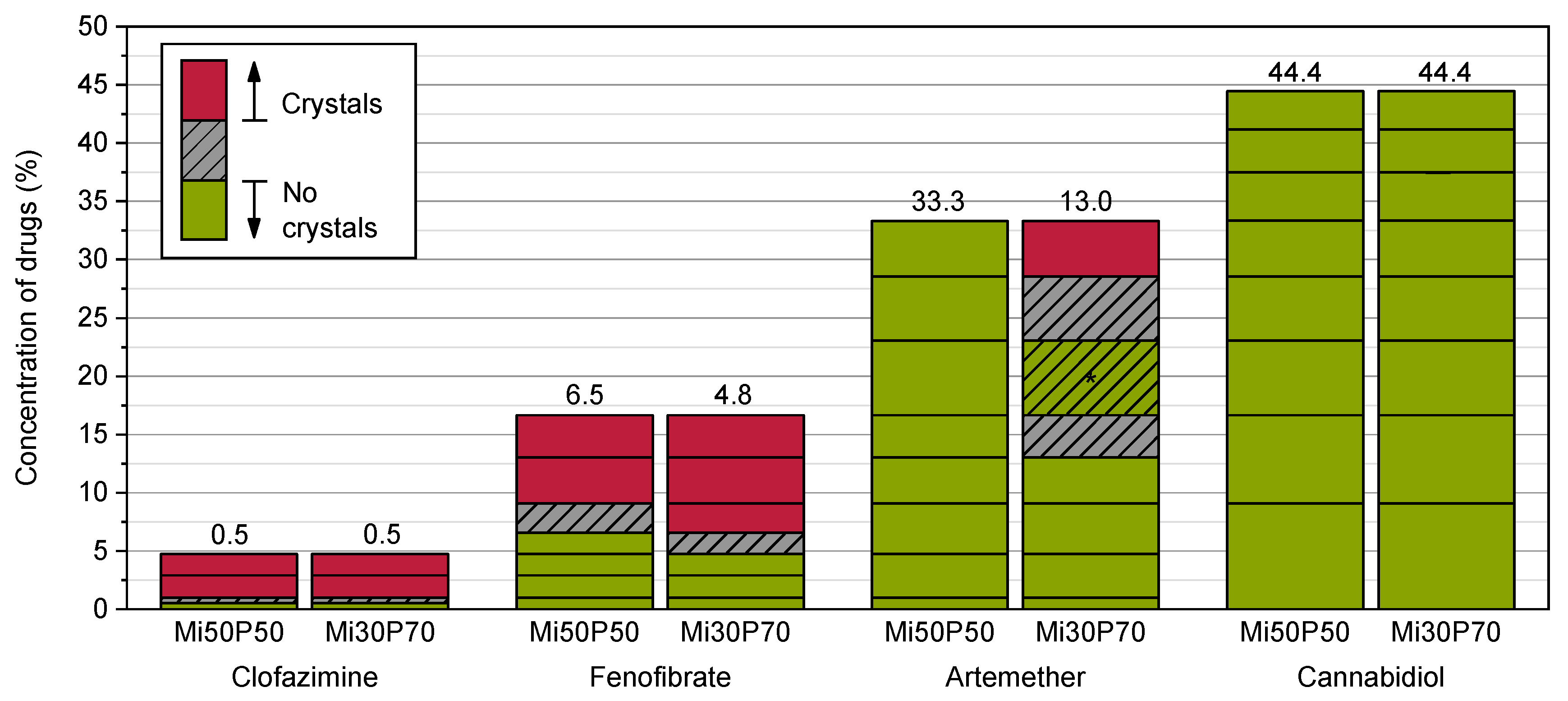
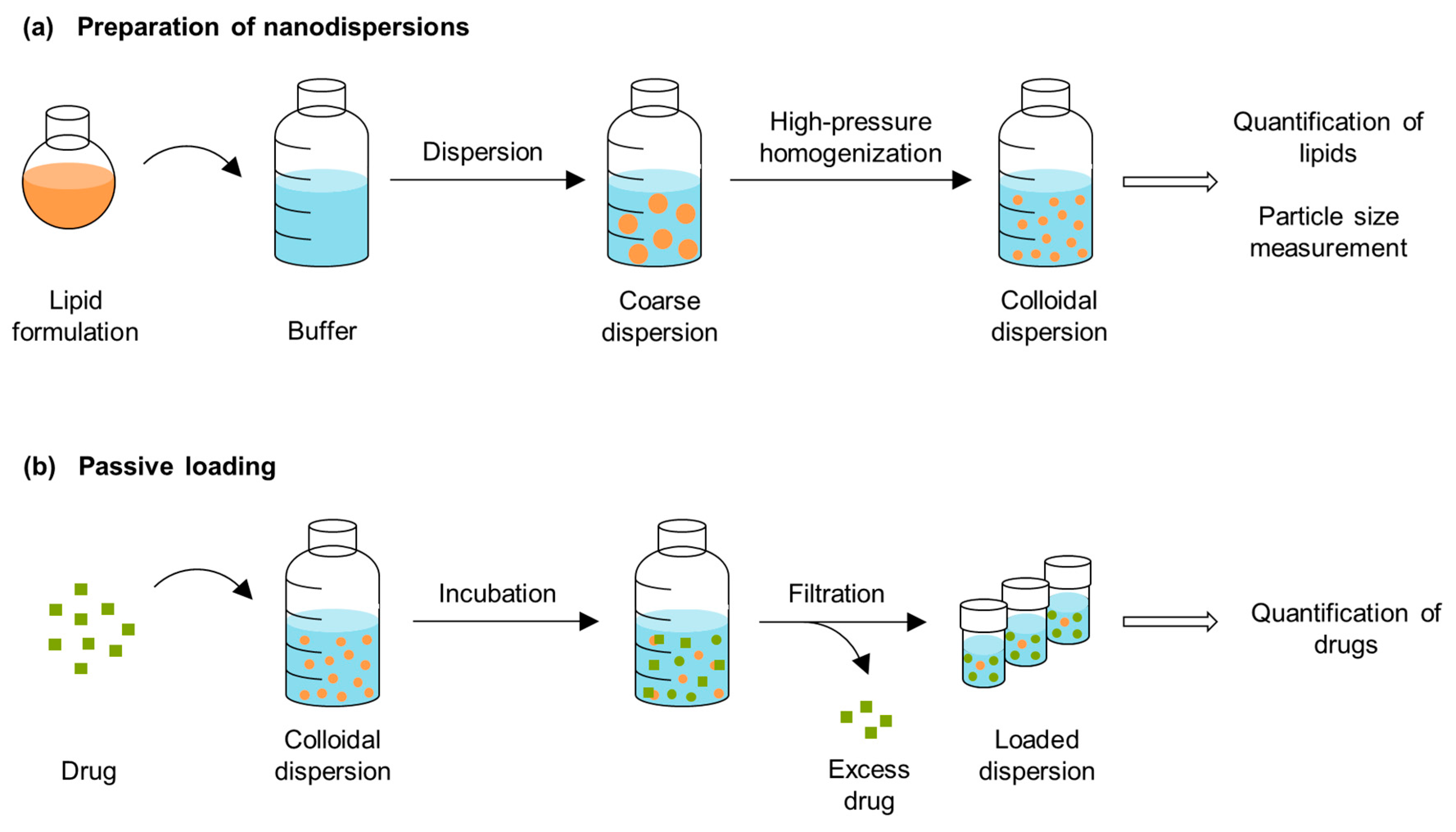
2.2. Passive Loading of Dispersed Formulations
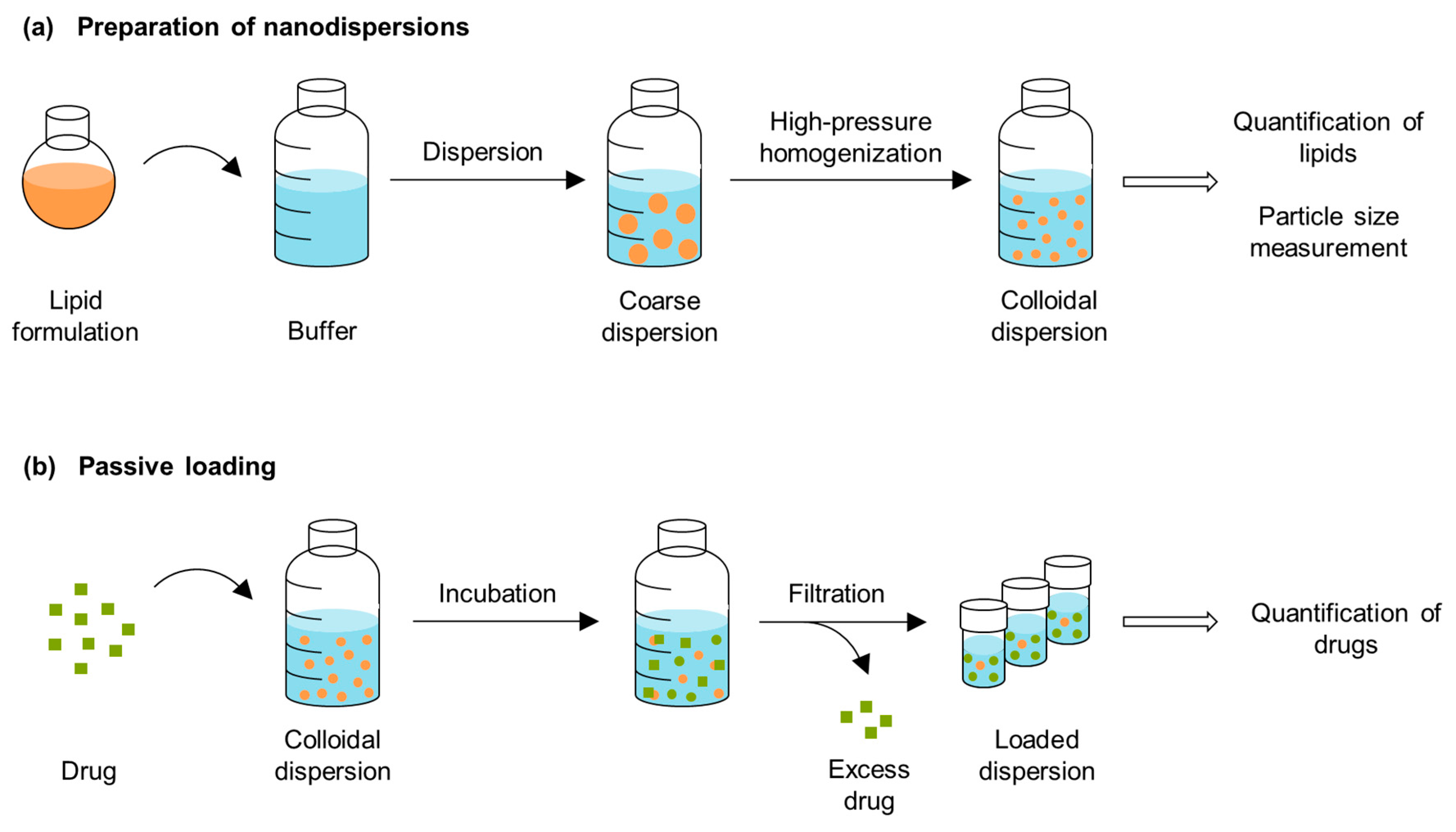
2.2.1. Preparation of Nanodispersions
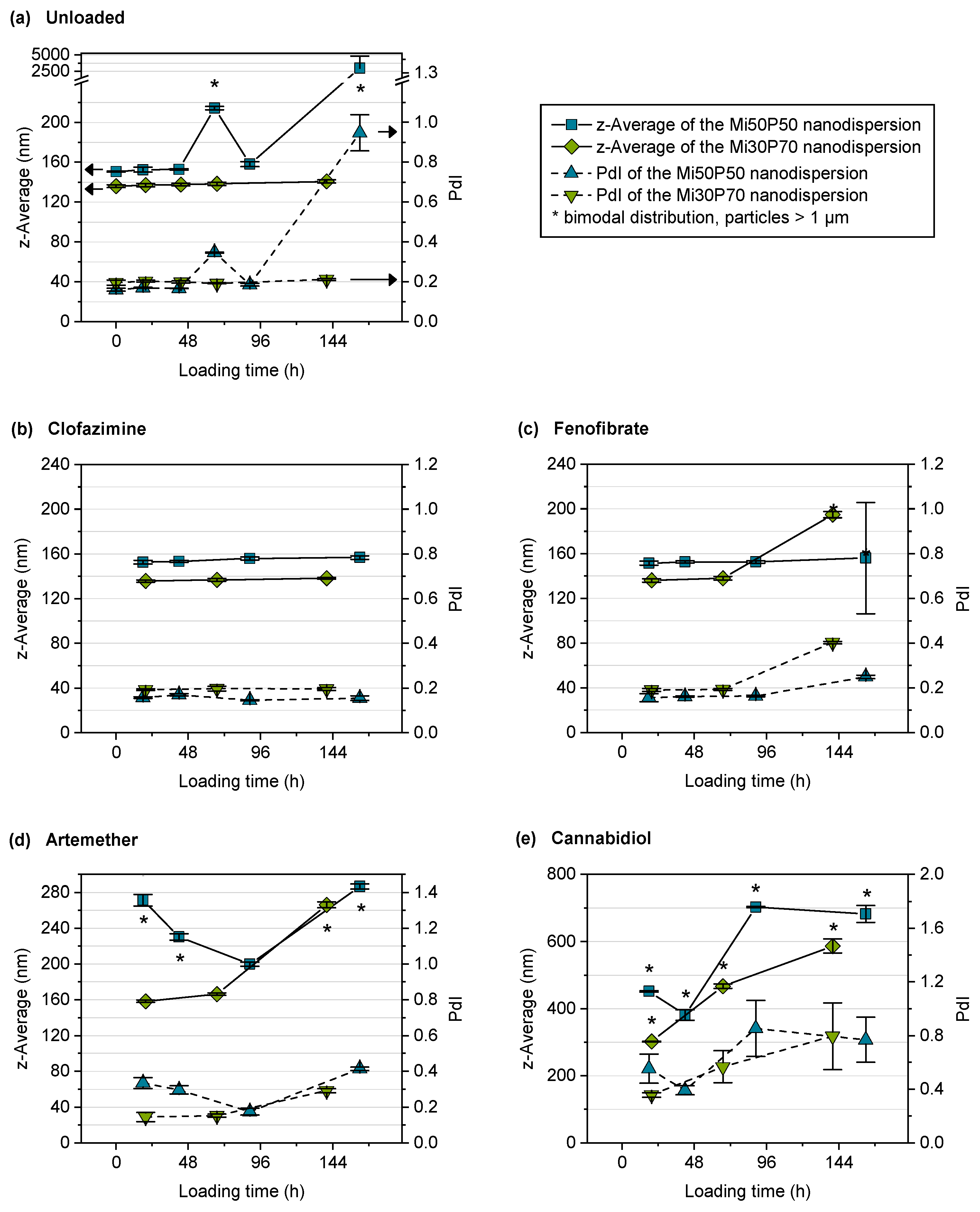
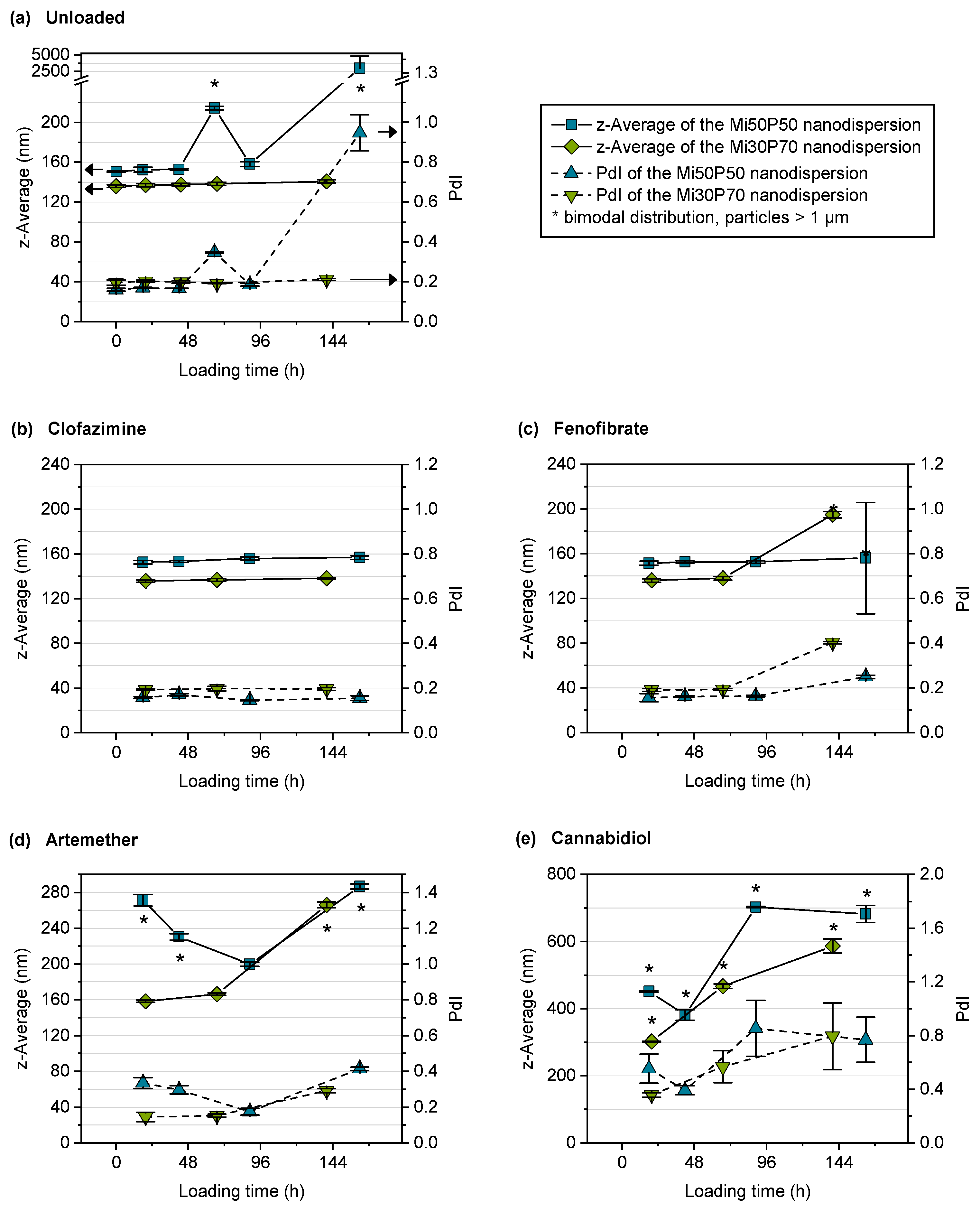
2.2.2. Stability of Unloaded Nanodispersions
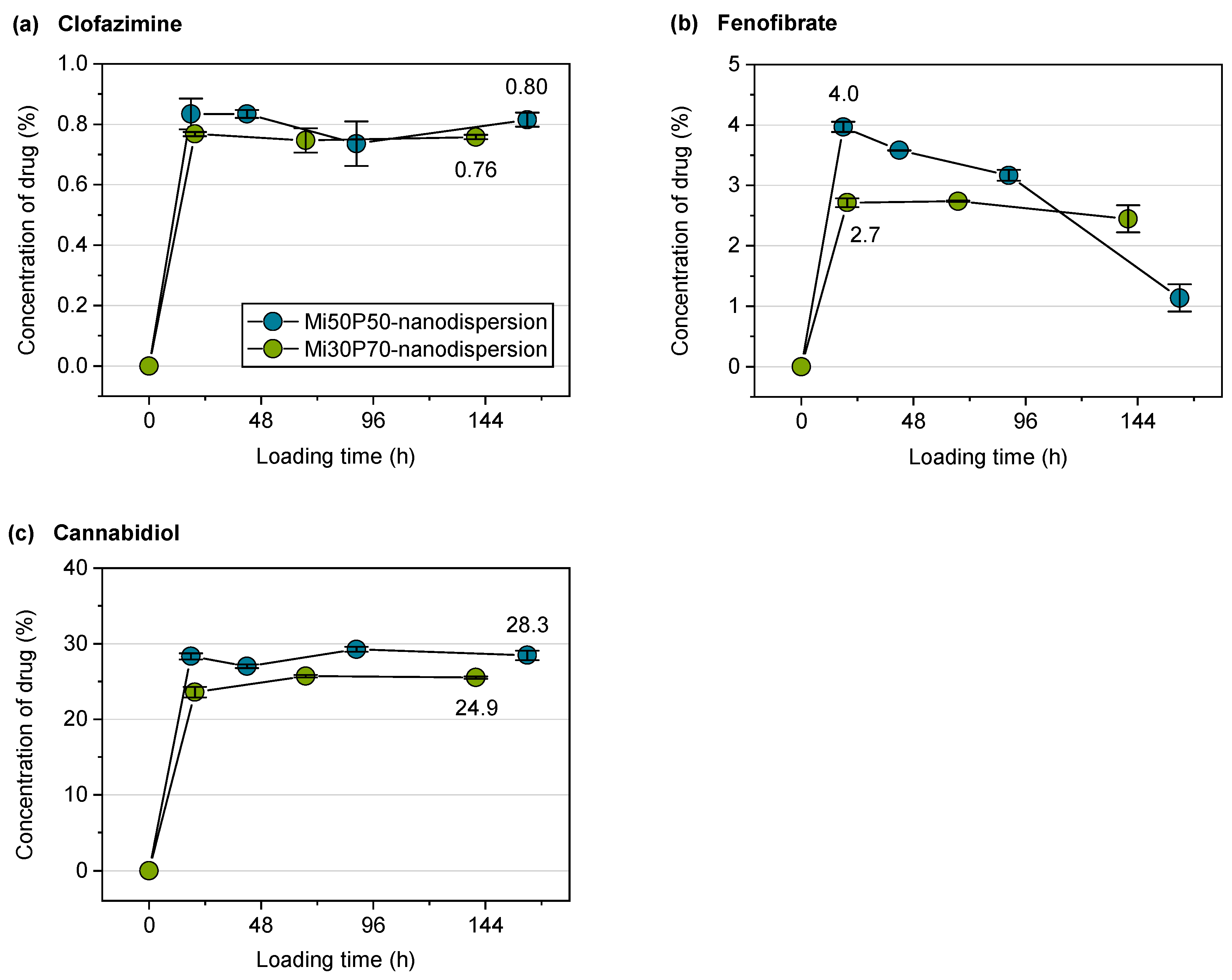
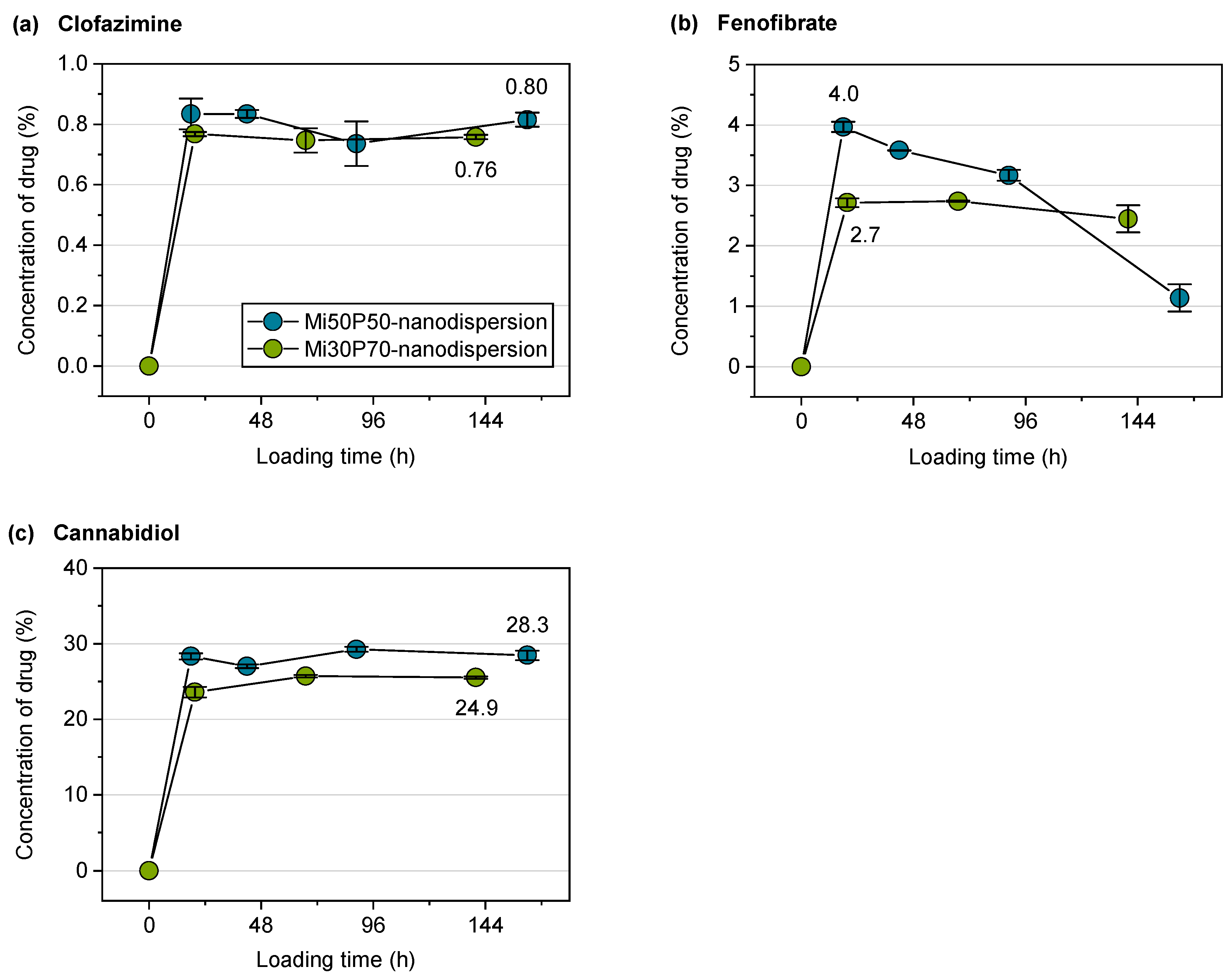
2.2.3. Loading of Nanodispersions with Fenofibrate, Artemether and Cannabidiol
2.2.4. Loading of Nanodispersions with Clofazimine
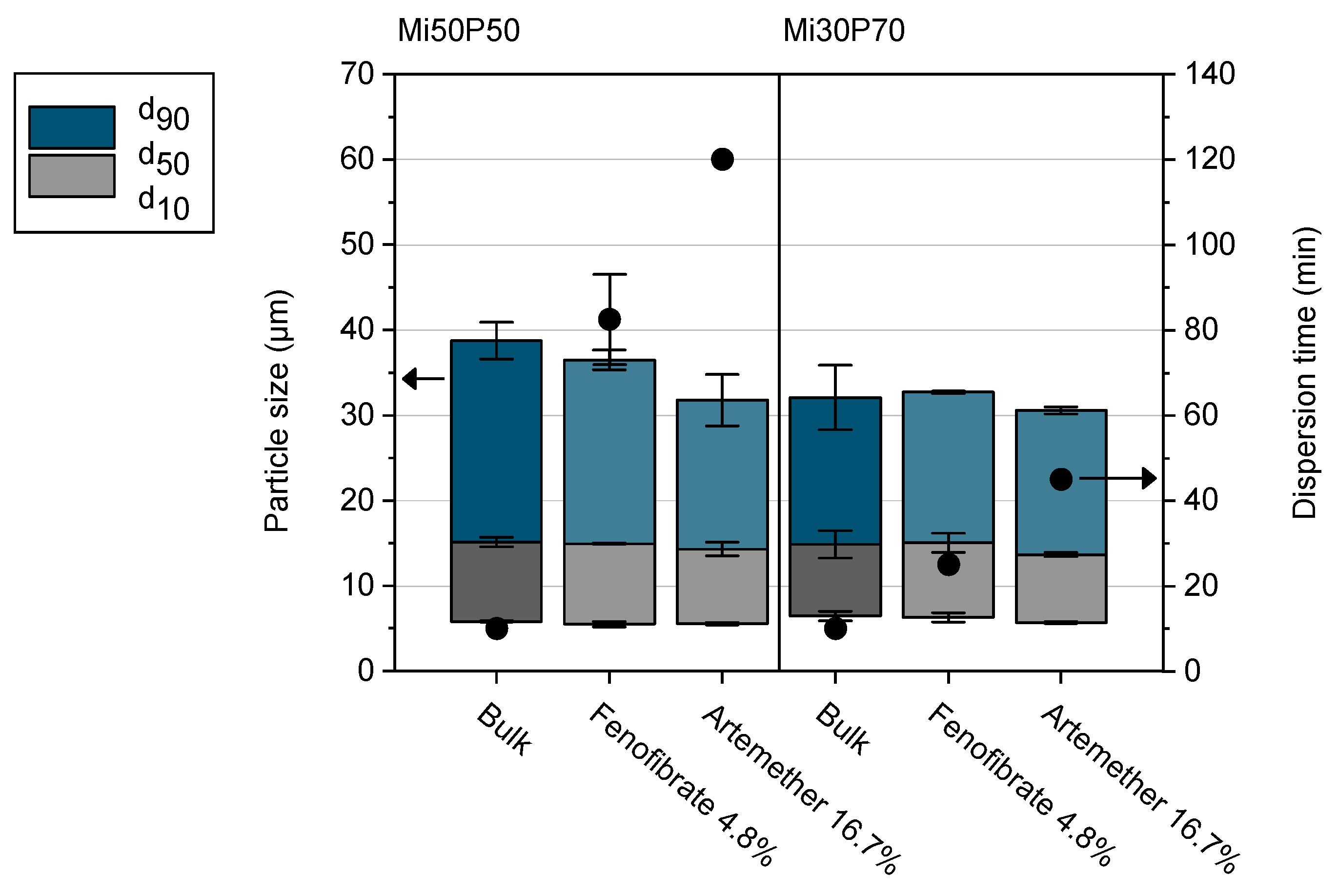
2.3. Testing of Self-Dispersing Properties
3. Materials and Methods
3.1. Materials
3.2. Preparation of Lipid Bulk Formulations
3.3. Direct Loading of Lipid Bulk Formulations with Drugs
3.3.1. Preparation of Loaded Formulations
3.3.2. Determination of the Solubility through the Detection of Drug Crystals
3.4. Passive Loading of the Dispersed Formulations
3.4.1. Preparation of the Nanodispersions
3.4.2. Particle Size Measurements
3.4.3. Passive Loading
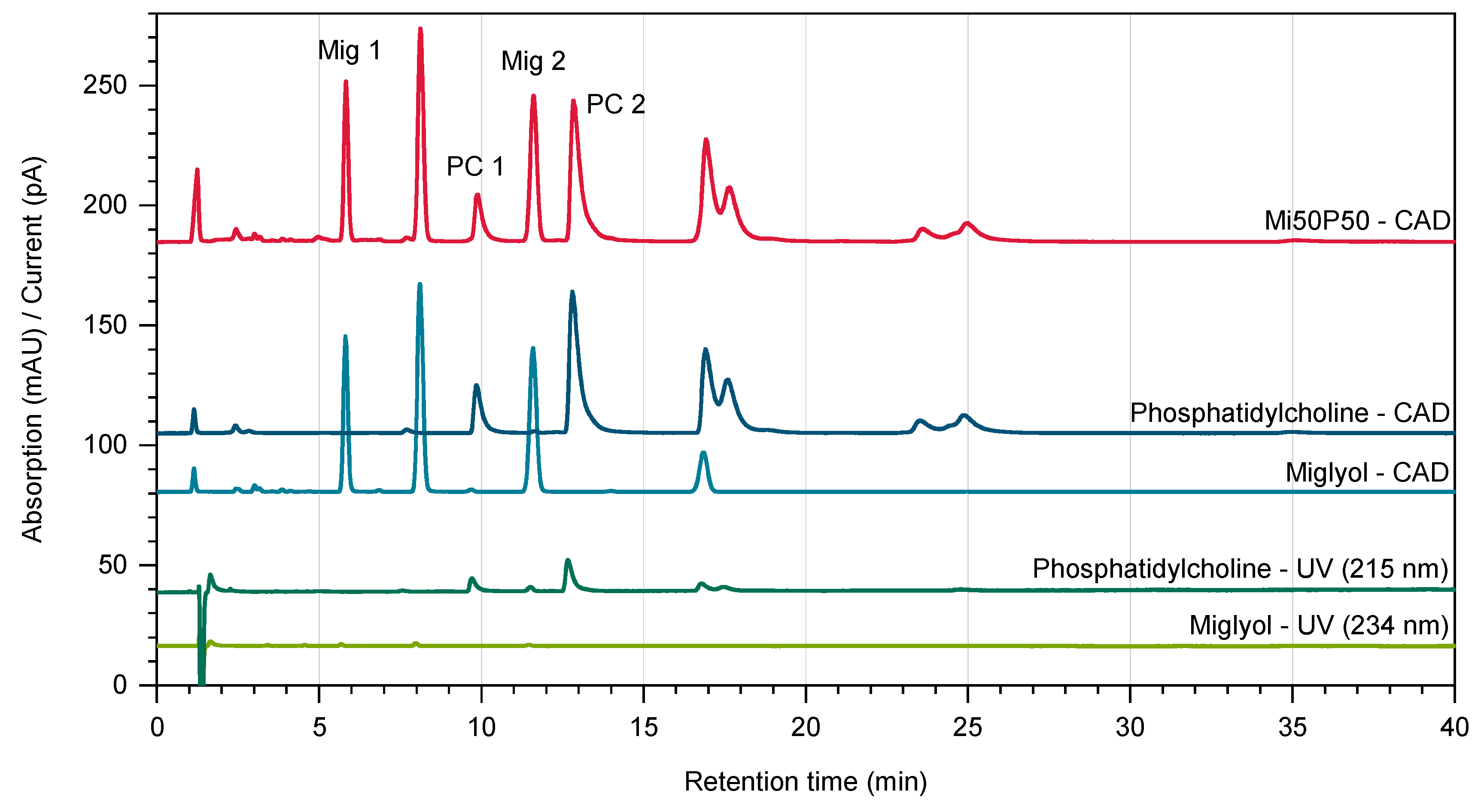
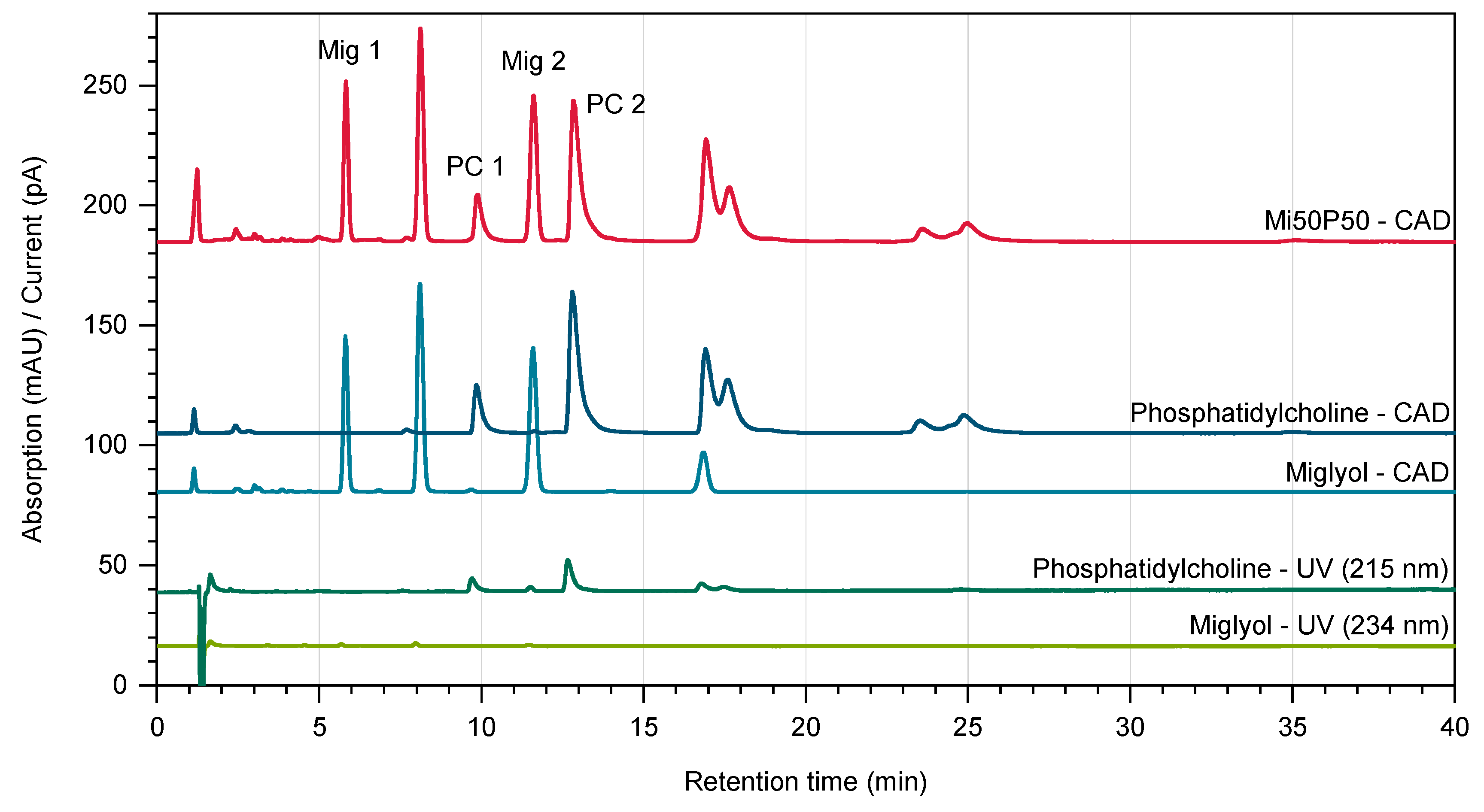
3.4.4. Quantification of Lipids
3.4.5. Quantification of the Drugs
3.5. Testing of Self-Dispersing Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.J.; Bergström, C.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, C.M.; Griffin, B.T. Biopharmaceutical challenges associated with drugs with low aqueous solubility-The potential impact of lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Ujhelyi, Z.; Fenyvesi, F.; Váradi, J.; Fehér, P.; Kiss, T.; Veszelka, S.; Deli, M.; Vecsernyés, M.; Bácskay, I. Evaluation of cytotoxicity of surfactants used in self-micro emulsifying drug delivery systems and their effects on paracellular transport in Caco-2 cell monolayer. Eur. J. Pharm. Sci. 2012, 47, 564–573. [Google Scholar] [CrossRef]
- Gloxhuber, C. Toxicological properties of surfactants. Arch. Toxicol. 1974, 32, 245–269. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. 21CFR184.1400 Revised as of April 1, 2019. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=184.1400 (accessed on 22 October 2020).
- Grüne, L.; Bunjes, H. Self-dispersing formulations for the delivery of poorly soluble drugs–Miscibility of phosphatidylcholines with oils and fats. Eur. J. Pharm. Biopharm. 2020, 151, 209–219. [Google Scholar] [CrossRef]
- Grüne, L.; Bunjes, H. Suitability of phosphatidylcholine-based formulations for liquid filling in hard capsules. Eur. J. Pharm. Sci. 2020, 153, 105470. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, M.T. Lipid-based formulations for oral delivery of lipophilic drugs. Drug Discov. Today Technol. 2012, 9, e97–e104. [Google Scholar] [CrossRef]
- Dai, W.-G.; Pollock-Dove, C.; Dong, L.C.; Li, S. Advanced screening assays to rapidly identify solubility-enhancing formulations: High-throughput, miniaturization and automation. Adv. Drug Deliv. Rev. 2008, 60, 657–672. [Google Scholar] [CrossRef]
- Khoo, S.-M.; Humberstone, A.J.; Porter, C.; Edwards, G.A.; Charman, W.N. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int. J. Pharm. 1998, 167, 155–164. [Google Scholar] [CrossRef]
- Nekkanti, V.; Rueda, J.; Wang, Z.; Betageri, G.V. Comparative evaluation of proliposomes and self micro-emulsifying drug delivery system for improved oral bioavailability of nisoldipine. Int. J. Pharm. 2016, 505, 79–88. [Google Scholar] [CrossRef]
- Tran, T.; Siqueira, S.; Amenitsch, H.; Müllertz, A.; Rades, T. In vitro and in vivo performance of monoacyl phospholipid-based self-emulsifying drug delivery systems. J. Control Release 2017, 255, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, M. Drug supersaturation during formulation digestion, including real-time analytical approaches. Adv. Drug Deliv. Rev. 2019, 142, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.; Porter, C. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Massing, U.; Ingebrigtsen, S.G.; Škalko-Basnet, N.; Holsæter, A.M. Dual centrifugation-A novel “in-vial” liposome processing technique. In Liposomes; Catala, A., Ed.; InTechOpen: Rijeka, Croatia, 2017; pp. 3–28. ISBN 978-953-51-3579-1. [Google Scholar]
- Tian, W.; Schulze, S.R.; Brandl, M.; Winter, G. Vesicular phospholipid gel-based depot formulations for pharmaceutical proteins: Development and in vitro evaluation. J. Control Release 2010, 142, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Andreas Hettich GmbH & Co. KG. Herstellung Von Sterilen Liposomen Mit Hoher Einschlusseffizienz. Available online: https://www.hettichlab.com/downloadcenter/Products/Catalogs_Brochures/HETTICH/Liposomen_Methode.pdf (accessed on 17 June 2020).
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Göke, K.; Bunjes, H. Drug solubility in lipid nanocarriers: Influence of lipid matrix and available interfacial area. Int. J. Pharm. 2017, 529, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, K.M.; Bunjes, H. Evaluation of the drug loading capacity of different lipid nanoparticle dispersions by passive drug loading. Eur. J. Pharm. Biopharm. 2017, 117, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kupetz, E.; Preu, L.; Kunick, C.; Bunjes, H. Parenteral formulation of an antileishmanial drug candidate–Tackling poor solubility, chemical instability, and polymorphism. Eur. J. Pharm. Biopharm. 2013, 85, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Andermark, V.; Göke, K.; Kokoschka, M.; Abu el Maaty, M.A.; Lum, C.T.; Zou, T.; Sun, R.W.-Y.; Aguiló, E.; Oehninger, L.; Rodríguez, L.; et al. Alkynyl gold(I) phosphane complexes: Evaluation of structure-activity-relationships for the phosphane ligands, effects on key signaling proteins and preliminary in-vivo studies with a nanoformulated complex. J. Inorg. Biochem. 2016, 160, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Göke, K.; Bunjes, H. Parameters influencing the course of passive drug loading into lipid nanoemulsions. Eur. J. Pharm. Biopharm. 2018, 126, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Hanus, L. Cannabidiol: An overview of some chemical and pharmacological aspects. Part I: Chemical aspects. Chem. Phys. Lipids 2002, 121, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.C.; Porter, C.J.H.; Charman, W.N.; Bergström, C.A.S. Computational prediction of drug solubility in lipid based formulation excipients. Pharm. Res. 2013, 30, 3225–3237. [Google Scholar] [CrossRef] [PubMed]
- Alskär, L.C.; Porter, C.J.H.; Bergström, C.A.S. Tools for early prediction of drug loading in lipid-based formulations. Mol. Pharmaceut. 2016, 13, 251–261. [Google Scholar] [CrossRef]
- Fachinfo-Service®: Fachinformationsverzeichnis Deutschland; Rote Liste® Service GmbH: Frankfurt/Main, Germany, 2020.
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Baker, M.T.; Naguib, M. Propofol: The challenges of formulation. Anesthesiology 2005, 103, 860–876. [Google Scholar] [CrossRef]
- Westesen, K.; Wehler, T. Physicochemical characterization of a model intravenous oil-in-water emulsion. J. Pharm. Sci. 1992, 81, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M.P.; Rolland, J.P.; Riess, J.G. Detrimental effect of excess lecithin on the stability of fluorocarbon/lecithin emulsions. J. Phys. Chem. 1991, 95, 5673–5676. [Google Scholar] [CrossRef]
- Francke, N.M.; Bunjes, H. Influence of drug loading on the physical stability of phospholipid-stabilised colloidal lipid emulsions. Int. J. Pharm. X 2020, 2, 100060. [Google Scholar] [CrossRef] [PubMed]
- Sznitowska, M.; Dabrowska, E.A.; Janicki, S. Solubilizing potential of submicron emulsions and aqueous dispersions of lecithin. Int. J. Pharm. 2002, 246, 203–206. [Google Scholar] [CrossRef]
- Simulated Gastric Fluid. In European Pharmacopoeia, 10th ed.; European Directorate for the Quality of Medicines of the Council of Europe, Ed.; Council of Europe: Strassbourg, France, 2019; Main Volume. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mi50P50 Nanodispersion | Mi30P70 Nanodispersion | |
|---|---|---|
| Fraction of Miglyol in the formulation (%) | 49.6 ± 1.4 | 31.3 ± 0.4 |
| Fraction of phosphatidylcholine in the formulation (%) | 50.4 ± 0.9 | 68.7 ± 0.8 |
| Concentration of lipids in the dispersion (%) | 4.9 ± 0.1 | 4.6 ± 0.1 |
| z-Average diameter of the dispersion (nm) | 150.6 ± 0.4 | 136.1 ± 1.3 |
| Polydispersity index of the dispersion | 0.160 ± 0.007 | 0.196 ± 0.013 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grüne, L.; Bunjes, H. Solubility of Poorly Soluble Drugs in Phosphatidylcholine-Based Drug Delivery Systems: Comparison of the Loading Capacity in the Bulk Formulation and Its Dispersed State. Pharmaceuticals 2024, 17, 400. https://doi.org/10.3390/ph17030400
Grüne L, Bunjes H. Solubility of Poorly Soluble Drugs in Phosphatidylcholine-Based Drug Delivery Systems: Comparison of the Loading Capacity in the Bulk Formulation and Its Dispersed State. Pharmaceuticals. 2024; 17(3):400. https://doi.org/10.3390/ph17030400
Chicago/Turabian StyleGrüne, Linda, and Heike Bunjes. 2024. "Solubility of Poorly Soluble Drugs in Phosphatidylcholine-Based Drug Delivery Systems: Comparison of the Loading Capacity in the Bulk Formulation and Its Dispersed State" Pharmaceuticals 17, no. 3: 400. https://doi.org/10.3390/ph17030400
APA StyleGrüne, L., & Bunjes, H. (2024). Solubility of Poorly Soluble Drugs in Phosphatidylcholine-Based Drug Delivery Systems: Comparison of the Loading Capacity in the Bulk Formulation and Its Dispersed State. Pharmaceuticals, 17(3), 400. https://doi.org/10.3390/ph17030400