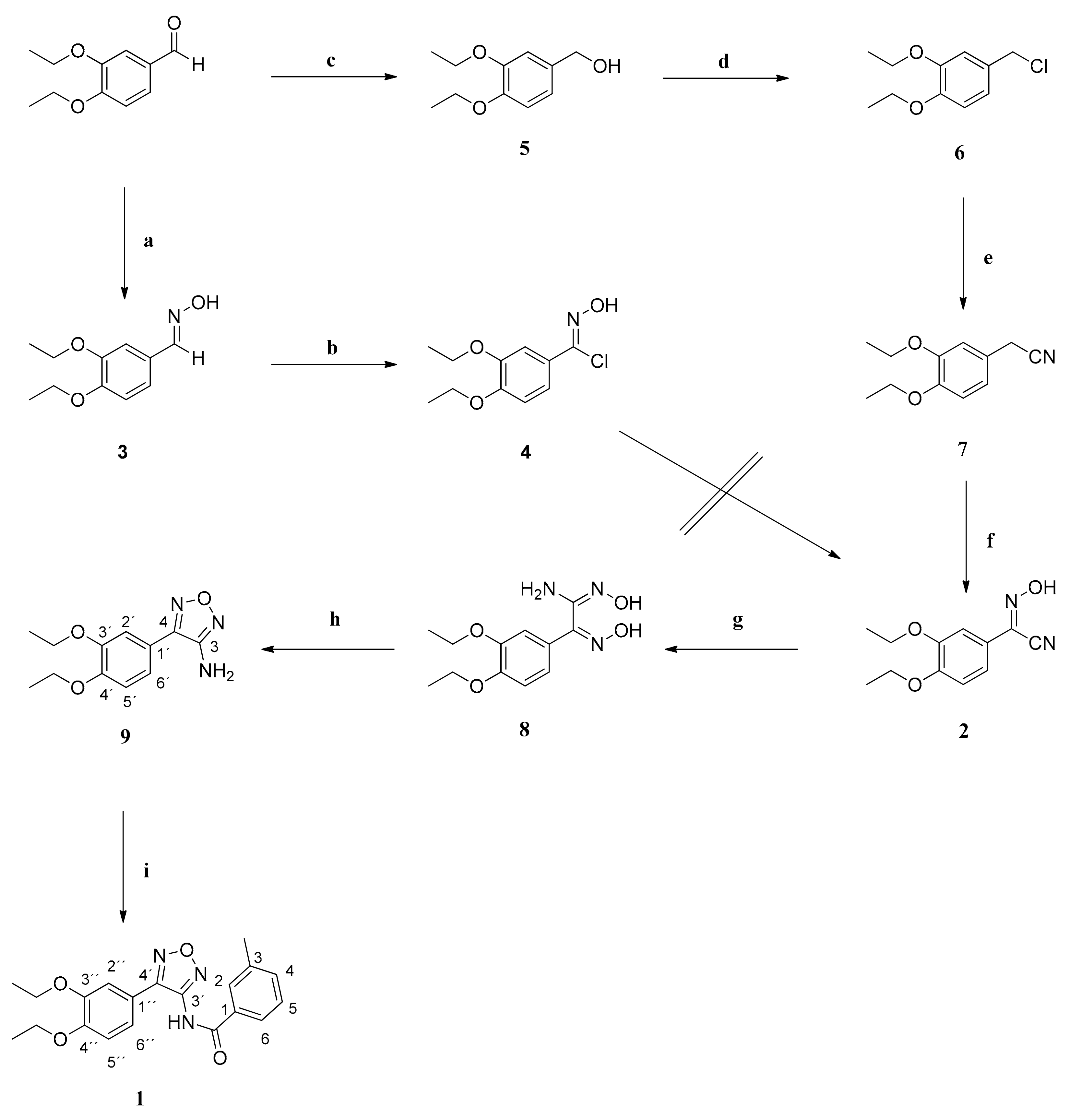
3.2. Syntheses
(3,4-Diethoxyphenyl)methanol (5): NaBH4 (0.57 g (15.00 mmol)) was added in portions to an ice-cooled solution of 3,4-diethoxybenzaldehyde (2.91 g (15.00 mmol)) in dry methanol (16 mL). After that, the ice bath was removed and the reaction mixture was stirred at 25 °C for 1 h. Then, the solvent was evaporated in vacuo and the residue was mixed with water and extracted with CH2Cl2. The combined organic phases were washed with water, dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo giving compound 5 as colorless oil (2.80 g (95%)), which was used without further purification.
NMR data were in accordance with literature data [
35].
4-(Chloromethyl)-1,2-diethoxybenzene (6): Thionyl chloride (4.14 g (34.80 mmol)) was added dropwise via a dropping funnel to an ice-cooled solution of benzyl alcohol 5 (2.36 g (12.00 mmol)) in dry CH2Cl2 (50 mL). The ice bath was removed and the reaction mixture was stirred at 25 °C for 20 h. Then, the reaction was quenched with 2N NaOH at 0 °C and the mixture was basified to a pH of 10–11. The aqueous and organic phases were separated and the aqueous phase was extracted with CH2Cl2. The combined organic phases were dried over anhydrous sodium sulfate and filtered and the solvent was removed in vacuo, giving compound 6 as brown oil (2.47 g (96%)), which was used without further purification. IR = 2977, 1604, 1510, 1476, 1428, 1392, 1256, 1232, 1135, 1038, 985, 805; 1H NMR (CDCl3, 400 MHz) δ = 1.42–1.47 (m, 6H, 2 CH3), 4.06–4.13 (m, 4H, 2 CH2), 4.55 (s, 2H,CH2Cl), 6.82 (d, J = 8.1 Hz, 1H, 5-H), 6.90 (dd, J = 8.1, 2.0 Hz, 1H, 6-H), 6.91 (d, J = 2.0 Hz, 1H, 2-H); 13C NMR (CDCl3, 100 MHz) δ = 14.76 (2 CH3), 46.71 (CH2Cl), 64.56 (2 CH2), 113.08 (C-5), 113.86 (C-2), 121.21 (C-6), 129.96 (C-1), 148.81 (C-3), 148.93 (C-4); HRMS (EI+) calcd for C11H15ClO2: 214.0761; found: 214.0755.
2-(3,4-Diethoxyphenyl)acetonitrile (7): Benzyl chloride 6 (2.15 g (10.00 mmol)) was dissolved in dry DMF (20 mL). KCN (1.30 g (20.00 mmol)) was added and the suspension was stirred at 100 °C for 4 h. After that, the solvent was evaporated in vacuo and the residue was mixed with water and extracted with ethyl acetate. The organic phases were combined and washed with water and brine, dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo, giving compound 7 as brown oil (1.95 g (95%)), which was used without further purification.
NMR data were in accordance with literature data [
36].
(3,4-Diethoxyphenyl)(hydroxyimino)acetonitrile (2): Sodium (0.37 g (12.00 mmol)) was dissolved in dry ethanol (15 mL) and then cooled to 0 °C with an ice bath. A solution of compound 7 (1.64 g (8.00 mmol)) in dry ethanol (10 mL) was added dropwise. Finally, isopentyl nitrite (1.41 g (12.00 mmol)) was added dropwise with a syringe through a septum. The ice bath was removed and the reaction mixture stirred at 25 °C for 20 h. The solution was diluted with ethyl acetate (80 mL) and washed with 2N HCl, 8% aq NaHCO3 and brine. The organic phase was dried over anhydrous sodium sulfate and filtered and the solvent was removed in vacuo, giving compound 2 as white amorphous solid (1.86 g (99%)), which was used without further purification. IR = 3364, 1601, 1512, 1438, 1337, 1273, 1210, 1146, 1076, 1006, 845, 806; 1H NMR (CDCl3, 400 MHz) δ = 1.42 (t, J = 6.9 Hz, 3H, CH3), 1.43 (t, J = 6.9 Hz, 3H, CH3), 4.08 (q, J = 6.9 Hz, 2H, OCH2), 4.10 (q, J = 6.9 Hz, 2H, OCH2), 7.00 (d, J = 8.5 Hz, 1H, 5-H), 7.26 (dd, J = 8.5, 1.9 Hz, 1H, 6-H), 7.33 (d, J = 1.9 Hz, 1H, 2-H); 13C NMR (CDCl3, 100 MHz) δ = 15.16 (CH3), 15.19 (CH3), 65.75 (OCH2), 65.89 (OCH2), 110.68 (C-2), 111.04 (CN), 114.03 (C-5), 121.22 (C-6), 124.25 (C-1), 132.87 (C=NOH), 150.49 (C-3), 152.73 (C-4); HRMS (EI+) calcd for C12H14N2O3: 234.1004; found: 234.0996.
2-(3,4-Diethoxyphenyl)-N’-hydroxy-2-(hydroxyimino)ethanimidamide (8): Oxime 2 (1.41 g (6.00 mmol)) was dissolved in methanol (18 mL) and a solution of NH2OH × HCl (0.50 g (7.20 mmol)) and NaHCO3 (0.61 g (7.20)) in water (9 mL) was added. The mixture was refluxed for 20 h and then the solvent was evaporated in vacuo. The residue was mixed with water (20 mL) and extracted with ethyl acetate. The organic phases were combined dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo, giving the raw amidoxime which was recrystallized in CH2Cl2 affording compound 8 as beige crystals (0.55 g (34%)). m.p. 148 °C. IR = 3370, 2981, 1661, 1598, 1516, 1444, 1393, 1270, 1201, 1144, 1040, 978, 810; 1H NMR (DMSO-d6, 400 MHz) δ = 1.33 (t, J = 7.0 Hz, 6H, 2 CH3), 3.97–4.07 (m, 4H, 2 OCH2), 5.67 (s, 2H, NH2), 6.95 (d, J = 8.4 Hz, 1H, 5-H), 7.06 (dd, J = 8.4, 1.9 Hz, 1H, 6-H), 7.22 (d, J = 1.9 Hz, 1H, 2-H), 9.39 (s, 1H, NOH), 11.34 (s, 1H, NOH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.66 (CH3), 14.72 (CH3), 63.75 (OCH2), 63.81 (OCH2), 110.59 (C-2), 112.59 (C-5), 120.24 (C-6), 127.24 (C-1), 146.64 (C(=NOH)NH2), 147.66 (C-3), 149.20 (C-4), 149.22 (C=NOH); HRMS (EI+) calcd for C12H17N3O4: 267.1219; found: 267.1230.
4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-amine (9): Amidoxime 8 (0.80 g (3.00 mmol)) was dissolved in 2N NaOH (30 mL) and refluxed for 20 h. The formed precipitate was filtered, washed with water and dried. The beige precipitate was pure product 9 (0.67 g (83%)) and was used without further purification. m.p. 155 °C. IR = 3374, 3322, 3244, 2984, 1593, 1536, 1501, 1396, 1327, 1305, 1253, 1215, 1141, 1111, 1041, 941, 862, 848, 815; 1H NMR (DMSO-d6, 400 MHz) δ = 1.35 (t, J = 6.9 Hz, 6H, 2 CH3), 4.10 (q, J = 6.9 Hz, 4H, 2 OCH2), 6.12 (s, 2H, NH2), 7.09 (d, J = 8.4 Hz, 1H, 5’-H), 7.25 (d, J = 1.8 Hz, 1H, 2’-H), 7.29 (dd, J = 8.4, 1.8 Hz, 1H, 6’-H); 13C NMR (DMSO-d6, 100 MHz) δ = 14.62 (CH3), 14.66 (CH3), 63.85 (2 OCH2), 112.20 (C-2’), 113.25 (C-5’), 117.65 (C-1’), 120.60 (C-6’), 146.69 (C-4), 148.35 (C-3’), 149.87 (C-4’), 155.21 (C-3); HRMS (EI+) calcd for C12H15N3O3: 249.1113; found: 249.1099.
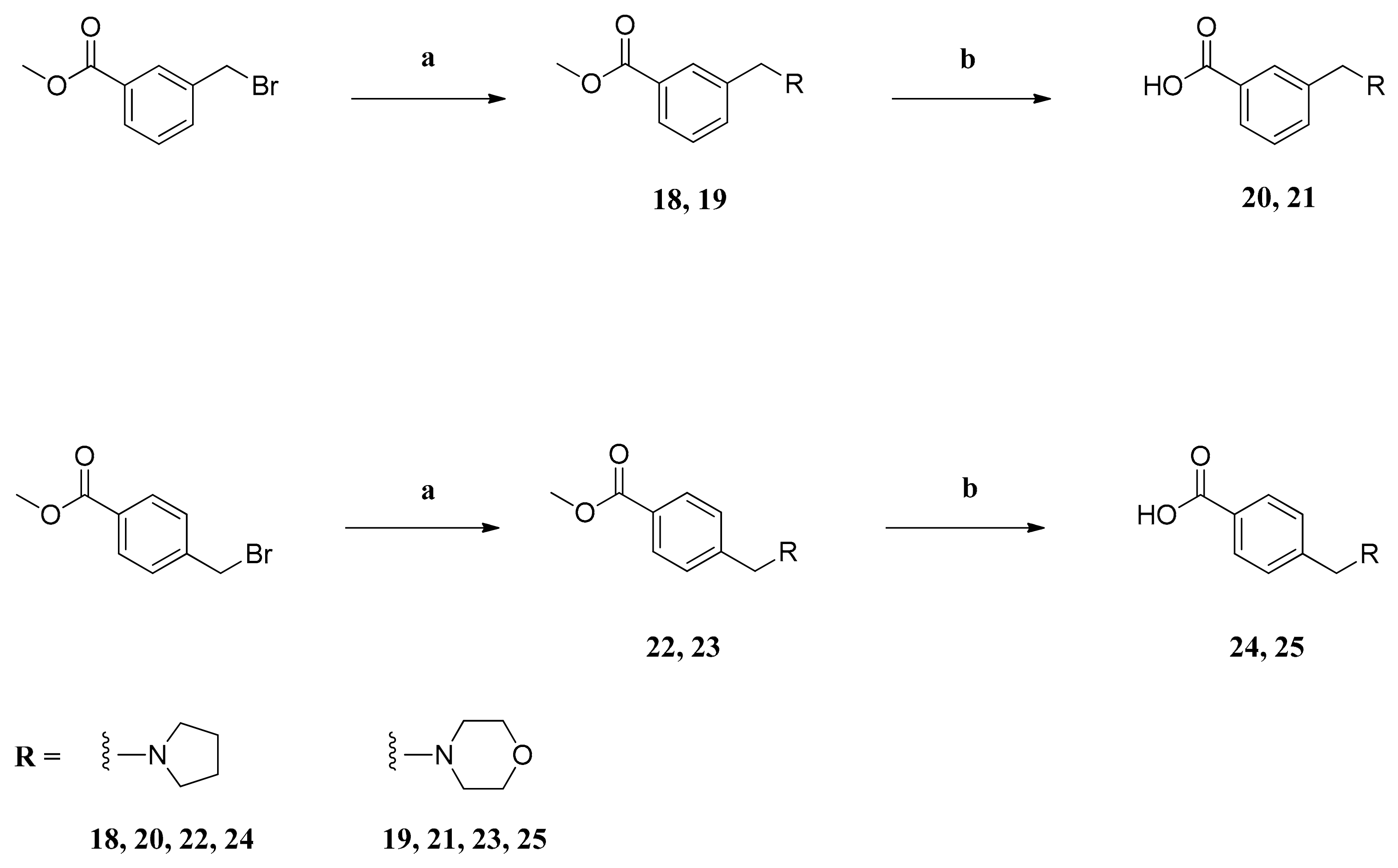
The general procedure for the synthesis of tertiary amines (18, 19, 22 and 23) is as follows: The respective heterocyclic amine (8.00 mmol), K2CO3 (8.00 mmol) and a catalytical amount of NaI (3.20 mmol) were added to a solution of the corresponding benzyl bromide (4.00 mmol) in dry acetonitrile (40 mL). The mixture was refluxed for 20 h. The suspension was cooled and filtered. The filtrate was evaporated in vacuo to dryness. The residue was mixed with water and the aqueous phase was extracted with CH2Cl2. The combined organic phases were washed with 8% aq NaHCO3 and brine. The organic phase was dried over anhydrous sodium sulfate and filtered and the solvent was removed in vacuo, giving pure amines 18, 19, 22 and 23, which were used without further purification.
Methyl 3-((pyrrolidin-1-yl)methyl)benzoate (18): The reaction of methyl 3-(bromomethyl)benzoate (0.92 g (4.00 mmol)), pyrrolidine (0.57 g (8.00 mmol)), K2CO3 (1.11 g (8.00 mmol)) and NaI (0.48 g (3.20 mmol)) in dry acetonitrile (40 mL) gave compound 18 as pale brown oil (0.87 g (99%)).
NMR data were in accordance with literature data [
37].
Methyl 3-((morpholin-4-yl)methyl)benzoate (19): The reaction of methyl 3-(bromomethyl)benzoate (0.92 g (4.00 mmol)), morpholine (0.70 g (8.00 mmol)), K2CO3 (1.11 g (8.00 mmol)) and NaI (0.48 g (3.20 mmol)) in dry acetonitrile (40 mL) gave compound 19 as yellow oil (0.93 g (99%)).
NMR data were in accordance with literature data [
38].
Methyl 4-((pyrrolidin-1-yl)methyl)benzoate (22): The reaction of methyl 4-(bromomethyl)benzoate (0.92 g (4.00 mmol)), pyrrolidine (0.57 g (8.00 mmol)), K2CO3 (1.11 g (8.00 mmol)) and NaI (0.48 g (3.20 mmol)) in dry acetonitrile (40 mL) gave compound 22 as pale brown oil (0.82 g (94%)).
NMR data were in accordance with literature data [
39].
Methyl 4-((morpholin-4-yl)methyl)benzoate (23): The reaction of methyl 4-(bromomethyl)benzoate (0.92 g (4.00 mmol)), morpholine (0.70 g (8.00 mmol)), K2CO3 (1.11 g (8.00 mmol)) and NaI (0.48 g (3.20 mmol)) in dry acetonitrile (40 mL) gave compound 23 as yellow oil (0.90 g (96%)).
NMR data were in accordance with literature data [
40].
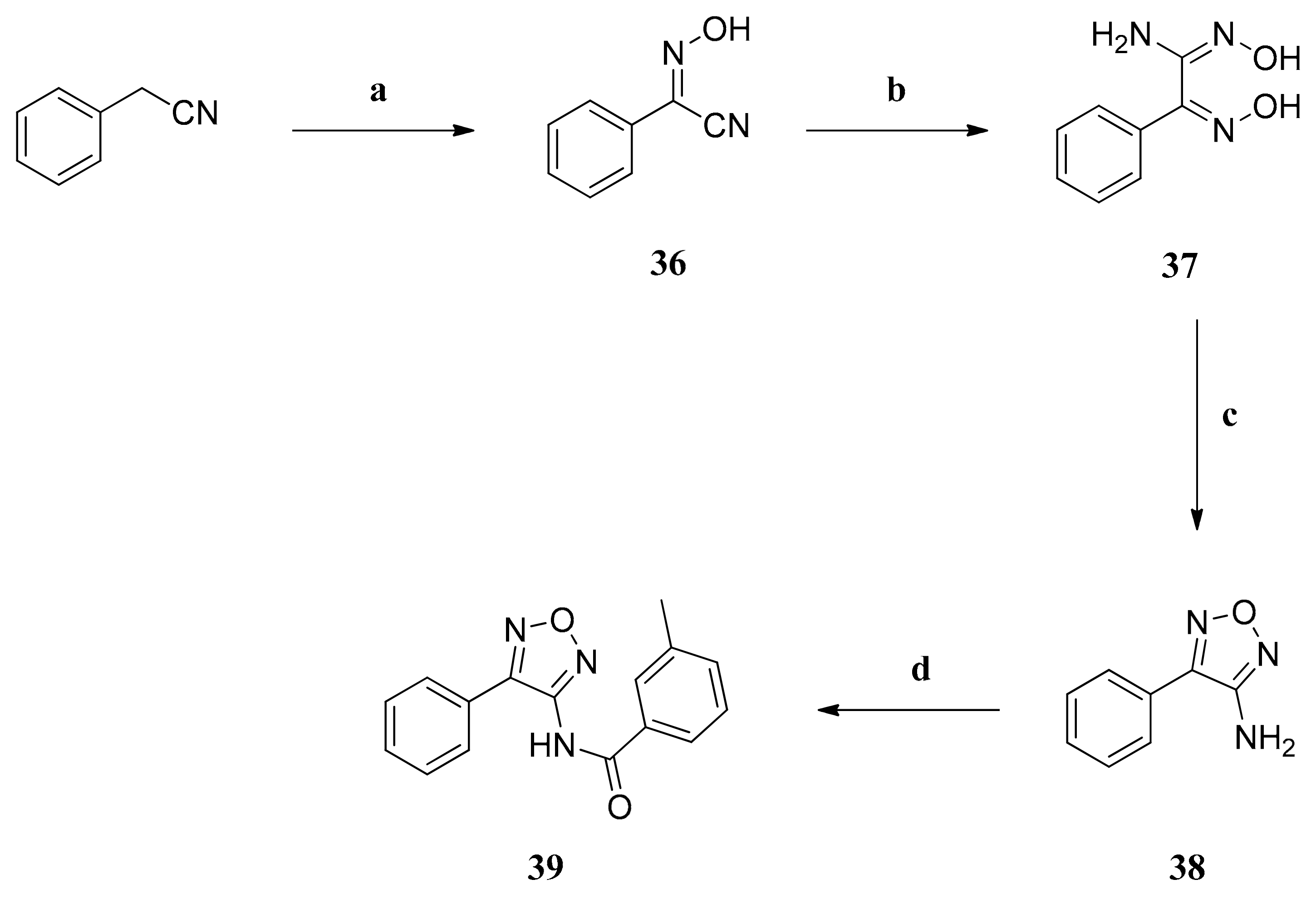
2-(Hydroxyimino)-2-phenylacetonitrile (36): Sodium (0.37 g (12.00 mmol)) was dissolved in dry ethanol (15 mL) and then cooled to 0 °C with an ice bath. A solution of benzyl cyanide (0.94 g (8.00 mmol)) in dry ethanol (10 mL) was added dropwise. Finally, isopentyl nitrite (1.41 g (12.00 mmol)) was added dropwise with a syringe through a septum. The ice bath was removed and the reaction mixture stirred at 25 °C for 20 h. The solution was diluted with ethyl acetate (80 mL) and washed with 2N HCl, 8% aq NaHCO3 and brine. The organic phase was dried over anhydrous sodium sulfate and filtered and the solvent was removed in vacuo, giving compound 36 as yellow amorphous solid (1.15 g (98%)), which was used without further purification.
NMR data were in accordance with literature data [
41].
N’-Hydroxy-2-(hydroxyimino)-2-phenylethanimidamide (37): Oxime 36 (0.88 g (6.00 mmol)) was dissolved in methanol (18 mL) and a solution of NH2OH × HCl (0.50 g (7.20 mmol)) and NaHCO3 (0.61 g (7.20)) in water (9 mL) was added. The mixture was refluxed for 20 h and then the solvent was evaporated in vacuo. The residue was mixed with water (20 mL) and extracted with ethyl acetate. The organic phases were combined, dried over anhydrous sodium sulfate and filtered. The solvent was evaporated in vacuo, giving the raw amidoxime which was recrystallized in CH2Cl2 affording compound 37 as white crystals (0.25 g (23%)).
NMR data were in accordance with literature data [
42].
4-Phenyl-1,2,5-oxadiazol-3-amine (38): Amidoxime 37 (0.54 g (3.00 mmol)) was dissolved in 2N NaOH (30 mL) and refluxed for 20 h. The formed precipitate was filtered, washed with water and dried. The white precipitate was pure product 38 (0.17 g (36%)) and was used without further purification.
NMR data were in accordance with literature data [
43].
The general procedure for the synthesis of carboxylic acids (20, 21, 24 and 25) is as follows: To a solution of the respective methyl ester (3.00 mmol) in methanol (10 mL), 2N NaOH (9.0 mL) was added. The mixture was stirred at 25 °C for 20 h and then the reaction mixture was acidified with conc HCl to a pH of 6. The solvent was evaporated in vacuo and the residue was mixed with CH2Cl2 (20 mL) and sonicated for 5 min. The suspension was filtered and the filtrate was evaporated in vacuo to dryness, giving pure carboxylic acids 20, 21, 24 and 25, which were used without further purification.
3-((Pyrrolidin-1-yl)methyl)benzoic acid (20): The reaction of methyl ester 18 (0.66 g (3.00 mmol)) and 2N NaOH (9 mL) in methanol (10 mL) gave compound 20 as white foam (0.17 g (27%)). IR = 2927, 2597, 1613, 1567, 1455, 1381, 1263; 1H NMR (CDCl3, 400 MHz) δ = 2.12 (br, s, 4H, (CH2)2), 3.26 (br, 4H, N(CH2)2), 4.20 (br, s, 2H, ArCH2), 7.40 (t, J = 7.7 Hz, 1H, 5-H), 7.55 (br, 1H, 4-H), 8.07 (d, J = 7.7 Hz, 1H, 6-H), 8.50 (br, s, 1H, 2-H); 13C NMR (CDCl3, 100 MHz) δ = 23.19 ((CH2)2), 52.71 (N(CH2)2), 58.49 (ArCH2), 128.33 (C-5), 130.39 (C-6), 130.75 (C-1), 132.47 (C-2), 132.55 (C-4), 136.70 (C-3), 170.88 (COOH); HRMS (EI+) calcd for C12H15NO2: 205.1103; found: 205.1088.
3-((Morpholin-4-yl)methyl)benzoic acid (21): The reaction of methyl ester 19 (0.71 g (3.00 mmol)) and 2N NaOH (9 mL) in methanol (10 mL) gave compound 21 as white amorphous solid (0.64 g (97%)).
NMR data were in accordance with literature data [
44].
4-((Pyrrolidin-1-yl)methyl)benzoic acid (24): The reaction of methyl ester 22 (0.66 g (3.00 mmol)) and 2N NaOH (9 mL) in methanol (10 mL) gave compound 24 as white foam (0.55 g (90%)).
NMR data were in accordance with literature data [
39].
4-((Morpholin-4-yl)methyl)benzoic acid (25): The reaction of methyl ester 23 (0.71 g (3.00 mmol)) and 2N NaOH (9 mL) in methanol (10 mL) gave compound 25 as yellow oil (0.62 g (94%)).
NMR data were in accordance with literature data [
40].
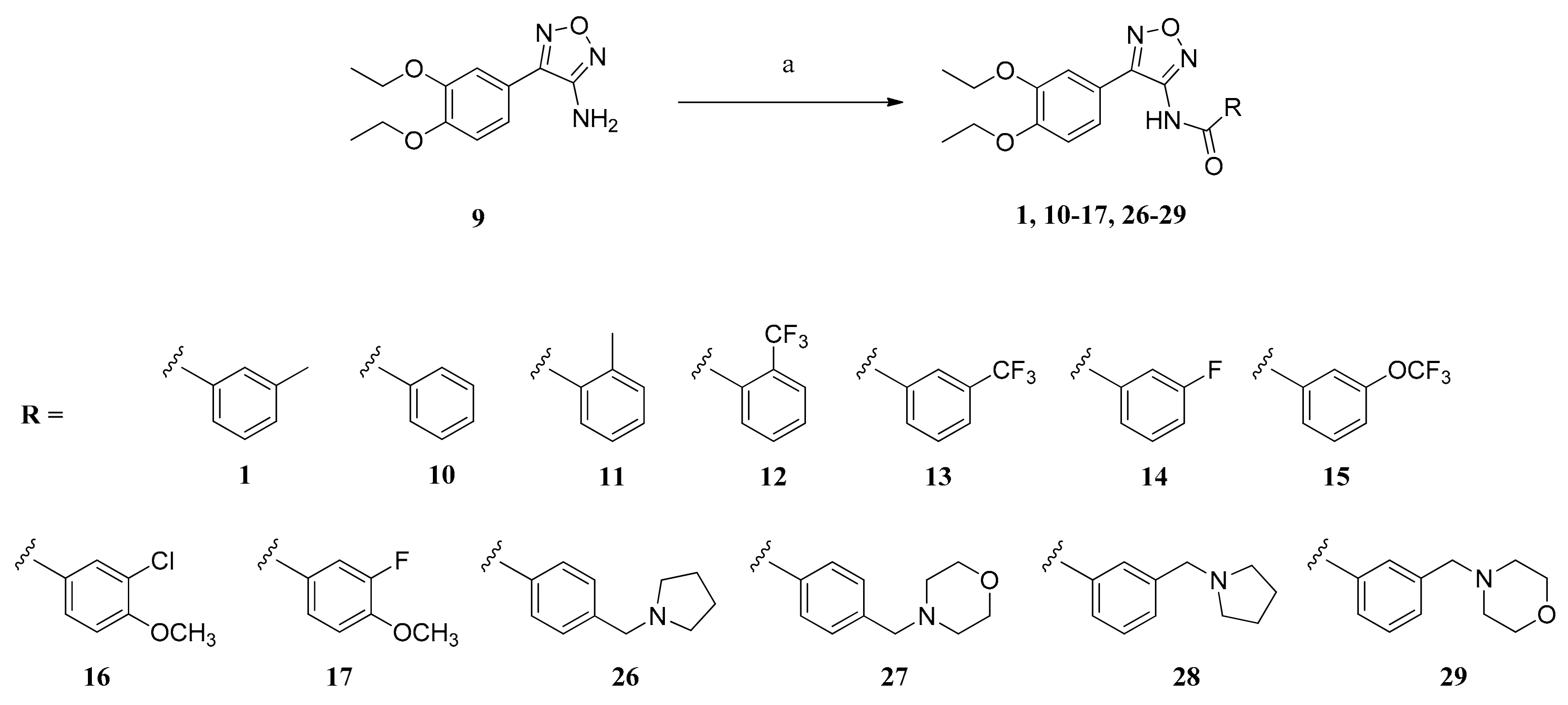
The general procedure for the synthesis of carboxamides (1, 10–17, 26–29) is detailed in the following subsections.
3.2.1. Method A
An ice-cooled suspension of NaH (60% dispersion in mineral oil; 2.00 mmol) in dry DMF (14 mL) was mixed with aminofurazan 9 (1.00 mmol) and stirred for 20 min. Then, a solution of acid chloride (1.30 mmol) in dry DMF (2 mL) was added dropwise and the reaction mixture was stirred at 60 °C for 20 h. Afterward, the mixture was quenched with water at 0 °C and the aqueous phase was extracted with CH2Cl2. The organic layer was washed with 8% aq NaHCO3 and brine, dried over anhydrous sodium sulfate and filtered, and the solvent was evaporated in vacuo, yielding the raw carboxamides 1 and 10–14, which were further purified by crystallization.
3.2.2. Method B
To an ice-cooled solution of carboxylic acid (1.50 mmol) in dry CH2Cl2 (14 mL), oxalyl dichloride 2 M in CH2Cl2 (1.90 mmol) was added dropwise under stirring. After 1 h, the ice bath was removed and the reaction batch was stirred 20 h at 25 °C in an atmosphere of Ar. Subsequently, the solvent was evaporated in vacuo and the crude acyl chloride was dissolved in dry DMF (9 mL). An ice-cooled suspension of NaH (60% dispersion in mineral oil; 2.00 mmol) in dry DMF (14 mL) was mixed with aminofurazan 9 (1.00 mmol) and stirred for 20 min. Then, the solution of acyl chloride in dry DMF was added dropwise and the reaction mixture was stirred at 60 °C for 48 h. Afterward, the mixture was quenched with water at 0 °C and the aqueous phase was extracted with CH2Cl2. The organic layer was washed with 8% aq NaHCO3 and brine, dried over anhydrous sodium sulfate and filtered, and the solvent was evaporated in vacuo, yielding the raw carboxamides 16, 17 and 26–29, which were further purified by column chromatography or crystallization.
3.2.3. Method C
A solution of benzoic acid (1.50 mmol), N-hydroxysuccinimide (1.58 mmol) and N,N′-dicyclohexylcarbodiimide (1.50 mmol) was dissolved in dry THF (10 mL) and stirred at 25 °C for 20 h. The formed precipitate was filtered and the filtrate was evaporated in vacuo to dryness to obtain the crude NHS ester which was used without further purification. An ice-cooled suspension of NaH (60% dispersion in mineral oil; 2.00 mmol) in dry DMF (14 mL) was mixed with aminofurazan 9 (1.00 mmol) and stirred for 20 min. Then, the solution of NHS ester in dry DMF (4 mL) was added dropwise and the reaction mixture was stirred at 60 °C for 20 h. Afterward, the mixture was quenched with water at 0 °C and the aqueous phase was extracted with CH2Cl2. The organic layer was washed with 8% aq NaHCO3 and brine, dried over anhydrous sodium sulfate and filtered, and the solvent was evaporated in vacuo, yielding the raw carboxamide 15, which was further purified by crystallization.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-methylbenzamide (1): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), 3-methylbenzoyl chloride (201 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 1 as white crystals (63 mg (17%)). m.p. 162 °C. IR = 3198, 2981, 1665, 1591, 1531, 1500, 1394, 1377, 1325, 1277, 1260, 1218, 1142, 1042, 940, 862, 810; 1H NMR (DMSO-d6, 400 MHz) δ = 1.20 (t, J = 6.9 Hz, 3H, CH3), 1.32 (t, J = 6.9 Hz, 3H, CH3), 2.40 (s, 3H, ArCH3), 3.90 (q, J = 6.9 Hz, 2H, OCH2), 4.05 (q, J = 6.9 Hz, 2H, OCH2), 7.07 (d, J = 8.4 Hz, 1H, 5”-H), 7.32-7.37 (m, 2H, 2”-H, 6”-H), 7.45 (t, J = 7.6, 1H, 5-H), 7.49 (d, J = 7.6 Hz, 1H, 4-H), 7.79 (br, d, J = 7.4 Hz, 1H, 6-H), 7.82 (br, s, 1H, 2-H), 11.17 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.47 (CH3), 14.56 (CH3), 20.86 (ArCH3), 63.79 (2 OCH2), 111.68 (C-2”), 113.21 (C-5”), 116.92 (C-1”), 120.44 (C-6”), 125.17 (C-6), 128.52 (C-2), 128.61 (C-5), 132.22 (C-1), 133.41 (C-4), 138.13 (C-3), 148.10 (C-3”), 150.06 (C-3’), 150.33 (C-4”), 151.23 (C-4’), 166.78 (C=O); HRMS (EI+) calcd for C20H21N3O4: 367.1532; found: 367.1526.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)benzamide (10): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), benzoyl chloride (183 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 10 as white crystals (81 mg (23%)). m.p. 199 °C. IR = 3212, 2980, 1660, 1591, 1530, 1501, 1468, 1395, 1378, 1324, 1275, 1258, 1216, 1141, 1040, 916, 852, 811; 1H NMR (DMSO-d6, 400 MHz) δ = 1.19 (t, J = 6.9 Hz, 3H, CH3), 1.31 (t, J = 6.9 Hz, 3H, CH3), 3.88 (q, J = 7.0 Hz, 2H, OCH2), 4.05 (q, J = 6.9 Hz, 2H, OCH2), 7.07 (d, J = 8.4 Hz, 1H, 5”-H), 7.31 (d, J = 1.7 Hz, 1H, 2”-H), 7.33 (dd, J = 8.4, 1.7 Hz, 1H, 6”-H), 7.58 (t, J = 7.4 Hz, 2H, 3-H, 5-H), 7.68 (t, J = 7.4 Hz, 1H, 4-H), 8.00 (d, J = 7.2 Hz, 2H, 2-H, 6-H), 11.24 (br s, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.45 (CH3), 14.57 (CH3), 63.78 (2 OCH2), 111.65 (C-2”), 113.19 (C-5”), 116.87 (C-1”), 120.43 (C-6”), 128.05 (C-2, C-6), 128.74 (C-3, C-5), 132.10 (C-1), 132.91 (C-4), 148.11 (C-3”), 149.95 (C-3’), 150.36 (C-4”), 151.24 (C-4’), 166.66 (C=O); HRMS (EI+) calcd for C19H19N3O4: 353.1375; found: 353.1373.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-2-methylbenzamide (11): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), 2-methylbenzoyl chloride (201 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 11 as white crystals (103 mg (28%)). m.p. 170 °C. IR = 3214, 2983, 1663, 1591, 1503, 1395, 1377, 1324, 1259, 1216, 1142, 1042, 918, 854, 803, 741, 687, 657, 618; 1H NMR (DMSO-d6, 400 MHz) δ = 1.28 (t, J = 7.0 Hz, 3H, CH3), 1.34 (t, J = 7.0 Hz, 3H, CH3), 2.34 (s, 3H, ArCH3), 3.99 (q, J = 7.0 Hz, 2H, OCH2), 4.08 (q, J = 7.0 Hz, 2H, OCH2), 7.11 (d, J = 8.4 Hz, 1H, 5”-H), 7.33–7.37 (m, 4H, 2”-H, 3-H, 5-H, 6”-H), 7.45 (t, J = 7.3 Hz, 1H, 4-H), 7.65 (d, J = 7.3 Hz, 1H, 6H), 11.12 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.61 (2 CH3), 19.56 (ArCH3), 63.84 (OCH2), 63.97 (OCH2), 112.06 (C-2”), 113.13 (C-5”), 116.96 (C-1”), 120.67 (C-6”), 125.79 (C-5), 127.89 (C-6), 130.92 (C-4), 131.10 (C-3), 133.97 (C-1), 136.63 (C-2), 148.21 (C-3”), 149.83 (C-3´), 150.34 (C-4”), 151.22 (C-4´), 168.70 (C=O); HRMS (EI+) calcd for C19H21N3O4: 367.1532; found: 367.1526.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-2-(trifluoromethyl)benzamide (12): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), 2-(trifluoromethyl)benzoyl chloride (271 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in methanol to yield compound 12 as white crystals (38 mg (9%)). m.p. 171 °C. IR = 3209, 2983, 1676, 1604, 1531, 1494, 1397, 1377, 1316, 1277, 1217, 1177, 1129, 1043, 921, 854, 811, 772, 651, 617; 1H NMR (DMSO-d6, 400 MHz) δ = 1.32 (t, J = 7.0 Hz, 3H, CH3), 1.34 (t, J = 7.0 Hz, 3H, CH3), 4.05 (q, J = 7.0 Hz, 2H, OCH2), 4.09 (q, J = 7.0 Hz, 2H, OCH2), 7.13 (d, J = 8.2 Hz, 1H, 5”-H), 7.34 (d, J = 1.8 Hz, 1H, 2”-H), 7.36 (dd, J = 8.2, 1.8 Hz, 1H, 6”-H), 7.76-7.84 (m, 3H, 4-H, 5-H, 6-H), 7.89 (d, J = 8.2 Hz, 1H, 3-H), 11.46 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.61 (CH3), 14.64 (CH3), 63.84 (OCH2), 63.96 (OCH2), 112.37 (C-2”), 113.09 (C-5”), 116.52 (C-1”), 120.94 (C-6”), 123.52 (q, J = 274 Hz, CF3), 126.25 (q, J = 31.7 Hz, C-2), 126.71 (q, J = 4.9 Hz, C-3), 128.69 (C-6), 131.05 (C-4), 132.69 (C-5), 134.03 (m, C-1), 148.23 (C-3”), 149.00 (C-3’), 150.41 (C-4”), 150.72 (C-4’), 166.47 (C=O); HRMS (EI+) calcd for C20H18F3N3O4: 421.1249; found: 421.1248.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-(trifluoromethyl)benzamide (13): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), 3-(trifluoromethyl)benzoyl chloride (271 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 13 as white crystals (131 mg (31%)). m.p. 153 °C. IR = 3203, 1675, 1498, 1378, 1336, 1260, 1217, 1172, 1121, 1042, 815; 1H NMR (DMSO-d6, 400 MHz) δ = 1.19 (t, J = 7.0 Hz, 3H, CH3), 1.32 (t, J = 7.0 Hz, 3H, CH3), 3.90 (q, J = 7.0 Hz, 2H, OCH2), 4.06 (q, J = 7.0 Hz, 2H, OCH2), 7.08 (d, J =8.3 Hz, 1H, 5”-H), 7.33 (d, J = 1.9 Hz, 1H, 2”-H), 7.34 (dd, J = 8.3, 1.9 Hz, 1H, 6”-H), 7.84 (t, J = 7.7 Hz, 1H, 5-H), 8.06 (d, J = 7.7 Hz, 1H, 4-H), 8.29 (d, J = 7.7 Hz, 1H, 6-H), 8.36 (s, 1H, 2-H), 11.53 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.40 (CH3), 14.56 (CH3), 63.77 (OCH2), 63.81 (OCH2), 111.72 (C-2”), 113.24 (C-5”), 116.83 (C-1”), 120.59 (C-6”), 123.82 (q, J = 272 Hz, CF3), 124.66 (q, J = 3.9 Hz, C-2), 129.33 (q, J = 4.4 Hz, C-4), 129.44 (q, J = 32.2 Hz, C-3), 130.18 (C-5), 132.27 (C-6), 133.21 (C-1), 148.11 (C-3”), 149.80 (C-3’), 150.36 (C-4”), 151.08 (C-4’), 165.25 (C=O); HRMS (EI+) calcd for C20H18F3N3O4: 421.1249; found: 421.1242.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-fluorobenzamide (14): Method A: The reaction of compound 9 (249 mg (1.00 mmol)), 3-fluorobenzoyl chloride (206 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 14 as beige crystals (186 mg (50%)). m.p. 250 °C. IR = 3216, 2983, 1663, 1591, 1505, 1364, 1275, 1215, 1142, 1039, 880, 857, 809; 1H NMR (DMSO-d6, 400 MHz) δ = 1.27 (t, J = 7.0 Hz, 3H, CH3), 1.33 (t, J = 7.0 Hz, 3H, CH3), 3.99 (q, J = 7.0 Hz, 2H, OCH2), 4.07 (q, J = 7.0 Hz, 2H, OCH2), 7.06 (d, J =8.4 Hz, 1H, 5”-H), 7.32 (td, J = 8.4, 2.1 Hz, 1H, 4-H), 7.48 (ddd, J = 8.4, 7.6, 5.7 Hz, 1H, 5-H), 7.63 (dd, J = 8.3, 1.4 Hz, 1H, 6”-H), 7.81-7.87 (m, 2H, 2-H, 2”-H), 7.93 (d, J = 7.6 Hz, 1H, 6-H); 13C NMR (DMSO-d6, 100 MHz) δ = 14.60 (CH3), 14.66 (CH3), 63.76 (2 OCH2), 112.34 (C-2”), 113.08 (C-5”), 114.69 (d, J = 22.1 Hz, C-2), 117.25 (d, J = 21.2 Hz, C-4), 119.08 (C-1”), 120.60 (C-6”), 124.25 (d, J = 2.7 Hz, C-6), 129.87 (d, J = 7.9 Hz, C-5), 140.35 (C-1), 147.84 (C-3”), 149.66 (C-4”), 150.19 (C-4’), 155.35 (C-3’), 161.94 (d, J = 242 Hz, C-3), 166.84 (C=O); HRMS (EI+) calcd for C19H18FN3O4: 371.1261; found: 371.1276.
3-Chloro-N-(4-(3,4-diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-4-methoxybenzamide (16): Method B: The reaction of 3-chloro-4-methoxybenzoic acid (280 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acid chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in toluene to yield compound 16 as white crystals (255 mg (61%)). m.p. 260 °C (decomp.). IR = 2978, 1663, 1600, 1501, 1470, 1395, 1360, 1271, 1215, 1142, 1057, 1040, 937, 855, 811; 1H NMR (DMSO-d6, 400 MHz) δ = 1.30 (t, J = 7.0 Hz, 3H, CH3), 1.34 (t, J = 7.0 Hz, 3H, CH3), 3.91 (s, 3H, OCH3), 4.02 (q, J = 7.0 Hz, 2H, OCH2), 4.07 (q, J = 7.0 Hz, 2H, OCH2), 7.06 (d, J = 8.4 Hz, 1H, 5”-H), 7.18 (d, J = 8.7 Hz, 1H, 5-H), 7.65 (dd, J = 8.4, 1.6 Hz, 1H, 6”-H), 7.92 (d, J = 1.6 Hz, 1H, 2”-H), 8.05 (dd, J = 8.7, 1.9 Hz, 1H, 6-H), 8.17 (d, J = 1.6 Hz, 1H, 2-H), 11.27 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.69 (2 CH3), 56.27 (OCH3), 63.72 (2 OCH2), 111.80 (C-5), 112.33 (C-2”), 112.99 (C-5”), 119.43 (C-1”), 120.18 (C-3), 120.55 (C-6”), 128.55 (C-6), 129.79 (C-2), 131.72 (C-1), 147.76 (C-3”), 149.48 (C-4”), 150.09 (C-4’), 153.07 (C-3’), 156.03 (C-4), 166.89 (C=O); HRMS (EI+) calcd for C20H20ClN3O5: 417.1092; found: 417.1081.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-fluoro-4-methoxybenzamide (17): Method B: The reaction of 3-fluoro-4-methoxybenzoic acid (255 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acid chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in toluene to yield compound 17 as beige crystals (124 mg (31%)). m.p. 232 °C (decomp.). IR = 3199, 2982, 1663, 1615, 1500, 1469, 1396, 1377, 1327, 1286, 1217, 1140, 1040, 853, 802; 1H NMR (DMSO-d6, 400 MHz) δ = 1.25 (t, J = 6.9 Hz, 3H, CH3), 1.33 (t, J = 6.9 Hz, 3H, CH3), 3.92 (s, 3H, OCH3), 3.96 (q, J = 6.9 Hz, 2H, OCH2), 4.07 (q, J = 6.9 Hz, 2H, OCH2), 7.06 (d, J = 8.6 Hz, 1H, 5”-H), 7.27 (t, J = 8.7 Hz, 1H, 5-H), 7.51 (dd, J = 8.4, 2.0 Hz, 1H, 6”-H), 7.62 (d, J = 2.0 Hz, 1H, 2”-H), 7.89 (d, J = 8.3 Hz, 1H, 6-H), 7.90 (s, 1H, 2-H), 11.29 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.55 (CH3), 14.63 (CH3), 56.23 (d, J = 2.3 Hz, OCH3), 63.74 (2 OCH2), 112.02 (C-2”), 113.06 (C-5, C-5”), 115.54 (d, J = 19.0 Hz, C-2), 118.23 (C-1”), 120.50 (C-6”), 125.30 (d, J = 3.1 Hz, C-6), 128.22 (C-1), 147.90 (C-3”), 149.86 (d, J = 23.6 Hz, C-4), 149.89 (C-4”), 150.61 (C-4’), 150.76 (d, J = 244 Hz, C-3), 153.19 (C-3’), 166.11 (C=O); HRMS (EI+) calcd for C20H20FN3O5: 401.1387; found: 401.1380.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-4-((pyrrolidin-1-yl)methyl)benzamide (26): Method B: The reaction of carboxylic acid 20 (308 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acid chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in toluene to yield compound 26 as light yellow crystals (140 mg (32%)). m.p. 251 °C (decomp.). IR = 2973, 2788, 1584, 1471, 1365, 1271, 1246, 1201, 1141, 1091, 1038, 997, 937, 880, 854, 809; 1H NMR (DMSO-d6, 400 MHz) δ = 1.32 (t, J = 7.0 Hz, 3H, CH3), 1.35 (t, J = 7.0 Hz, 3H, CH3), 1.69 (br, s, 4H, (CH2)2), 2.43 (br, s, 4H, N(CH2)2), 3.59 (s, 2H, ArCH2), 4.05 (q, J = 7.0 Hz, 2H, OCH2), 4.08 (q, J = 7.0 Hz, 2H, OCH2), 7.07 (d, J = 8.5 Hz, 1H, 5”-H), 7.27 (d, J = 7.8 Hz, 2H, 3-H, 5-H), 7.92 (dd, J = 7.8, 1.9 Hz, 1H, 6”-H), 8.11 (d, J = 7.8 Hz, 2H, 2-H, 6-H), 8.24 (d, J = 1.9 Hz, 1H, 2”-H); 13C NMR (DMSO-d6, 100 MHz) δ = 14.74 (CH3), 14.79 (CH3), 23.14 ((CH2)2), 53.54 (N(CH2)2), 59.54 (ArCH2), 63.68 (2 OCH2), 112.67 (C-2”), 112.86 (C-5”), 120.64 (C-1”), 120.72 (C-6”), 127.37 (C-3, C-5), 128.24 (C-2, C-6), 139.78 (C-1), 140.34 (C-4), 147.60 (C-3”), 149.12 (C-4”), 149.56 (C-4’), 158.93 (C-3’), 169.56 (C=O); HRMS (EI+) calcd for C24H28N4O4: 436.2111; found: 436.2102.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-4-((morpholin-4-yl)methyl)benzamide (27): Method B: The reaction of carboxylic acid 25 (332 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acyl chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in toluene to yield compound 27 as pale yellow crystals (258 mg (57%)). m.p. 269 °C (decomp.). IR = 2977, 1585, 1499, 1472, 1360, 1271, 1202, 1141, 1109, 1035, 867; 1H NMR (DMSO-d6, 400 MHz) δ = 1.32 (t, J = 7.0 Hz, 3H, CH3), 1.35 (t, J = 7.0 Hz, 3H, CH3), 2.36 (br, s, 4H, N(CH2)2), 3.49 (s, 2H, ArCH2), 3.58 (t, J = 4.5 Hz, 4H, O(CH2)2), 4.05 (q, J = 7.0 Hz, 2H, OCH2), 4.08 (q, J = 7.0 Hz, 2H, OCH2), 7.07 (d, J = 8.6 Hz, 1H, 5”-H), 7.28 (d, J = 8.2 Hz, 2H, 3-H, 5-H), 7.92 (dd, J = 8.6, 1.6 Hz, 1H, 6”-H), 8.12 (d, J = 7.8 Hz, 2H, 2-H, 6-H), 8.23 (d, J = 1.6 Hz, 1H, 2”-H); 13C NMR (DMSO-d6, 100 MHz) δ = 14.75 (CH3), 14.79 (CH3), 53.22 (N(CH2)2), 62.36 (ArCH2), 63.69 (2 OCH2), 66.24 (O(CH2)2), 112.66 (C-2”), 112.86 (C-5”), 120.64 (C-1”), 120.73 (C-6”), 127.86 (C-3, C-5), 128.28 (C-2, C-6), 138.60 (C-1), 140.08 (C-4), 147.61 (C-3”), 149.12 (C-4”), 149.56 (C-4’), 158.97 (C-3’), 169.47 (C=O); HRMS (EI+) calcd for C24H28N4O5: 452.2060; found: 452.2043.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-((pyrrolidin-1-yl)methyl)benzamide (28): Method B: The reaction of carboxylic acid 20 (308 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acid chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by column chromatography (silica gel, CH2Cl2/toluene/MeOH, 20:1:1) to yield compound 28 as yellow oil (262 mg (60%)). IR = 3212, 2974, 2799, 1661, 1591, 1500, 1473, 1395, 1377, 1259, 1217, 1140, 1041, 849, 800; 1H NMR (CDCl3, 400 MHz) δ = 1.40 (t, J = 7.0 Hz, 3H, CH3), 1.46 (t, J = 7.0 Hz, 3H, CH3), 1.78–1.81 (m, 4H, (CH2)2), 2.52–2.56 (m, 4H, N(CH2)2), 3.69 (s, 2H, ArCH2), 4.04 (q, J = 7.0 Hz, 2H, OCH2), 4.11 (q, J = 7.0 Hz, 2H, OCH2), 6.91 (d, J = 8.2 Hz, 1H, 5”-H), 7.23 (dd, J = 8.2, 1.9 Hz, 1H, 6”-H), 7.25 (d, J = 1.9 Hz, 1H, 2”-H), 7.42 (t, J = 7.7 Hz, 1H, 5-H), 7.57 (d, J = 7.6 Hz, 1H, 4-H), 7.75 (d, J = 7.8 Hz, 1H, 6-H), 7.87 (s, 1H, 2-H); 13C NMR (CDCl3, 100 MHz) δ = 14.64 (CH3), 14.65 (CH3), 23.42 ((CH2)2), 54.20 (N(CH2)2), 60.09 (ArCH2), 64.44 (OCH2), 64.58 (OCH2), 112.36 (C-2”), 112.94 (C-5”), 117.28 (C-1”), 120.33 (C-6”), 126.40 (C-6), 127.82 (C-2), 128.92 (C-5), 132.39 (C-1), 133.45 (C-4), 140.34 (C-3), 148.88 (C-3’), 149.08 (C-3”), 149.83 (C-4’), 150.82 (C-4”), 165.69 (C=O); HRMS (EI+) calcd for C24H28N4O4: 436.2111; found: 436.2086.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-((morpholin-4-yl)methyl)benzamide (29): Method B: The reaction of carboxylic acid 21 (332 mg (1.50 mmol)) and oxalyl dichloride 2 M in CH2Cl2 (0.95 mL (1.90 mmol)) in dry CH2Cl2 (14 mL) gave the raw acid chloride which was suspended in dry DMF (9 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in toluene to yield compound 29 as white crystals (113 mg (25%)). m.p. 199 °C. IR = 2978, 1605, 1502, 1468, 1361, 1271, 1209, 1141, 1114, 1036, 854; 1H NMR (CD3OD, 400 MHz) δ = 1.28 (t, J = 7.0 Hz, 3H, CH3), 1.40 (t, J = 7.0 Hz, 3H, CH3), 2.48 (br, s, 4H, N(CH2)2), 3.58 (s, 2H, ArCH2), 3.70 (t, J = 4.5 Hz, 4H, O(CH2)2), 3.94 (q, J = 7.0 Hz, 2H, OCH2), 4.09 (q, J = 7.0 Hz, 2H, OCH2), 6.98 (d, J = 8.3 Hz, 1H, 5”-H), 7.36 (t, J = 7.7 Hz, 1H, 5-H), 7.43 (d, J = 7.7 Hz, 1H, 4-H), 7.64 (dd, J = 8.3, 1.6 Hz, 1H, 6”-H), 7.72 (d, J = 1.6 Hz, 1H, 2”-H), 8.00 (d, J = 7.7 Hz, 1H, 6-H), 8.03 (s, 1H, 2-H); 13C NMR (CD3OD, 100 MHz) δ = 15.23 (2 CH3), 54.77 (N(CH2)2), 64.49 (ArCH2), 65.72 (OCH2), 65.76 (OCH2), 67.92 (O(CH2)2), 114.03 (C-2”), 114.47 (C-5”), 121.81 (C-1”), 122.20 (C-6”), 128.81 (C-6), 128.95 (C-5), 131.01 (C-2), 132.31 (C-4), 137.99 (C-3), 140.85 (C-1), 149.92 (C-3”), 151.48 (C-4”), 151.79 (C-4’), 160.84 (C-3’), 173.71 (C=O); HRMS (EI+) calcd for C24H28N4O5: 452.2060; found: 452.2042.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-(trifluoromethoxy)benzamide (15): Method C: The reaction of 3-(trifluoromethoxy)benzoic acid (309 mg (1.50 mmol)), N-hydroxysuccinimide (182 mg (1.58 mmol)) and N,N′-dicyclohexylcarbodiimide (310 mg (1.50 mmol)) in dry THF (10 mL) gave the raw NHS ester. It was suspended in dry DMF (4 mL) and added to a suspension of compound 9 (249 mg (1.00 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (14 mL), giving the raw carboxamide which was purified by recrystallization in CH2Cl2 to yield compound 15 as white crystals (136 mg (31%)). m.p. 145–147 °C. IR = 3209, 1675, 1590, 1501, 1378, 1260, 1216, 1170, 1042, 809, 633; 1H NMR (DMSO-d6, 400 MHz) δ = 1.19 (t, J = 7.0 Hz, 3H, CH3), 1.32 (t, J = 7.0 Hz, 3H, CH3), 3.90 (q, J = 7.0 Hz, 2H, OCH2), 4.05 (q, J = 7.0 Hz, 2H, OCH2), 7.06 (d, J = 8.3 Hz, 1H, 5”-H), 7.39–7.44 (m, 2H, 2”-H, 6”-H), 7.63 (d, J = 8.3 Hz, 1H, 4-H), 7.68 (t, J = 7.8 Hz, 1H, 5-H), 7.96 (s, 1H, 2-H), 8.06 (d, J = 7.5 Hz, 1H, 6-H), 11.32 (br, 1H, NH); 13C NMR (DMSO-d6, 100 MHz) δ = 14.45, 14.62 (2 CH3), 63.72, 63.78 (2 OCH2), 111.79 (C-2”), 113.13 (C-5”), 117.50 (C-1”), 120.08 (q, J = 257 Hz, CF3), 120.50 (C-2), 120.56 (C-6”), 124.68 (C-4), 127.27 (C-6), 130.75 (C-5), 136.13 (C-1), 148.01 (C-3”), 148.34 (C-3’), 150.14 (C-4”), 150.84 (C-4’), 151.53 (C-3), 165.46 (C=O); HRMS (EI+) calcd for C20H18F3N3O5: 437.1198; found: 437.1171.
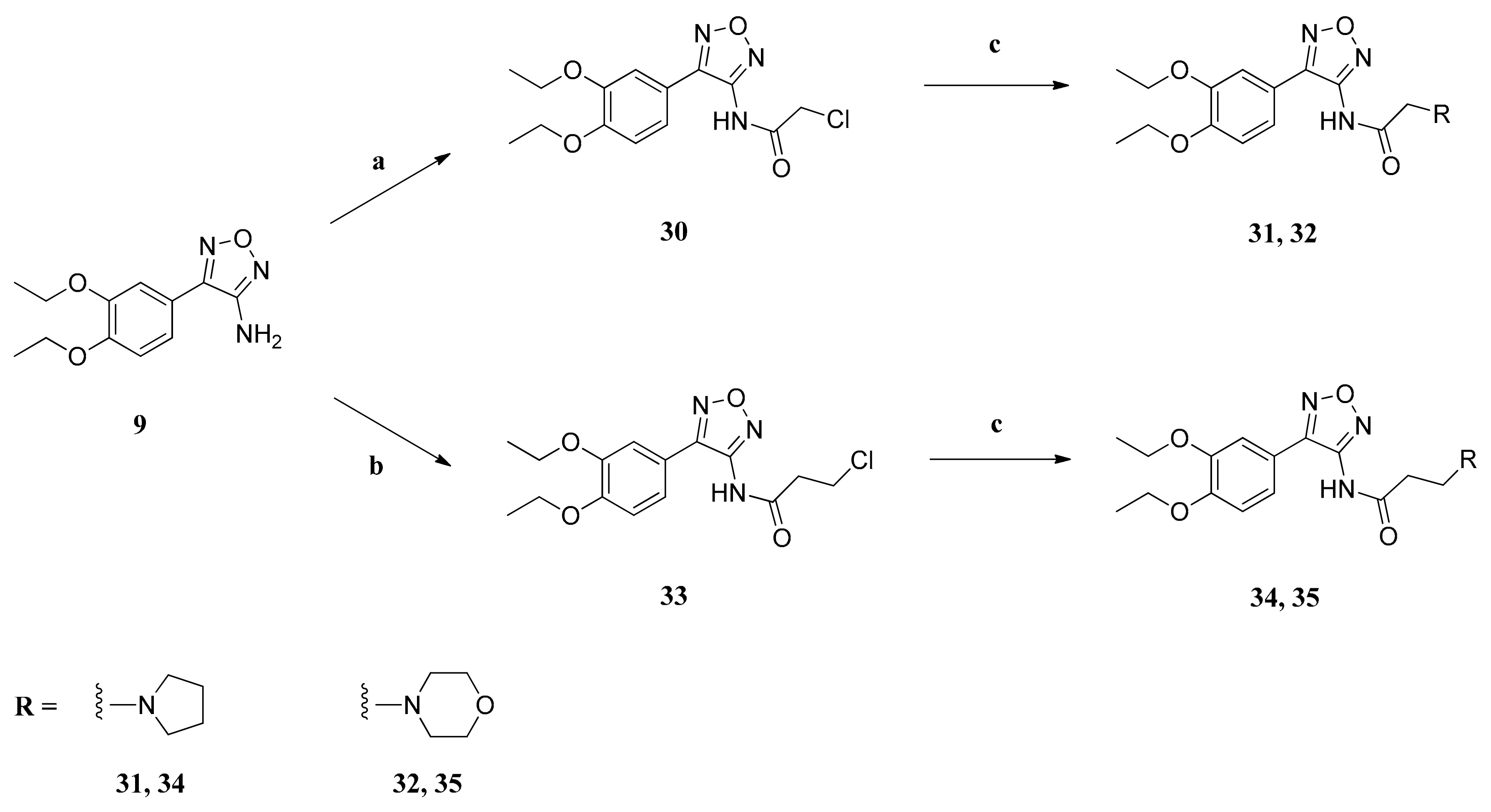
3.2.4. General Procedure for the Synthesis of Compounds 31, 32, 34 and 35
Amino furazan 9 (1.00 mmol) was dissolved in a mixture of dry diethyl ether (4 mL) and dry benzene (16 mL). Dry pyridine (1.13 mmol) was added and then a solution of chloroacyl chloride (1.10 mmol) in dry diethyl ether (4 mL) was added dropwise. The mixture was refluxed for 20 h and then the solvent was evaporated in vacuo. The residue was mixed with water (35 mL) and the aqueous phase was extracted with CH2Cl2. The combined organic layers were dried over anhydrous sodium sulfate and filtered, and the solvent was evaporated in vacuo, yielding the raw carboxamide which was used without further purification. It was dissolved in dry acetonitrile (20 mL), and the respective amine (2.00 mmol), K2CO3 (2.00 mmol) and a catalytical amount of NaI (0.65 mmol) were added. The mixture was stirred at 70 °C for 20 h and then cooled to ambient temperature. The suspension was filtered and the filtrate was evaporated in vacuo to dryness. The residue was mixed with water (20 mL) and the aqueous phase was extracted with CH2Cl2. The organic layer was washed with 8% aqueous NaHCO3 and brine, dried over anhydrous sodium sulfate and filtered, and the solvent was evaporated in vacuo, yielding the carboxamide, which was purified by column chromatography.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-2-(pyrrolidin-1-yl)acetamide (31): The reaction of amino furazan 9 (249 mg (1.00 mmol)), chloroacetyl chloride (124 mg (1.10 mmol)) and pyridine (89 g (1.13 mmol)) in a mixture of dry diethyl ether (8 mL) and dry benzene (16 mL) gave the raw carboxamide. This was mixed with pyrrolidine (142 mg (2.00 mmol)), K2CO3 (276 mg (2.00 mmol)) and NaI (97 mg (0.65 mmol)) in dry acetonitrile (20 mL), giving the tertiary amine which was purified by column chromatography (silica gel, CH2Cl2/MeOH, 100:1) to yield compound 31 as pale yellow amorphous solid (87 mg (24%)). IR = 2983, 1726, 1593, 1545, 1519, 1461, 1256, 1216, 1149, 1042, 844, 811; 1H NMR (CDCl3, 400 MHz) δ = 1.48 (t, J = 7.0 Hz, 3H, CH3), 1.50 (t, J = 7.0 Hz, 3H, CH3), 1.79–1.85 (m, 4H, (CH2)2), 2.68–2.73 (m, 4H, N(CH2)2), 3.38 (s, 2H, CH2CO), 4.13 (q, J = 7.0 Hz, 2H, OCH2), 4.17 (q, J = 7.0 Hz, 2H, OCH2), 6.97 (d, J = 8.4 Hz, 1H, 5”-H), 7.12 (dd, J = 8.4, 2.1 Hz, 1H, 6”-H), 7.22 (d, J = 2.1 Hz, 1H, 2”-H), 9.73 (br, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 14.63 (CH3), 14.70 (CH3), 24.04 ((CH2)2), 54.62 (N(CH2)2), 58.76 (CH2), 64.63, 64.88 (2 OCH2), 113.07 (C-5”), 113.13 (C-2”), 117.07 (C-1”), 120.06 (C-6”), 147.54 (C-3’), 148.36 (C-4’), 149.53 (C-3”), 151.07 (C-4”), 168.87 (C=O); HRMS (EI+) calcd for C18H24N4O4: 360.1797; found: 360.1782.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-2-(morpholin-4-yl)acetamide (32): The reaction of amino furazan 9 (249 mg (1.00 mmol)), chloroacetyl chloride (124 mg (1.10 mmol)) and pyridine (89 g (1.13 mmol)) in a mixture of dry diethyl ether (8 mL) and dry benzene (16 mL) gave the raw carboxamide. It was mixed with morpholine (174 mg (2.00 mmol)), K2CO3 (276 mg (2.00 mmol)) and NaI (97 mg (0.65 mmol)) in dry acetonitrile (20 mL), giving the tertiary amine which was purified by column chromatography (silica gel, CH2Cl2/MeOH, 40:1) to yield compound 32 as white amorphous solid (233 mg (62%)). IR = 3326, 2982, 1731, 1595, 1542, 1520, 1462, 1328, 1254, 1216, 1146, 1112, 1041, 1009, 867, 809; 1H NMR (CDCl3, 400 MHz) δ = 1.46–1.53 (m, 6H, 2 CH3), 2.64 (t, J = 4.6 Hz, 4H, N(CH2)2), 3.24 (s, 2H, 2-H), 3.68 (br, t, J = 4.6 Hz, 4H, O(CH2)2), 4.12–4.20 (m, 4H, 2 OCH2), 7.00 (d, J = 8.4 Hz, 1H, 5”-H), 7.11 (dd, J = 8.4, 2.0 Hz, 1H, 6”-H), 7.23 (d, J = 2.0 Hz, 1H, 2”-H), 9.75 (br, s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 14.62 (CH3), 14.71 (CH3), 53.79 (N(CH2)2), 61.76 (C-2), 64.69, 65.01 (2 OCH2), 66.85 (O(CH2)2), 113.05 (C-5”), 113.57 (C-2”), 116.84 (C-1”), 120.00 (C-6”), 147.42 (C-3’), 148.30 (C-4’), 149.65 (C-3”), 151.29 (C-4”), 167.80 (C=O); HRMS (EI+) calcd for C18H24N4O5: 376.1747; found: 376.1743.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-(pyrrolidin-1-yl)propanamide (34): The reaction of amino furazan 9 (249 mg (1.00 mmol)), 3-chloropropanoyl chloride (140 mg (1.10 mmol)) and pyridine (89 g (1.13 mmol)) in a mixture of dry diethyl ether (8 mL) and dry benzene (16 mL) gave the raw carboxamide. It was mixed with pyrrolidine (142 mg (2.00 mmol)), K2CO3 (276 mg (2.00 mmol)) and NaI (97 mg (0.65 mmol)) in dry acetonitrile (20 mL), giving the tertiary amine which was purified by column chromatography (silica gel, CH2Cl2/MeOH, 15:1) to yield compound 34 as yellow amorphous solid (300 mg (80%)). IR = 2977, 1719, 1551, 1476, 1434, 1396, 1323, 1274, 1250, 1217, 1149, 1043, 916, 845; 1H NMR (CDCl3, 400 MHz) δ = 1.46–1.51 (m, 6H, 2 CH3), 1.53–1.57 (m, 4H, (CH2)2), 2.51–2.56 (m, 4H, N(CH2)2), 2.63 (t, J = 5.9 Hz, 2H, 2-H), 2.82 (t, J = 5.9 Hz, 2H, 3-H), 4.10–4.18 (m, 4H, 2 OCH2), 6.96 (d, J = 8.3 Hz, 1H, 5”-H), 7.11 (dd, J = 8.3, 1.8 Hz, 1H, 6”-H), 7.19 (d, J = 1.8 Hz, 1H, 2”-H), 12.02 (br, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 14.68 (CH3), 14.72 (CH3), 23.35 ((CH2)2), 34.06 (C-2), 51.07 (C-3), 53.08 (N(CH2)2), 64.89, 64.65 (2 OCH2), 112.95 (C-5”), 113.50 (C-2”), 117.36 (C-1”), 120.46 (C-6”), 148.42 (C-3’), 148.71 (C-4’), 149.27 (C-3”), 150.88 (C-4”), 170.64 (C=O); HRMS (EI+) calcd for C19H26N4O4: 374.1954; found: 374.1973.
N-(4-(3,4-Diethoxyphenyl)-1,2,5-oxadiazol-3-yl)-3-(morpholin-4-yl)propanamide (35): The reaction of amino furazan 9 (249 mg (1.00 mmol)), 3-chloropropanoyl chloride (140 mg (1.10 mmol)) and pyridine (89 g (1.13 mmol)) in a mixture of dry diethyl ether (8 mL) and dry benzene (16 mL) gave the raw carboxamide. It was mixed with morpholine (174 mg (2.00 mmol)), K2CO3 (276 mg (2.00 mmol)) and NaI (97 mg (0.65 mmol)) in dry acetonitrile (20 mL), giving the tertiary amine which was purified by column chromatography (silica gel, CH2Cl2/MeOH, 40:1) to yield compound 35 as white amorphous solid (223 mg (57%)). IR = 3205, 2978, 1677, 1537, 1472, 1375, 1274, 1215, 1142, 1119, 1042, 870, 814; 1H NMR (CDCl3, 400 MHz) δ = 1.48 (q, J = 6.9 Hz, 6H, 2 CH3), 2.49 (br, t, J = 4.6 Hz, 4H, N(CH2)2), 2.64 (t, J = 5.7 Hz, 2H, 2-H), 2.72 (t, J = 5.7 Hz, 2H, 3-H), 3.36–3.41 (m, 4H, O(CH2)2), 4.10–4.17 (m, 4H, 2 OCH2), 6.96 (d, J = 8.2 Hz, 1H, 5”-H), 7.15 (dd, J = 8.2, 2.1 Hz, 1H, 6”-H), 7.19 (d, J = 2.1 Hz, 1H, 2”-H), 11.28 (br, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 14.62 (CH3), 14.70 (CH3), 31.56 (C-2), 52.70 (N(CH2)2), 53.56 (C-3), 64.63 (OCH2), 64.98 (OCH2), 66.17 (O(CH2)2), 113.09 (C-5”), 113.56 (C-2”), 117.09 (C-1”), 120.83 (C-6”), 148.22 (C-3’), 148.95 (C-4’), 149.28 (C-3”), 151.14 (C-4”), 170.19 (C=O); HRMS (EI+) calcd for C19H26N4O5: 390.1903; found: 390.1892.
3-Methyl-N-(4-phenyl-1,2,5-oxadiazol-3-yl)benzamide (39): Method A: The reaction of compound 38 (161 mg (1.00 mmol)), 3-methylbenzoyl chloride (201 mg (1.30 mmol)) and NaH (60% dispersion in mineral oil) (80 mg (2.00 mmol)) in dry DMF (16 mL) gave the raw carboxamide which was purified by column chromatography (aluminium oxide basic, cyclohexane/ethyl acetate, 3:1) to yield compound 39 as white amorphous solid (75 mg (27%)). IR = 3288, 1663, 1588, 1566, 1521, 1481, 1383, 1284, 885, 807; 1H NMR (CDCl3, 400 MHz) δ = 2.41 (s, 3H, CH3), 7.38 (t, J = 7.5 Hz, 1H, 5-H), 7.42 (d, J = 7.5 Hz, 1H, 4-H), 7.45–7.53 (m, 3H, 3”-H, 4”-H, 5”-H), 7.63 (d, J = 7.5 Hz, 1H, 6-H), 7.69 (s, 1H, 2-H), 7.70 (d, J = 7.5 Hz, 2H, 2”-H, 6”-H), 8.29 (s, 1H, NH); 13C NMR (CDCl3, 100 MHz) δ = 21.32 (CH3), 124.45 (C-6), 125.29 (C-1”), 127.57 (C-2”, C-6”), 128.39 (C-2), 128.88 (C-5), 129.32 (C-3”, C-5”), 130.81 (C-4”), 131.99 (C-1), 133.95 (C-4), 139.13 (C-3), 148.73 (C-3’), 149.94 (C-4’), 165.38 (CO); HRMS (EI+) calcd for C16H13N3O2: 279.1008; found: 279.0997.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}