3.2. Chemical Synthesis
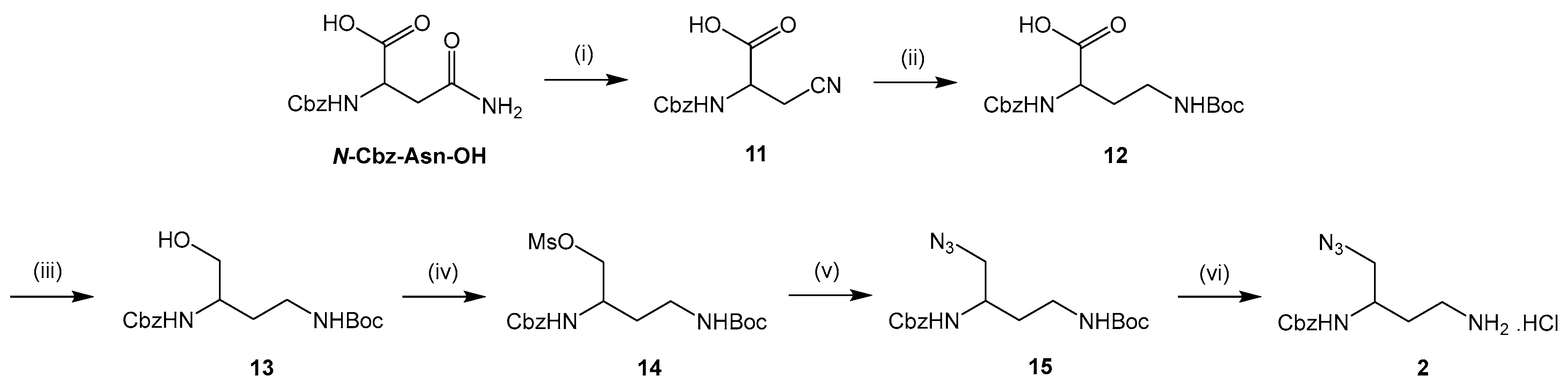
3.2.1. Synthesis of New Diamine Building Block 2
2-(((Benzyloxy)carbonyl)amino)-3-cyanopropanoic acid (11). To a solution of N-Cbz-Asn-OH (10 mmol) in THF (10 mL) cooled at −10 °C were added trifluoroacetic anhydride (1.2 equiv) and pyridine (5 equiv). The mixture was stirred at rt for 18 h. After evaporating THF under reduced pressure, a 1 N NaOH solution (20 mL) was added and pyridine was extracted with Et2O (2 × 20 mL). The aqueous layer was acidified to pH 2 with a 6 N HCl solution and extracted with EtOAc (2 × 20 mL). The organic phase was washed with brine (2 × 20 mL), dried over Na2SO4 and concentrated in vacuo to give compound 11 as a white solid (2.3 g, 92%). mp 145 °C. νmax: 3410, 3331, 3220, 1693, 1533, 1262, 1060, 735, 606 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.38–7.29 (m, 5H), 5.13 (s, 2H), 4.97 (br s, 1H), 4.51 (t, 3J = 6.7 Hz, 1H), 3.08–2.91 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 171.8 (np), 137.9 (np), 129.4 (2C), 128.9, 128.6 (2C), 118.2 (np), 67.7 (np), 51.6, 21.1 (np). HRMS (ESI): m/z calculated for C12H12N2O4Na, [M + Na]+ 271.0695, found 271.0687.
2-(((Benzyloxy)carbonyl)amino)-4-((tert-butoxycarbonyl)amino)butanoic acid (12). To a solution of compound 11 (8 mmol) in dry methanol (60 mL) cooled at 0 °C were added Boc2O (2 equiv) and NiCl2.6H2O (0.1 equiv). NaBH4 (7 equiv) was then introduced in several portions over a period of 45 min. The mixture was stirred at rt for 1 h and diethylenetriamine (1 equiv) was added to quench NiCl2.6H2O. After stirring further for 1 h, methanol was evaporated and the resulting product was extracted with Et2O (2 × 80 mL). The organic phase was washed with a saturated NaHCO3 solution (2 × 80 mL) and brine (2 × 80 mL), dried over Na2SO4 and concentrated in vacuo to give compound 12 as a yellow oil (2.7 g, 96%). νmax: 3314, 2975, 1689, 1515, 1247, 1162, 1046, 741, 623 cm−1. 1H NMR (400 MHz, chloroform-d): δ 8.49 (br s, 1H), 7.34–7.31 (m, 5H), 5.85 (br s, 1H), 5.2 (br s, 1H), 5.09 (s, 2H), 4.40 (m, 1H), 3.38–3.05 (m, 2H), 2.27–2.02 (m, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, chloroform-d): δ 175.0, 158.1, 156.8, 136.3, 128.7 (2C), 128.3 (2C), 128.2, 80.1, 67.2, 51.6, 45.0, 36.3, 28.4 (3C). HRMS (ESI): m/z calculated for C17H24N2O6Na, [M + Na]+ 375.1532, found 375.1519.
Benzyl tert-butyl (4-hydroxybutane-1,3-diyl)dicarbamate (13). To a solution of compound 12 (7.4 mmol) in dry THF (15 mL) cooled at −10 °C were added N-methylmorpholine (1 equiv) and ethyl chloroformate (1 equiv). After stirring for 10 min, NaBH4 (2.7 equiv) was introduced in one portion. The mixture was diluted in methanol (75 mL) and further stirred at 0 °C for 1 h. THF and methanol were evaporated and the resulting product was extracted with EtOAc (2 × 100 mL). The organic phase was washed with a saturated NaHCO3 solution (2 × 100 mL) and brine (2 × 100 mL), dried over Na2SO4 and concentrated in vacuo to give compound 13 as a white pasty solid (1.6 g, 62%) after purification by column chromatography on silica gel (EtOAc/Cyclohexane 60:40). νmax: 3343, 2939, 1679, 1526, 1247, 1166, 1061, 628 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.29–7.27 (m, 5H), 5.38 (br s, 1H), 5.19 (br s, 1H), 5.10 (s, 2H), 3.65–3.49 (m, 3H), 3.23 (br s, 1H), 3.08–3.04 (m, 1H), 2.94–2.86 (m, 1H), 1.67–1.50 (m, 2H), 1.35 (s, 9H). 13C NMR (101 MHz, chloroform-d): δ 156.9, 156.3, 136.4, 128.6 (2C), 128.2, 128.0 (2C), 79.4, 66.9, 64.9, 50.5, 37.2, 32.0, 28.4 (3C). HRMS (ESI): m/z calculated for C17H26N2O5Na, [M + Na]+ 361.1739, found 361.1747.
2-(((Benzyloxy)carbonyl)amino)-4-((tert-butoxycarbonyl)amino)butyl methanesulfonate (14). Compound 13 (4.4 mmol) was dissolved in CH2Cl2 (40 mL) and the solution was cooled at 0 °C. Triethylamine (1.1 equiv) and mesyl chloride (1.1 equiv) were then added. The mixture was stirred at 0 °C for 1 h and the resulting product was extracted with CH2Cl2 (2 × 60 mL). The organic phase was washed with a 0,5 N HCl solution (2 × 60 mL), a 5% NaHCO3 solution (2 × 60 mL) and brine (2 × 60 mL), dried over Na2SO4 and concentrated in vacuo to give compound 14 as a brown oil (1.8 g, 99%). νmax: 3250, 3059, 2947, 1726, 1154, 1037, 938, 772 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.31–7.26 (m, 5H), 5.20 (br s, 1H), 5.03 (s, 2H), 4.93 (br s, 1H), 4.14–4.20 (m, 2H), 3.93–3.89 (m, 1H), 3.31–3.27 (m, 1H), 3.07–3.03 (m, 1H), 2.95 (s, 3H), 1.69–1.55 (m, 2H), 1.36 (s, 9H). 13C NMR (101 MHz, chloroform-d): δ 156.3, 156.1, 136.2, 128.6 (2C), 128.3, 128.1 (2C), 79.5, 71.0, 67.0, 48.0, 37.3, 34.5, 31.8, 28.4 (3C). HRMS (ESI): m/z calculated for C18H28N2O7SNa, [M + Na]+ 439.1515, found 439.1523.
Benzyl tert-butyl (4-azidobutane-1,3-diyl)dicarbamate (15). Compound 14 (3.8 mmol) was dissolved in DMF (5 mL). Sodium azide (2 equiv) was then added and the mixture was heated at 60 °C for 5 h under argon. The solvent was removed and the residue was taken up in EtOAc (3 × 25 mL). The organic phase was washed with brine (3 × 25 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (EtOAc/Cyclohexane 50:50) to give compound 15 as a white pasty solid (1.1 g, 79%). νmax: 3346, 2096, 1680, 1522, 1251, 1162, 1067, 696, 626 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.39–7.33 (m, 5H), 5.20 (br s, 1H), 5.12 (s, 2H), 5.05 (br s, 1H), 3.89–3.86 (m, 1H), 3.49–3.36 (m, 3H), 3.02–2.95 (m, 1H), 1.73–1.56 (m, 2H), 1.45 (s, 9H). Q-DEPT NMR (101 MHz, chloroform-d): δ 156.3 (np), 156.1 (np), 136.3 (np), 128.6 (2C), 128.2, 128.1 (2C), 79.4 (np), 67.1 (np), 54.8 (np), 48.5, 37.0 (np), 32.8 (np), 28.4 (3C). HRMS (ESI): m/z calculated for C17H25N5O4Na, [M + Na]+ 386.1804, found 386.1814.
Benzyl (4-amino-1-azidobutan-2-yl)carbamate hydrochloride (2). To a solution of compound 15 (0.1 mmol) dissolved in 1,4-dioxane (3 mL) was added 4 N HCl solution in 1,4-dioxane (30 equiv). The mixture was stirred at rt for 36 h and concentrated under reduced pressure. The residue was washed with Et2O (2 × 3 mL) that was then evaporated in vacuo to give compound 2 as a white pasty solid (18 mg, 61%). νmax: 3298, 3031, 2939, 2893, 2098, 1692, 1525, 1251, 1048, 698 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.41–7.29 (m, 5H), 5.23 (s, 2H), 3.83–3.79 (m, 1H), 3.60 (br s, 1H), 3.42 (d, 3J = 5.8 Hz, 2H), 3.01–2.94 (m, 2H), 1.93–1.74 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 158.7 (np), 138.0 (np), 129.5 (2C), 129.0, 128.7 (2C), 67.7 (np), 55.3 (np), 50.0, 37.8 (np), 31.2 (np). HRMS (ESI): m/z calculated for C12H18N5O2, [M + H]+ 264.1461, found 264.1453.
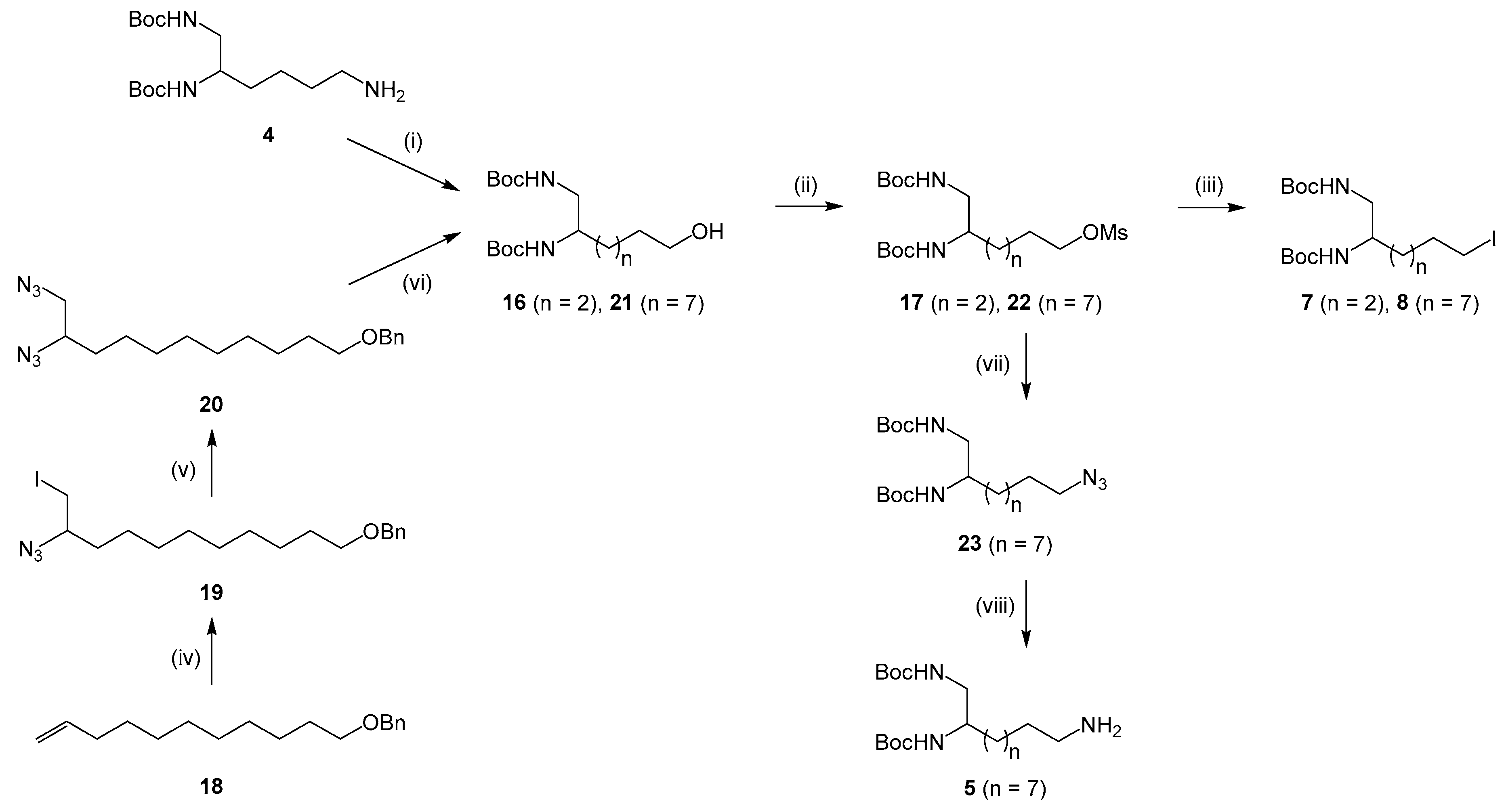
3.2.2. Synthesis of New Diamine Building Blocks 5, 7 and 8
For compound
18, synthetic procedure and NMR spectra were in full accordance with those reported in the literature [
31].
Synthesis of Intermediates 17 and 22
(((10-azido-11-iodoundecyl)oxy)methyl)benzene (19). A solution of iodine chloride (1.3 equiv.) in acetonitrile (10 mL) was added dropwise over 10 min to a stirred suspension of sodium azide (3.3 equiv.) in acetonitrile (10 mL) at −15 °C under argon. The compound 18 (10 mmol) was then added at −15 °C over a period of 20 min and the mixture was warmed to rt. Stirring was continued for 2 h and the mixture was worked up by addition of water (20 mL) and extraction with cyclohexane (2 × 40 mL). The organic layer was washed with a 10% aqueous sodium thiosulfate (2 × 40 mL) and brine (2 × 40 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (EtOAc/Cyclohexane 5:95) to give compound 19 as a yellow oil (3 g, 70%). νmax: 2924, 2852, 2099, 1456, 1272, 1101, 736, 698, 616 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.35–7.30 (m, 4H), 7.30–7.26 (m, 1H), 4.51 (s, 2H), 3.47 (t, 3J = 8.0 Hz, 2H), 3.41–3.36 (m, 1H), 3.30–3.22 (m, 2H), 1.69–1.56 (m, 8H), 1.38–1.36 (m, 6H), 1.27–1.25 (m, 2H). 13C NMR (101 MHz, chloroform-d): δ 139.0, 128.6 (2C), 127.9 (2C), 127.7, 73.1, 70.8, 63.0, 34.7, 30.0 (2C), 29.7, 29.6, 29.4, 26.4, 26.1, 8.8. HRMS (ESI): m/z calculated for C18H28N3ONaI, [M + Na]+ 452.1175, found 452.1174.
(((10,11-diazidoundecyl)oxy)methyl)benzene (20). Compound 19 (6 mmol) was dissolved in DMSO (7 mL) and sodium azide (3 equiv) was added. The mixture was stirred at rt for 20 h under argon and then diluted in EtOAc (3 × 25 mL). The organic phase was washed with brine (3 × 25 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (EtOAc/Cyclohexane 5:95) to give compound 20 as colorless oil (1.6 g, 78%). νmax: 2931, 2856, 2326, 2103, 1717, 1277, 890, 796, 715, 631 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.35–7.30 (m, 4H), 7.30–7.27 (m, 1H), 4.51 (s, 2H), 3.47 (t, 3J = 8.0 Hz, 2H), 3.44–3.28 (m, 3H), 1.65–1.58 (m, 3H), 1.57–1.51 (m, 3H), 1.45–1.43 (m, 2H), 1.38–1.35 (m, 8H). 13C NMR (101 MHz, chloroform-d): δ 139.0, 128.6 (2C), 127.9 (2C), 127.7, 73.1, 70.7, 62.3, 55.1, 32.0, 30.0, 29.7 (2C), 29.6, 29.5, 26.4, 26.1. HRMS (ESI): m/z calculated for C18H28N6ONa, [M + Na]+ 367.2222, found 367.2223.
di-tert-butyl (6-hydroxyhexane-1,2-diyl)dicarbamate (16). Compound 4 (2 mmol) was dissolved in 1,4-dioxane/H2O 1:1 (40 mL) and the mixture was heated to 65 °C. Sodium nitroprusside (2.5 equiv) was added portionwise over a period of 30 min and the reaction mixture was heated for 16 h. After cooling to rt, 1,4-dioxane was removed and the resulting product was extracted with EtOAc (3 × 80 mL). The organic phase was washed with brine (3 × 80 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (EtOAc) to give compound 16 as a white solid (160 mg, 24%). mp 55 °C. νmax: 3363, 2919, 2852, 1682, 1523, 1448, 1367, 1247, 1160, 1071, 1039, 986, 884, 641 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.99 (br s, 1H), 4.77 (br s, 1H), 3.61–3.57 (m, 3H), 3.16–3.12 (m, 2H), 2.29 (br s, 1H), 1.60–1.51 (m, 2H), 1.49–1.44 (m, 4H), 1.40 (s, 18H). 13C NMR (101 MHz, chloroform-d): δ 156.9, 156.5, 79.6 (2C), 62.4, 51.6, 44.8, 32.7, 32.6, 28.6 (6C), 22.2. HRMS (ESI): m/z calculated for C16H32N2O5Na, [M + Na]+ 355.2209, found 355.2216.
di-tert-butyl (11-hydroxyundecane-1,2-diyl)dicarbamate (21). To a solution of compound 20 (4.4 mmol) in dry methanol (50 mL) was added Pd/C (10% w/w) and a 1 N aqueous HCl solution (2 equiv). The mixture was placed under H2 and stirred for 24 h at rt. After filtering the catalyst and evaporating methanol, the residue was dissolved in 1,4-dioxane (20 mL) before adding Boc2O (1.6 equiv) and triethylamine (6.9 equiv). The mixture was stirred for further 2 h at rt. The solvent was removed under reduced pressure and the resulting product was extracted with EtOAc (2 × 40 mL). The organic phase was washed with brine (3 × 40 mL), dried over Na2SO4 and concentrated in vacuo to give compound 21 as a colorless oil (1.3 g, 75%). νmax: 3343, 2926, 2855, 1679, 1522, 1366, 1248, 1163, 1056, 857, 607 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.92 (br s, 1H), 4.63 (br s, 1H), 3.61 (t, 3J = 6.6 Hz, 2H), 3.58–3.50 (m, 1H), 3.16–3.12 (m, 2H), 1.79 (br s, 1H), 1.54 (quint, 3J = 6.6 Hz, 2H), 1.41 (s, 18H), 1.31–1.25 (m, 14H). 13C NMR (101 MHz, chloroform-d): δ 156.8, 156.5, 79.5 (2C), 63.1, 51.5, 45.2, 33.3, 33.0 (2C), 29.6 (3C), 28.6 (6C), 25.9 (2C). HRMS (ESI): m/z calculated for C21H42N2O5Na, [M + Na]+ 425.2991, found 425.2982.
General procedure for the synthesis of mesylates 17 and 22: Compound 16 or 21 (1.4–3 mmol) was dissolved in CH2Cl2 (15–30 mL) and the solution was cooled at 0 °C. Triethylamine (1.1 equiv) and mesyl chloride (1.1 equiv) were then added. The mixture was stirred at 0 °C for 2 h. After evaporating CH2Cl2, the resulting product was extracted with EtOAc (2 × 30–60 mL). The organic phase was washed with a 0,5 N HCl solution (2 × 30–60 mL), a 5% NaHCO3 solution (2 × 30–60 mL) and brine (2 × 30–60 mL), dried over Na2SO4 and concentrated in vacuo to give compound 17 or 22.
5,6-bis((tert-butoxycarbonyl)amino)hexyl methanesulfonate (17). Compound 17 was obtained from compound 16 (1.4 mmol) according to the general procedure as a white solid (455 mg, 80%) after purification by column chromatography on silica gel (CH2Cl2/MeOH 98:2). mp 70 °C. νmax: 3363, 2980, 2936, 1682, 1519, 1443, 1349, 1245, 1162, 976, 828, 626 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.89 (br s, 1H), 4.70 (br s, 1H), 4.19 (t, 3J = 6.4 Hz, 2H), 3.62–3.58 (m, 1H), 3.16–3.12 (m, 2H), 2.98 (s, 3H), 1.78–1.69 (m, 2H), 1.49–1.44 (m, 4H), 1.41 (s, 18H). Q-DEPT NMR (101 MHz, chloroform-d): δ 156.8 (np), 156.4 (np), 79.7 (np, 2C), 69.9 (np), 51.4, 44.8 (np), 37.6, 32.5 (np), 29.1 (np), 28.6 (6C), 22.1 (np). HRMS (ESI): m/z calculated for C17H34N2O7SNa, [M + Na]+ 433.1984, found 433.2000.
10,11-bis((tert-butoxycarbonyl)amino)undecyl methanesulfonate (22). Compound 22 was obtained from compound 21 (3 mmol) according to the general procedure as a white solid (1.3 g, 91%). mp 62 °C. νmax: 3362, 2927, 2857, 1685, 1523, 1352, 1251, 1165, 941, 831, 627 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.87 (br s, 1H), 4.58 (br s, 1H), 4.20 (t, 3J = 6.6 Hz, 2H), 3.60–3.56 (m, 1H), 3.26–3.09 (m, 2H), 2.98 (s, 3H), 1.72 (quint, 3J = 6.6 Hz, 2H), 1.42 (s, 18H), 1.37–1.26 (m, 14H). 13C NMR (101 MHz, chloroform-d): δ 156.7, 156.5, 79.5 (2C), 70.4, 51.5, 45.2, 37.6, 33.2, 29.6 (2C), 29.5, 29.3, 29.2, 28.6 (6C), 26.0, 25.6. HRMS (ESI): m/z calculated for C22H44N2O7SNa, [M + Na]+ 503.2767, found 503.2767.
Synthesis of Precursor 5
di-tert-butyl (11-azidoundecane-1,2-diyl)dicarbamate (23). Compound 22 (1.2 mmol) was dissolved in DMF (8 mL). Sodium azide (2.5 equiv) was then added and the mixture was heated overnight at 60 °C under argon. The solvent was removed and the residue was taken up in EtOAc (3 × 20 mL). The organic phase was washed with brine (3 × 20 mL), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (EtOAc/Cyclohexane 2:98) to give compound 23 as a white solid (420 mg, 82%). mp 56 °C. νmax: 3341, 2975, 2925, 2855, 2091, 1675, 1526, 1457, 1366, 1247, 1163, 880, 755, 634 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.88 (br s, 1H), 4.59 (br s, 1H), 3.60–3.56 (m, 1H), 3.23 (t, 3J = 7.0 Hz, 2H), 3.16–3.12 (m, 2H), 1.56 (quint, 3J = 7.1 Hz, 2H), 1.41 (s, 18H), 1.30-1.23 (m, 14H). Q-DEPT NMR (101 MHz, chloroform-d): δ 156.8 (np), 156.5 (np), 79.5 (np, 2C), 51.7 (np), 51.5, 45.2 (np), 33.3 (np), 29.9 (np), 29.7 (np), 29.6 (np), 29.3 (np), 29.1 (np), 28.6 (6C), 26.9 (np), 26.1 (np). HRMS (ESI): m/z calculated for C21H41N5O4Na, [M + Na]+ 450.3056, found 450.3052.
di-tert-butyl (11-aminoundecane-1,2-diyl)dicarbamate (5). To a solution of compound 23 (0.4 mmol) in dry methanol (20 mL) was added Pd/C (10% w/w). The mixture was placed under H2 and stirred for 2 h at rt. After filtering the catalyst, the methanol was evaporated to give compound 5 as a yellow oil (155 mg, 88%). νmax: 3342, 2976, 2927, 2856, 2090, 1684, 1520, 1365, 1248, 1165, 1069, 732, 630cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.95 (br s, 1H), 4.65 (br s, 1H), 3.58–3.54 (m, 1H), 3.15–3.11 (m, 2H), 2.64 (t, 3J = 7.0 Hz, 1H), 2.57 (t, 3J = 7.4 Hz, 1H), 1.87 (br s, 2H), 1.55–1.42 (m, 2H), 1.40 (s, 18H), 1.30–1.20 (m, 14H). Q-DEPT NMR (101 MHz, chloroform-d): δ 156.8 (np), 156.5 (np), 79.4 (np, 2C), 51.5, 45.1 (np), 42.4 (np), 33.9 (np), 33.2 (np), 29.7 (2C, np), 28.6 (6C), 27.6 (np), 27.1 (np), 26.1 (np), 25.6 (np). HRMS (ESI): m/z calculated for C21H44N3O4, [M + H]+ 402.3332, found 402.3332.
Synthesis of Precursors 7 and 8
General procedure: Compound 17 or 22 (1–1.3 mmol) was dissolved in acetone (15–20 mL) and sodium iodide (5 equiv) was added. The mixture was stirred at rt for 10–16 h. The solvent was removed and the residue was taken up in EtOAc (3 × 30–40 mL). The organic phase was washed with brine (3 × 30–40 mL), dried over Na2SO4 and concentrated in vacuo to give compound 7.
di-tert-butyl (6-iodohexane-1,2-diyl)dicarbamate (7). Compound 7 was obtained from compound 17 (1 mmol) according to the general procedure (16 h) as a white solid (230 mg, 53%). mp 68 °C. νmax: 3359, 2978, 2921, 1680, 1521, 1445, 1366, 1246, 1155, 1049, 880, 635 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.84 (br s, 1H), 4.64 (br s, 1H), 3.63–3.59 (m, 1H), 3.16 (t, 3J = 6.9 Hz, 2H), 1.86–1.77 (m, 2H), 1.58–1.49 (m, 4H), 1.43 (s, 18H), 1.39–1.32 (m, 2H). 13C NMR (101 MHz, chloroform-d): δ 156.8, 156.4, 79.7 (2C), 51.5, 45.0, 33.4, 32.2, 30.0, 28.7 (6C), 27.1. HRMS (ESI): m/z calculated for C16H31N2O4INa, [M + Na]+ 465.1226, found 465.1228.
di-tert-butyl (11-iodoundecane-1,2-diyl)dicarbamate (8). Compound 8 was obtained from compound 22 (1.3 mmol) according to the general procedure (10 h) as a white solid (518 mg, 79%). mp 69 °C. νmax: 3361, 2977, 2923, 2851, 1681, 1522, 1462, 1363, 1245, 1166, 869, 622 cm−1. 1H NMR (400 MHz, chloroform-d): δ 4.93 (br s, 1H), 4.66 (br s, 1H), 3.59–3.55 (m, 1H), 3.13 (t, 3J = 7.2 Hz, 2H), 1.80–1.73 (m, 4H), 1.38 (s, 18H), 1.22–1.15 (m, 14H). 13C NMR (101 MHz, chloroform-d): δ 156.7, 156.4, 79.4 (2C), 51.5, 45.1, 33.7, 33.2, 30.7, 29.6 (2C), 29.5, 28.7 (2C), 28.6 (6C), 26.0. HRMS (ESI): m/z calculated for C21H41N2O4INa, [M + Na]+ 535.2009, found 535.2004.
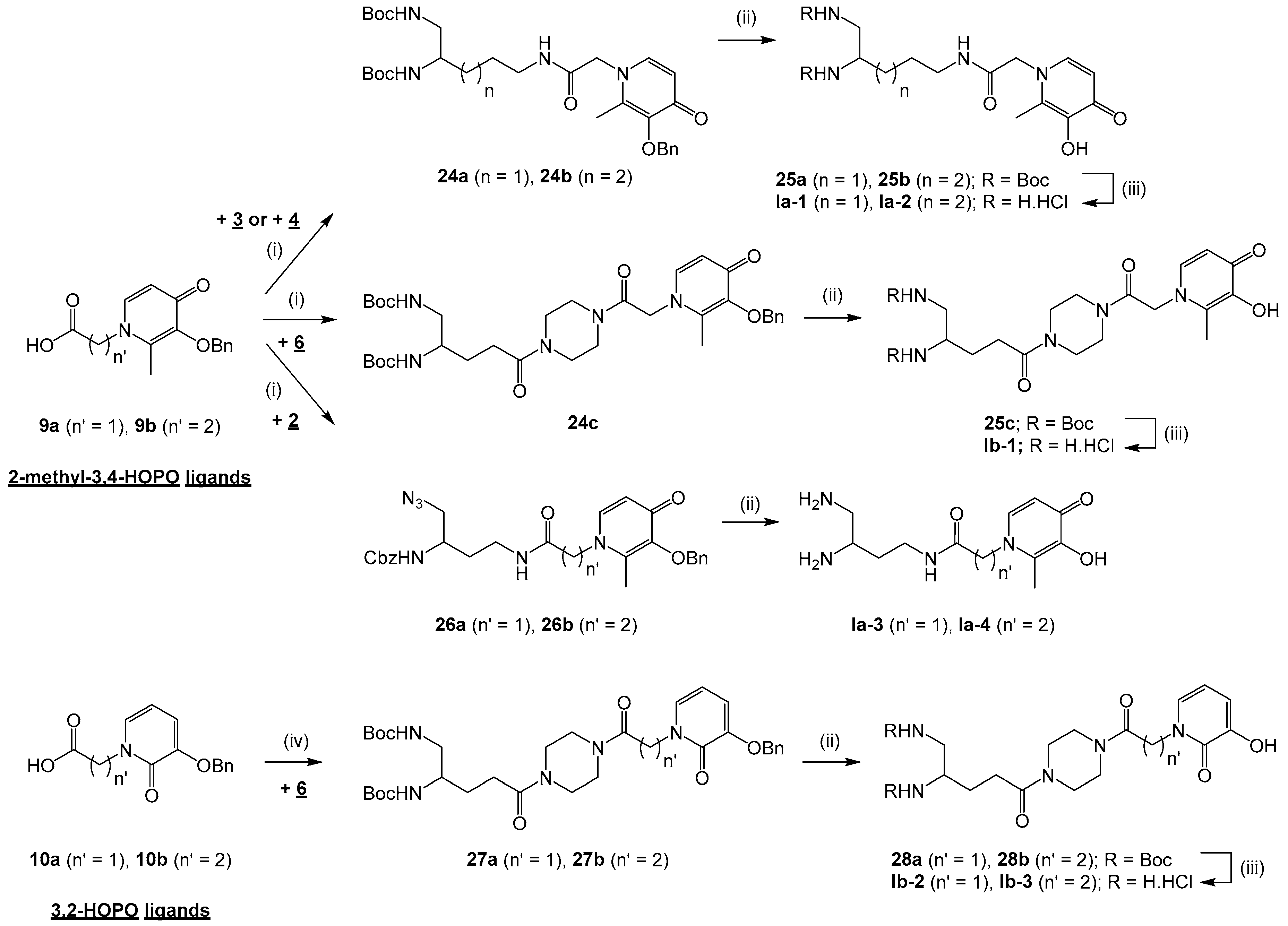
3.2.3. Pseudopeptidic Coupling of Diamine Building Blocks 2–4 and 6 with HOPO Ligands 9a-b and 10a-b in the Series Ia-b
The synthetic routes to prepare compounds
3,
4,
6,
9b,
10b,
27b,
28b and
Ib-3 have already been described by our research group [
8].
For compound
9a and
10a, synthetic pathways and NMR spectra were in full accordance with those reported in the literature [
10,
11].
General procedure for the synthesis of compounds 24a-c and 26a-b: To a solution of compound 9a or 9b (0.7–1.4 mmol) in CH2Cl2, THF or THF/DMF 8:1 to 11:1 (20–45 mL) cooled at 0 °C were added 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC, 1.2–1.4 equiv) and 1-hydroxybenzotriazole monohydrate (HOBt, 1–1.2 equiv). After pre-stirring at 0 °C for 30 min, compound 2, 3, 4 or 6 (1 equiv) and triethylamine (0–1.2 equiv) were added in the mixture that was further stirred at rt for 6–24 h. The solvent was evaporated under reduced pressure and the residue was diluted in EtOAc (2 × 50–100 mL). The organic phase was washed with brine (3 × 50–100 mL) or a 1 N KHCO3 solution (2 × 50–100 mL), a 5% NaHCO3 solution (2 × 50–100 mL) and brine (2 × 50–100 mL), dried over Na2SO4 and concentrated in vacuo to give compound 24a-c or 26a-b.
di-tert-butyl5-(2-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)acetamido)pentane-1,2-diyl)dicarbamate (24a). Compound 24a was obtained by coupling compounds 9a (1.4 mmol) and 3 according to the general procedure (THF/DMF 8:1, 24 h) as an off-white solid (520 mg, 65%) after purification by column chromatography on silica gel (EtOAc/MeOH 80:20). mp 101 °C. 1H NMR (300 MHz, methanol-d4): δ 7.61 (d, 3J = 7.5 Hz, 1H), 7.41–7.31 (m, 5H), 6.46 (d, 3J = 7.5 Hz, 1H), 5.05 (s, 2H), 4.68 (s, 2H), 3.58–3.54 (m, 1H), 3.23–3.18 (m, 2H), 3.12–2.97 (m, 2H), 2.07 (s, 3H), 1.58–1.30 (m, 22H). Q-DEPT NMR (75 MHz, methanol-d4): δ 175.6 (np), 168.5 (np), 163.7 (np), 158.7 (np), 147.3 (np), 145.9 (np), 143.0, 138.8 (np), 130.4 (2C), 129.8 (2C), 129.6, 117.4, 80.4 (np), 80.3 (np), 75.0 (np), 57.2 (np), 52.2, 45.8 (np), 40.9 (np), 31.2 (np), 29.1 (6C), 27.2 (np), 13.3. HRMS (ESI): m/z calculated for C30H44N4O7Na, [M + Na]+ 595.3108, found 595.3121.
di-tert-butyl(6-(2-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)acetamido)hexane-1,2-diyl)dicarbamate (24b). Compound 24b was obtained by coupling compounds 9a (1 mmol) and 4 according to the general procedure (THF/DMF 8:1, 24 h) as an off-white solid (364 mg, 62%) after purification by column chromatography on silica gel (EtOAc/MeOH 80:20). mp 79 °C. νmax: 1680, 1519, 1364, 1249, 1162, 999, 746 cm−1. 1H NMR (300 MHz, methanol-d4): δ 7.62 (d, 3J = 7.2 Hz, 1H), 7.42–7.32 (m, 5H), 6.47 (d, 3J = 7.2 Hz, 1H), 5.06 (s, 2H), 4.68 (s, 2H), 3.56–3.54 (m, 1H), 3.23–3.18 (m, 2H), 3.14–2.98 (m, 2H), 2.08 (s, 3H), 1.49–1.35 (m, 24H). Q-DEPT NMR (75 MHz, methanol-d4): δ 175.3 (np), 168.2 (np), 161.1 (np), 158.7 (np), 147.0 (np), 145.6 (np), 142.7, 138.5 (np), 130.2 (2C), 129.5 (2C), 129.3, 117.1, 80.1 (np), 80.0 (np), 74.7 (np), 56.9 (np), 52.1, 45.5 (np), 40.6 (np), 33.1 (np), 30.1 (np), 28.9 (6C), 24.4 (np), 13.0. HRMS (ESI): m/z calculated for C31H46N4O7Na, [M + Na]+ 609.3264, found 609.3271.
di-tert-butyl(5-(4-(2-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)acetyl)piperazin-1-yl)-5-oxopentane -1,2-diyl)dicarbamate (24c). Compound 24c was obtained by coupling compounds 9a (1 mmol) and 6 according to the general procedure (CH2Cl2, 18 h) as a white solid (348 mg, 51%) after purification by column chromatography on silica gel (CH2Cl2/MeOH 95:5). mp 105 °C. νmax: 3333, 2976, 1698, 1629, 1511, 1434, 1245, 1163, 1018, 742 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.35–7.28 (m, 6H), 6.42 (d, 3J = 7.2 Hz, 1H), 5.17 (s, 2H), 5.03 (s, 2H), 4.83 (br s, 2H), 3.60–3.42 (m, 9H), 3.21–3.17 (m, 2H), 2.43–2.37 (m, 2H), 2.04 (s, 3H), 1.88–1.67 (m, 2H), 1.43 (s, 18H). Q-DEPT NMR (101 MHz, chloroform-d): δ 173.1 (np), 171.4 (np), 164.5 (np), 157.0 (np), 156.3 (np), 145.6 (np), 144.1 (np), 140.8, 137.6 (np), 129.0 (2C), 128.7 (2C), 128.4, 116.3, 79.6 (np, 2C), 73.8 (np), 55.3 (np), 51.6, 45.1 (np), 44.7 (np), 42.5 (np), 41.7 (np), 41.4 (np), 29.7 (np), 28.6 (6C), 28.0 (np), 13.0. HRMS (ESI): m/z calculated for C34H49N5O8Na, [M + Na]+ 678.3479, found 678.3464.
benzyl(1-azido-4-(2-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)acetamido)butan-2-yl)carbamate (26a). Compound 26a was obtained by coupling compounds 9a (0.7 mmol) and 2 according to the general procedure (THF/DMF 11:1, 6 h) as a yellow pasty solid (312 mg, 86%) after purification by column chromatography on silica gel (EtOAc/MeOH 50:50). νmax: 3263, 3034, 2098, 1678, 1626, 1549, 1254, 1064, 738, 698 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.57 (d, 3J = 7.4 Hz, 1H), 7.36 (d, 3J = 7.7 Hz, 2H), 7.31–7.25 (m, 8H), 6.42 (d, 3J = 7.4 Hz, 1H), 5.03 (s, 2H), 5.00 (s, 2H), 4.63 (s, 2H), 3.75–3.72 (m, 1H), 3.31–3.21 (m, 4H), 2.05 (s, 3H), 1.72–1.56 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 173.9 (np), 166.9 (np), 157.2 (np), 145.6 (np), 144.2 (np), 141.5, 137.1 (np), 137.0 (np), 128.7 (3C), 128.1 (2C), 127.9 (2C), 127.6, 127.3 (2C), 115.7, 73.3 (np), 66.2 (np), 55.4 (np), 54.4 (np), 48.8, 35.9 (np), 31.1 (np), 11.9. HRMS (ESI): m/z calculated for C27H30N6O5Na, [M + Na]+ 541.2175, found 541.2186.
benzyl(1-azido-4-(3-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)propanamido)butan-2-yl)carbamate (26b). Compound 26b was obtained by coupling compounds 9b (1.1 mmol) and 2 according to the general procedure (THF, 6 h) as a yellow pasty solid (410 mg, 70%) after purification by column chromatography on silica gel (EtOAc/MeOH 50:50). νmax: 3262, 3063, 2942, 2097, 1623, 1548, 1246, 1062, 735, 698 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.89 (br s, 1H), 7.30–7.24 (m, 10H), 7.17 (d, 3J = 7.5 Hz, 1H), 6.21 (d, 3J = 7.4 Hz, 1H), 5.92 (br s, 1H), 5.04 (s, 2H), 5.01 (s, 2H), 4.00–3.96 (m, 2H), 3.71–3.67 (m, 1H), 3.37–3.32 (m, 2H), 3.30–3.05 (m, 2H), 2.37–2.34 (m, 2H), 2.06 (s, 3H), 1.66–1.63 (m, 2H). Q-DEPT NMR (101 MHz, chloroform-d): δ 173.2 (np), 168.9 (np), 156.4 (np), 145.8 (np), 141.9 (np), 139.1, 137.2 (np), 137.1 (np), 128.9 (2C), 128.5 (3C), 128.4 (2C), 128.2 (2C), 128.1, 116.9, 73.4 (np), 68.8 (np), 54.8 (np), 50.1 (np), 49.1, 36.8 (np), 35.8 (np), 31.7 (np), 12.3. HRMS (ESI): m/z calculated for C28H32N6O5Na, [M + Na]+ 555.2332, found 555.2332.
di-tert-butyl(5-(4-(2-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)acetyl)piperazin-1-yl)-5-oxopentane-1,2-diyl) dicarbamate (27a). To a solution of compound 10a (1.9 mmol) in 1,4-dioxane (25 mL) were added NHS (1.5 equiv) and DCC (1.2 equiv). The mixture was stirred overnight at rt. After filtering off DCC urea and evaporating 1,4-dioxane, the activated intermediate (1.2 mmol) was added with a pre-stirred solution of compound 6 (1.2 equiv) and triethylamine (3 equiv) in CH2Cl2 (25 mL). The mixture was stirred at rt for 48 h and diluted with additional CH2Cl2 (2 × 25 mL). The organic phase was washed with 1 N HCl (2 × 25 mL), saturated NaHCO3 solution (2 × 25 mL) and brine (3 × 25 mL), dried over Na2SO4 and concentrated in vacuo to give compound 27a as a white solid (448 mg, 56%) yield after purification by column chromatography on silica gel (CH2Cl2/MeOH 95:5). mp 97 °C. νmax: 3324, 2978, 2936, 2851, 1697, 1651, 1601, 1436, 1247, 1163, 1046, 741 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.38 (d, 3J = 7.1 Hz, 2H), 7.32 (t, 3J = 7.5 Hz, 2H), 7.26 (t, 3J = 7.8 Hz, 1H), 7.27 (m, 1H), 6.65 (d, 3J = 7.5 Hz, 1H), 6.05 (t, 3J = 7.0 Hz, 1H), 5.06 (s, 2H), 5.03 (br s, 2H), 4.76–4.69 (m, 2H), 3.75–3.41 (m, 9H), 3.17–3.14 (m, 2H), 2.40–2.34 (m, 2H), 1.85–1.63 (m, 2H), 1.39 (s, 18H). Q-DEPT NMR (101 MHz, chloroform-d): δ 171.3 (np), 165.6 (np, 2C), 158.0 (np), 156.1 (np), 148.3 (np), 135.9 (np), 129.6, 128.5 (2C), 127.9, 127.2 (2C), 115.9, 104.8, 79.3 (np, 2C), 70.7 (np), 51.4, 49.2 (np), 45.2 (np), 44.8 (np), 44.4 (np), 42.0 (np), 41.4 (np), 29.7 (np), 28.3 (6C), 27.8 (np). HRMS (ESI): m/z calculated for C33H47N5O8Na, [M + Na]+ 664.3322, found 664.3324.
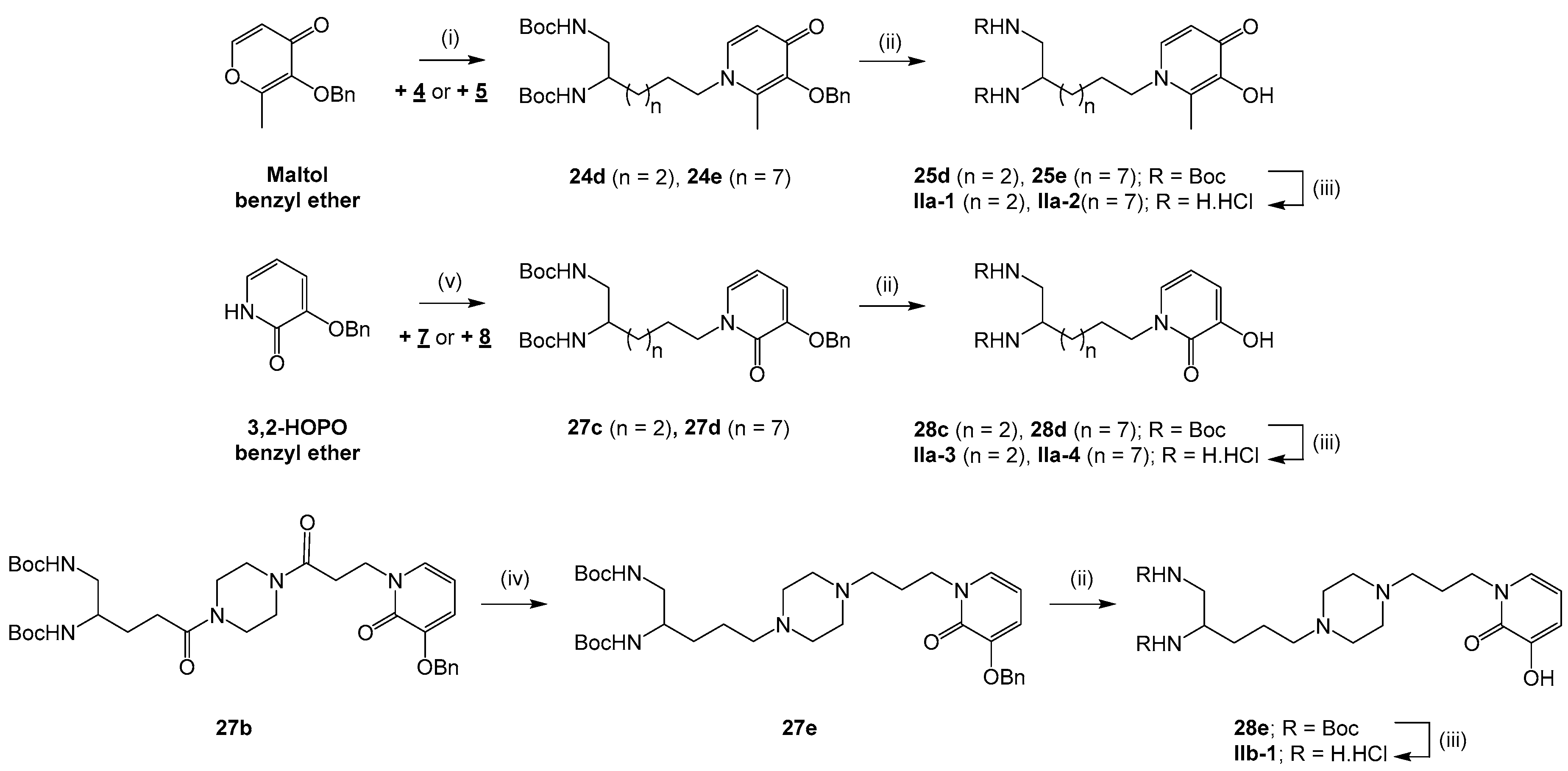
3.2.4. Condensation of Maltol Benzyl Ether with Amino Starting Blocks 4 and 5 in the Series IIa
For maltol benzyl ether, synthetic procedure and NMR spectra were in full accordance with those reported in the literature [
10,
12].
General procedure: To a solution of NaOH (0.5–2.6 equiv) in EtOH/H2O 1:1 to 1:2 (5–120 mL) were added compound 4 or 5 (0.3–3.4 mmol) and maltol benzyl ether (1.1–1.5 equiv). The mixture was heated under reflux for 18–24 h. After evaporating ethanol under reduced pressure, the aqueous phase was neutralized with a 6 N HCl solution and the resulting product was extracted with EtOAc (3 × 20–250 mL). The organic phase was washed with brine (3 × 20–250 mL), dried over Na2SO4 and concentrated in vacuo to give compound 24d or 24e.
di-tert-butyl (6-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)hexane-1,2-diyl)dicarbamate (24d). Compound 24d was obtained by coupling compound 4 (3.4 mmol) and maltol benzyl ether (1.5 equiv) according to the general procedure (NaOH (0.5 equiv), EtOH/H2O 1:1, 24 h) as a yellow pasty solid (1.1 g, 61%) after purification by column chromatography on silica gel (EtOAc/MeOH 90:10). 1H NMR (400 MHz, chloroform-d): δ 7.38 (d, 3J = 6.7 Hz, 2H), 7.32–7.26 (m, 3H), 7.16 (d, 3J = 7.5 Hz, 1H), 6.39 (d, 3J = 7.5 Hz, 1H), 5.03 (s, 2H), 4.94 (br s, 2H), 3.71–3.76 (m, 2H), 3.61–3.57 (m, 1H), 3.163.12 (m, 2H), 2.06 (s, 3H), 1.66–1.55 (m, 2H), 1.40–1.35 (m, 22H). Q-DEPT NMR (101 MHz, chloroform-d): δ 173.5 (np), 156.6 (np, 2C), 146.4 (np), 141.0 (np), 138.3, 137.9 (np), 129.3 (2C), 128.4 (2C), 128.1, 117.5, 79.7 (np, 2C), 73.1 (np), 53.9 (np), 51.2, 44.7 (np), 32.6 (np), 30.9 (np), 28.5 (6C), 23.0 (np), 12.5. HRMS (ESI): m/z calculated for C29H44N3O6, [M + H]+ 530.3230, found 530.3245.
di-tert-butyl(11-(3-(benzyloxy)-2-methyl-4-oxopyridin-1(4H)-yl)undecane-1,2-diyl)dicarbamate (24e). Compound 24e was obtained by coupling compound 5 (0.3 mmol) and maltol benzyl ether (1.1 equiv) according to the general procedure (NaOH (2.6 equiv), EtOH/H2O 1:2, 18 h) as an orange oil (113 mg, 63%) after purification by column chromatography on silica gel (EtOAc/MeOH 90:10). νmax: 2975, 2928, 2856, 1695, 1625, 1514, 1364, 1245, 1164, 1061, 730, 625 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.38 (dd, 3J = 7.6 Hz, 4J = 1.8 Hz, 2H), 7.29–7.26 (m, 3H), 7.16 (d, 3J = 7.5 Hz, 1H), 6.40 (d, 3J = 7.5 Hz, 1H), 5.20 (s, 2H), 4.96 (br s, 1H), 4.70 (br s, 1H), 3.69 (t, 3J = 6.1 Hz, 2H), 3.60–3.56 (m, 1H), 3.17–3.13 (m, 2H), 2.05 (s, 3H), 1.62-1.57 (m, 2H), 1.41 (s, 18H), 1.34–1.23 (m, 14H). 13C NMR (101 MHz, chloroform-d): δ 173.5, 156.8, 156.5, 146.3, 140.8, 138.3, 137.9, 129.4 (2C), 128.4 (2C), 128.2, 117.5, 79.5 (2C), 73.2, 54.1, 51.5, 45.1, 33.2, 31.0, 29.6, 29.5, 29.4, 29.3, 28.6 (6C), 26.5, 26.0, 12.6. HRMS (ESI): m/z calculated for C34H54N3O6, [M + H]+ 600.4013, found 600.4024.
3.2.5. Nucleophilic Substitution of Iodo Starting Blocks 7 and 8 by 3,2-HOPO Benzyl Ether in the Series IIa
For 3,2-HOPO benzyl ether, synthetic procedure and NMR spectra were in full accordance with those reported in the literature [
13].
General procedure: To a solution of 3,2-HOPO benzyl ether (0.3–0.6 mmol) in DMF (2–3 mL) cooled at 0 °C under argon was added a NaH 60% dispersion in mineral oil (1 equiv). After stirring at rt for 20 min, the mixture was cooled back to 0 °C and compound 7 or 8 (1 equiv) was introduced. The mixture was further stirred at rt for 2 h. The reaction was quenched with a saturated aqueous NH4Cl solution (5–10 mL). After evaporating DMF, the resulting product was extracted with EtOAc (3 × 15 mL). The organic phase was washed with brine (3 × 15 mL), dried over Na2SO4 and concentrated in vacuo to give compound 27c or 27d.
di-tert-butyl (6-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)hexane-1,2-diyl)dicarbamate (27c). Compound 27c was obtained by coupling 3,2-HOPO benzyl ether (0.3 mmol) and compound 7 according to the general procedure as a yellow oil (93 mg, 72%) after purification by column chromatography on silica gel (CH2Cl2/MeOH 95:5). νmax: 3316, 2974, 2930, 1697, 1651, 1599, 1514, 1365, 1248, 1169, 1057, 911, 734, 634 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.34 (d, 3J = 7.3 Hz, 2H), 7.26 (t, 3J = 6.1 Hz, 2H), 7.20 (t, 3J = 7.2 Hz, 1H), 6.80 (dd, 3J = 6.9 Hz, 4J = 1.6 Hz, 1H), 6.55 (dd, 3J = 7.4 Hz, 4J = 1.6 Hz, 1H), 5.92 (t, J = 7.1 Hz, 1H), 5.01 (s, 2H), 4.99 (br s, 1H), 4.82 (br s, 1H), 3.86 (t, J = 7.1 Hz, 2H), 3.53–3.49 (m, 1H), 3.09–3.05 (m, 2H), 1.69–1.66 (m, 2H), 1.52–1.43 (m, 2H), 1.33 (s, 18H), 1.32–1.25 (m, 2H). 13C NMR (101 MHz, chloroform-d): δ 158.2, 156.8, 156.4, 149.0, 136.5, 129.1, 128.6 (2C), 128.1, 127.4 (2C), 115.7, 104.8, 79.4 (2C), 70.8, 51.4, 49.6, 44.7, 32.5, 29.1, 28.5 (6C), 23.0. HRMS (ESI): m/z calculated for C28H41N3O6Na, [M + Na]+ 538.2893, found 538.2889.
di-tert-butyl (11-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)undecane-1,2-diyl)dicarbamate (27d). Compound 27d was obtained by coupling 3,2-HOPO benzyl ether (0.6 mmol) and compound 8 according to the general procedure as an off-white solid (232 mg, 62%) after purification by column chromatography on silica gel (EtOAc/Cyclohexane 80:20). mp 72 °C. νmax: 3360, 2980, 2922, 2853, 1682, 1649, 1595, 1525, 1456, 1367, 1239, 1163, 1051, 751, 647 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.41 (d, 3J = 7.3 Hz, 2H), 7.32 (t, 3J = 7.2 Hz, 2H), 7.26 (t, 3J = 7.2 Hz, 1H), 6.86 (dd, 3J = 6.9 Hz, 4J = 1.7 Hz, 1H), 6.61 (dd, 3J = 7.3 Hz, 4J = 1.1 Hz, 1H), 5.98 (t, 3J = 6.7 Hz, 1H), 5.09 (s, 2H), 4.92 (br s, 1H), 4.62 (br s, 1H), 3.92 (t, 3J = 2.8 Hz, 2H), 3.59–3.55 (m, 1H), 3.16–3.12 (m, 2H), 1.74–1.70 (m, 2H), 1.41 (s, 20H), 1.28–1.23 (m, 12H). 13C NMR (101 MHz, chloroform-d): δ 158.3, 156.7, 156.4, 149.1, 136.7, 129.3, 128.7 (2C), 128.1, 127.5 (2C), 115.7, 104.7, 79.4 (2C), 70.9, 50.1, 45.1, 33.2, 29.6, 29.5 (2C), 29.4, 29.3, 28.8, 28.6 (6C), 26.8, 26.0. HRMS (ESI): m/z calculated for C33H51N3O6Na, [M + Na]+ 608.3676, found 608.3682.
3.2.6. Synthesis of Piperazine-1,4-Dialkyl Intermediate 27e from Piperazine-1,4-Diamide Analog 27b in the Series IIb
di-tert-butyl(5-(4-(3-(3-(benzyloxy)-2-oxopyridin-1(2H)-yl)propyl)piperazin-1-yl)pentane-1,2-diyl) dicarbamate (27e). To a solution of compound 27b (1 mmol) in THF (100 mL) was added dropwise a 2 M BH3.Me2S solution (5 equiv) in THF. The mixture was heated under reflux for 2 h. After cooling to rt, the reaction was quenched with methanol (50 mL) and the mixture was further heated under reflux for 18 h. The solvents were evaporated and the residue was taken up in EtOH/1 N NaOH 5:1 (30 mL). After heating under reflux for 2 h, the ethanol was removed under reduced pressure and the aqueous phase was extracted with EtOAc (3 × 50 mL). The organic phase was washed with brine (3 × 50 mL), dried over Na2SO4 and concentrated in vacuo to give compound 27e as a colorless oil (276 mg, 44%) after purification by column chromatography on silica gel (EtOAc/MeOH 50:50). νmax: 3330, 2933, 1691, 1650, 1515, 1364, 1246, 1161, 739 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.40 (d, 3J = 7.3 Hz, 2H), 7.32 (t, 3J = 7.2 Hz, 2H), 7.26 (t, 3J = 7.8 Hz, 1H), 6.93 (d, 3J = 6.8 Hz, 1H), 6.61 (d, 3J = 7.4 Hz, 1H), 5.96 (t, 3J = 7.1 Hz, 1H), 5.27 (s, 2H), 5.25 (br s, 1H), 5.10–5.06 (m, 2H), 4.98 (br s, 1H), 3.99 (t, 3J = 6.8 Hz, 2H), 3.59–3.55 (m, 1H), 3.16–3.12 (m, 2H), 2.43–2.31 (m, 10H), 1.92 (t, 3J = 6.8 Hz, 2H), 1.54–1.52 (m, 2H), 1.40 (m, 20H). Q-DEPT NMR (101 MHz, chloroform-d): δ 158.5 (np, 2C), 149.0 (np), 136.8 (np), 129.8, 128.7 (2C), 128.1, 127.5 (2C), 115.7, 104.5, 79.4 (2C), 70.9 (np), 58.2 (np), 54.8 (np, 2C), 53.4 (np), 53.0 (np), 51.2, 48.2 (np), 45.1 (np), 30.7 (np), 29.9 (np), 28.6 (6C), 26.0 (np), 23.2 (np). HRMS (ESI): m/z calculated for C34H54N5O6, [M + H]+ 628.4074, found 628.4080.
3.2.7. Debenzylation of Hydroxy Group of Derivatives 24a-e and 27a-e and Synthesis of Final Products Ia-3 and Ia-4 by Catalytic Hydrogenation
General procedure: To a solution of compound 24a-e, 27a-e or 26a-b (0.2–1.4 mmol) in dry methanol (5–20 mL) was added Pd/C or Pd(OH)2/C (10% w/w). The mixture was placed under H2 and stirred for 3–48 h at rt. After filtering the catalyst, the methanol was evaporated to give compound 25a-e, 28a-e, Ia-3 or Ia-4.
di-tert-butyl(5-(2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetamido)pentane-1,2-diyl)dicarbamate (25a). Compound 25a was obtained from compound 24a (0.7 mmol) according to the general procedure (Pd/C, 6 h) as a white solid (263 mg, 78%). mp 138 °C. νmax: 3350, 2976, 1682, 1508, 1246, 1162, 666 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.49 (d, 3J = 4.8 Hz, 1H), 6.33 (d, 3J = 4.8 Hz, 1H), 4.70 (s, 2H), 3.53–3.51 (m, 1H), 3.19–3.17 (m, 2H), 3.07–2.91 (m, 2H), 2.26 (s, 3H), 1.561.44 (m, 4H), 1.42 (s, 18H). Q-DEPT NMR (400 MHz, chloroform-d): δ 169.9 (np), 167.1 (np), 157.3 (np), 157.0 (np), 145.5 (np), 139.0, 131.9 (np), 111.1, 78.6 (np, 2C), 55.5 (np), 50.5, 44.1 (np), 39.2 (np), 29.6 (np), 27.4 (6C), 25.4 (np), 10.7. HRMS (ESI): m/z calculated for C23H38N4O7Na, [M + Na]+ 505.2638, found 505.2636.
di-tert-butyl(6-(2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetamido)hexane-1,2-diyl)dicarbamate (25b). Compound 25b was obtained from compound 24b (0.4 mmol) according to the general procedure (Pd/C, 6 h) as a white solid (141 mg, 71%). mp 115 °C. νmax: 3346, 2979, 2934, 1676, 1510, 1245, 1161, 653 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.57 (d, 3J = 4.8 Hz, 1H), 6.41 (d, 3J = 4.8 Hz, 1H), 4.77 (s, 2H), 3.59–3.55 (m, 1H), 3.14–3.11 (m, 2H), 3.02–2.98 (m, 2H), 2.34 (s, 3H), 1.58–1.49 (m, 2H), 1.42–1.38 (m, 20H), 1.39–1.35 (m, 2H). Q-DEPT NMR (400 MHz, chloroform-d): δ 169.9 (np), 167.1 (np), 157.3 (np), 157.0 (np), 145.5 (np), 139.0, 131.9 (np), 111.1, 78.6 (np, 2C), 55.5 (np), 50.7, 44.0 (np), 39.1 (np), 31.6 (np), 28.7 (np), 27.4 (6C), 22.9 (np), 10.7. HRMS (ESI): m/z calculated for C24H40N4O7Na, [M + Na]+ 519.2795, found 519.2789.
di-tert-butyl(5-(4-(2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetyl)piperazin-1-yl)-5-oxopentane -1,2-diyl)dicarbamate (25c) Compound 25c was obtained from compound 24c (0.4 mmol) according to the general procedure (Pd/C, 48 h) as a white solid (218 mg, 100%). mp 96 °C. νmax: 3295, 1631, 1511, 1435, 1363, 1243, 1163, 1017, 753 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.31–7.27 (m, 1H), 6.37–6.33 (m, 1H), 5.98 (br s, 2H), 5.13 (br s, 1H), 4.88 (s, 2H), 3.67–3.43 (m, 9H), 3.16–3.12 (m, 2H), 2.37–2.33 (m, 2H), 2.17 (s, 3H), 1.80–1.63 (m, 2H), 1.40 (s, 18H). Q-DEPT NMR (101 MHz, chloroform-d): δ 171.6 (np), 168.9 (np), 168.6 (np), 164.5 (np), 156.9 (np), 142.8 (np), 138.6, 128.5 (np), 111.5, 79.6 (np, 2C), 55.0 (np), 50.7, 45.2 (np), 44.9 (np), 44.6 (np), 42.1 (np), 41.5 (np), 29.9 (np), 28.4 (6C), 28.1 (np), 12.2. HRMS (ESI): m/z calculated for C27H43N5O8Na, [M + Na]+ 588.3009, found 588.3002.
di-tert-butyl (6-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)hexane-1,2-diyl)dicarbamate (25d). Compound 25d was obtained from compound 24d (1.4 mmol) according to the general procedure (Pd/C, 8 h) as an orange solid (560 mg, 91%). mp 60 °C. νmax: 3318, 2975, 1687, 1506, 1363, 1242, 1161, 1038, 826 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.88–7.84 (m, 1H), 7.03–7.00 (m, 2H), 5.19 (br s, 1H), 5.11 (br s, 1H), 4.21–4.17 (m, 2H), 3.61–3.57 (m, 1H), 3.15–3.11 (m, 2H), 2.51 (s, 3H), 1.84–1.78 (m, 2H), 1.40–1.37 (m, 22H). 13C NMR (101 MHz, chloroform-d): δ 161.5, 157.1, 156.6, 144.5, 138.4, 137.5, 112.3, 79.6 (2C), 56.3, 51.2, 44.6, 32.3, 30.3, 28.6 (6C), 22.8, 12.8. HRMS (ESI): m/z calculated for C22H38N3O6, [M + H]+ 440.2761, found 440.2749.
di-tert-butyl (11-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)undecane-1,2-diyl)dicarbamate (25e). Compound 25e was obtained from compound 24e (0.2 mmol) according to the general procedure (Pd/C, 4 h) as a yellow oil (64 mg, 84%). νmax: 2928, 2855, 1694, 1575, 1507, 1454, 1363, 1242, 1162, 1036, 830, 776, 670, 629 cm−1. 1H NMR (400 MHz, chloroform-d): δ 7.29 (d, 3J = 5.6 Hz, 1H), 6.45 (d, 3J = 7.6 Hz, 1H), 5.49 (br s, 1H), 5.08 (br s, 1H), 4.80 (br s, 1H), 3.88 (t, 3J = 7.9 Hz, 2H), 3.64–3.60 (m, 1H), 3.19–3.15 (m, 2H), 2.40 (s, 3H), 1.74–1.59 (m, 2H), 1.43 (s, 18H), 1.31–1.25 (m, 14H). 13C NMR (101 MHz, chloroform-d): δ 169.0, 156.5, 156.3, 146.4, 136.9, 129.1, 111.5, 79.5 (2C), 54.4, 51.4, 45.0, 33.2, 31.1, 29.5, 29.4 (2C), 29.2, 28.6 (6C), 26.5, 26.0, 12.2. HRMS (ESI): m/z calculated for C27H48N3O6, [M + H]+ 510.3543, found 510.3540.
di-tert-butyl(5-(4-(2-(3-hydroxy-2-oxopyridin-1(2H)-yl)acetyl)piperazin-1-yl)-5-oxopentane-1,2-diyl)dicarbamate (28a). Compound 28a was obtained from compound 27a (0.5 mmol) according to the general procedure (Pd/C, 48 h) as an off-white solid (246 mg, 95%). mp 104 °C. νmax: 3333, 2933, 1648, 1598, 1601, 1439, 1246, 1161, 1027, 871, 740, 607 cm−1. 1H NMR (400 MHz, chloroform-d): δ 6.82–6.78 (m, 2H), 6.18 (t, 3J = 7.1 Hz, 1H), 5.03 (br s, 2H), 4.854.71 (m, 2H), 3.86–3.38 (m, 10H), 3.21–3.15 (m, 2H), 2.46–2.32 (m, 2H), 1.92–1.64 (m, 2H), 1.40 (s, 18H). Q-DEPT NMR (101 MHz, chloroform-d): δ 171.6 (np), 165.4 (np), 158.9 (np), 157.2 (np), 156.6 (np), 146.7 (np), 128.1, 114.9, 107.2, 79.8 (np, 2C), 51.6, 49.9 (np), 45.2 (np), 44.8 (np), 42.5 (np), 41.5 (np), 34.1 (np), 30.0 (np), 28.6 (6C), 28.2 (np). HRMS (ESI): m/z calculated for C26H41N5O8Na, [M + Na]+ 574.2853, found 574.2832.
di-tert-butyl (6-(3-hydroxy-2-oxopyridin-1(2H)-yl)hexane-1,2-diyl)dicarbamate (28c). Compound 28c was obtained from compound 27c (0.2 mmol) according to the general procedure (Pd/C, 3 h) as a yellow pasty solid (56 mg, 66%). νmax: 3348, 2978, 2931, 1680, 1591, 1525, 1243, 1161, 1061, 738, 643 cm−1. 1H NMR (400 MHz, chloroform-d): δ 6.79 (d, 3J = 7.1 Hz, 2H), 6.14 (t, 3J = 7.1 Hz, 1H), 5.02 (br s, 1H), 4.91 (br s, 1H), 4.05–3.92 (m, 2H), 3.60–3.56 (m, 1H), 3.18–3.14 (m, 2H), 1.81–1.74 (m, 2H), 1.73–1.50 (m, 2H), 1.44–1.40 (m, 20H). 13C NMR (101 MHz, chloroform-d): δ 158.9 (2C), 156.5, 146.8, 127.1, 113.8, 107.2, 79.6 (2C), 51.4, 49.4, 45.0, 30.0, 29.4, 28.7 (6C), 22.8. HRMS (ESI): m/z calculated for C21H35N3O6Na, [M + Na]+ 448.2424, found 448.2426.
di-tert-butyl (11-(3-hydroxy-2-oxopyridin-1(2H)-yl)undecane-1,2-diyl)dicarbamate (28d). Compound 28d was obtained from compound 27d (0.3 mmol) according to the general procedure (Pd/C, 3 h) as a brown oil (103 mg, 78%). νmax: 3344, 2926, 2856, 1690, 1596, 1516, 1365, 1245, 1162, 1052, 755 cm−1. 1H NMR (400 MHz, chloroform-d): δ 6.80–6.76 (m, 2H), 6.13 (t, 3J = 7.2 Hz, 1H), 4.95 (br s, 1H), 4.69 (br s, 1H), 3.97–3.93 (m, 2H), 3.60–3.56 (m, 1H), 3.16–3.12 (m, 2H), 1.75–1.71 (m, 2H), 1.43–1.39 (m, 26H), 1.31–1.27 (m, 6H). 13C NMR (101 MHz, chloroform-d): δ 158.7, 156.8, 156.5, 146.8, 127.2, 113.8, 106.9, 79.5 (2C), 51.5, 50.3, 45.2, 33.2, 29.7, 29.6 (2C), 29.5, 29.4, 29.3, 28.6 (6C), 26.7, 26.0. HRMS (ESI): m/z calculated for C26H45N3O6Na, [M + Na]+ 518.3206, found 518.3207.
di-tert-butyl(5-(4-(3-(3-hydroxy-2-oxopyridin-1(2H)-yl)propyl)piperazin-1-yl)pentane-1,2-diyl)dicarbamate (28e). Compound 28e was obtained from compound 27e (0.4 mmol) according to the general procedure (Pd/C, 6 h) as a brown pasty solid (202 mg, 94%). νmax: 3330, 2975, 1686, 1517, 1364, 1246, 1161, 752 cm−1. 1H NMR (400 MHz, chloroform-d): δ 6.83 (d, 3J = 6.8 Hz, 1H), 6.76 (d, 3J = 7.3 Hz, 1H), 6.11 (t, 3J = 7.1 Hz, 1H), 5.17 (br s, 1H), 5.08 (br s, 1H), 4.02 (t, 3J = 6.9 Hz, 2H), 3.58–3.54 (m, 1H), 3.15–3.12 (m, 4H), 2.91–2.83 (m, 6H), 2.57–2.56 (m, 2H), 2.14–2.10 (m, 2H), 1.80–1.78 (m, 2H), 1.39–1.35 (m, 22H). Q-DEPT NMR (101 MHz, chloroform-d): δ 159.0 (np), 157.2 (np), 156.3 (np), 147.0 (np), 127.4, 114.3, 107.1, 79.7 (np, 2C), 56.9 (np), 54.4 (np, 2C), 51.1, 50.2 (np), 48.4 (np, 2C), 44.7 (np), 30.1 (np), 28.6 (6C), 27.3 (np), 25.5 (np), 21.2 (np). HRMS (ESI): m/z calculated for C27H48N5O6, [M + H]+ 538.3605, found 538.3611.
N-(3,4-diaminobutyl)-2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetamide dihydrochloride (Ia-3). Compound Ia-3 was obtained from compound 26a (0.2 mmol) according to the general procedure (Pd/C, 48 h) as a colorless oil (82 mg, 100%). νmax: 3335, 3265, 3210, 3112, 2941, 1658, 1631, 1556, 1500, 1379, 1247, 639 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.54 (d, 3J = 7.0 Hz, 1H), 6.39 (d, 3J = 7.2 Hz, 1H), 4.83–4.75 (m, 2H), 3.40–3.14 (m, 3H), 2.94–2.53 (m, 2H), 2.31 (s, 3H), 1.75–1.47 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 171.3 (np), 168.8 (np), 147.0 (np), 140.3, 133.5 (np), 112.6, 56.9 (np), 50.5, 39.1 (np), 37.6 (np), 35.7 (np), 12.1. HRMS (ESI): m/z calculated for C12H21N4O3, [M + H]+ 269.1614, found 269.1617. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity > 86%.
N-(3,4-diaminobutyl)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)propanamide dihydrochloride (Ia-4). Compound Ia-4 was obtained from compound 26b (0.4 mmol) according to the general procedure (Pd(OH)2/C, 48 h) as a white pasty solid (104 mg, 98%). νmax: 3322, 3244, 3073, 2927, 1626, 1555, 1500, 1353, 1238, 824, 613 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.52 (d, 3J = 7.2 Hz, 1H), 6.32 (d, 3J = 7.2 Hz, 1H), 4.30 (t, 3J = 6.5 Hz, 2H), 3.30–3.12 (m, 3H), 2.63–2.59 (m, 2H), 2.53–2.44 (m, 2H), 2.42 (s, 3H), 1.54–1.25 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 171.8 (np), 171.0 (np), 147.8 (np), 139.0, 132.9 (np), 112.7, 51.3, 48.1 (np), 37.7 (np), 37.1 (np), 35.4 (np), 12.0. HRMS (ESI): m/z calculated for C13H23N4O3, [M + H]+ 283.1770, found 283.1762. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity >77%.
3.2.8. Final N,N’-di-Boc Removal of Compounds 25a-e and 28a-e
General procedure: To a solution of compound 25a-e or 28a-e (0.1–0.4 mmol) dissolved in 1,4–dioxane (2–5 mL) was added a 4 N HCl solution (20 equiv) in 1,4-dioxane. The mixture was stirred at rt for 2–48 h and concentrated under reduced pressure. The residue was washed with Et2O (2 × 2–5 mL) that was then evaporated in vacuo to give compound Ia-1, Ia-2, Ib-1, Ib-2, IIa-1 to IIa-4 or IIb-1 as precipitate.
N-(4,5-diaminopentyl)-2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetamide dihydrochloride (Ia-1). Compound Ia-1 was obtained from compound 25a (0.2 mmol) according to the general procedure (4 h) as a white solid (99 mg, 100%). mp 249 °C. νmax: 3299, 3205, 3056, 2929, 2866, 1678, 1568, 1508, 1340, 1270, 1039, 833, 633 cm−1. 1H NMR (400 MHz, methanol-d4): δ 8.22 (d, 3J = 5.7 Hz, 1H), 7.16 (d, 3J = 5.8 Hz, 1H), 5.29 (s, 2H), 3.66–3.61 (m, 1H), 3.37–3.32 (m, 4H), 2.54 (s, 3H), 1.95–1.60 (m, 4H). Q-DEPT NMR (400 MHz, methanol-d4): δ 167.1 (np), 160.4 (np), 144.8 (np), 144.0 (np), 149.0, 111.4, 59.1 (np), 50.6, 42.1 (np), 39.9 (np), 28.9 (np), 25.8 (np), 13.1. HRMS (ESI): m/z calculated for C13H23N4O3, [M + H]+ 283.1770, found 283.1772. LCMS: tr 0.8 min (98% A/2% B), 190 nm: purity >99%.
N-(5,6-diaminohexyl)-2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)acetamide dihydrochloride (Ia-2). Compound Ia-2 was obtained from compound 25b (0.2 mmol) according to the general procedure (48 h) as a white pasty solid (76 mg, 100%). νmax: 3214, 3044, 2917, 2856, 1667, 1632, 1502, 1338, 1254, 1117, 1034, 869, 614 cm−1. 1H NMR (400 MHz, methanol-d4): δ 8.19 (d, 3J = 6.7 Hz, 1H), 7.16 (d, 3J = 6.6 Hz, 1H), 5.26 (s, 2H), 3.62–3.60 (m, 1H), 3.34–3.29 (m, 4H), 2.52 (s, 3H), 1.90–1.72 (m, 2H), 1.66–1.61 (m, 2H), 1.59–1.53 (m, 2H). Q-DEPT NMR (400 MHz, methanol-d4): δ 166.9 (np), 160.4 (np), 144.8 (np), 144.0 (np), 140. 9, 111.4, 59.2 (np), 50.9, 42.1 (np), 40.2 (np), 31.1 (np), 29.7 (np), 23.2 (np), 13.1. HRMS (ESI): m/z calculated for C14H25N4O3, [M + H]+ 297.1927, found 297.1913. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity > 99%.
1-(2-(4-(4,5-diaminopentanoyl)piperazin-1-yl)-2-oxoethyl)-3-hydroxy-2-methylpyridin-4(1H)-one dihydrochloride (Ib-1). Compound Ib-1 was obtained from compound 25c (0.3 mmol) according to the general procedure (36 h) as a white solid (158 mg, 100%). mp 177 °C. νmax: 3289, 2909, 2854, 2655, 1628, 1441, 1336, 1224, 1117, 1018, 869, 812 cm−1. 1H NMR (400 MHz, water-d2): δ 7.96 (d, 3J = 6.8 Hz, 1H), 7.15 (d, 3J = 6.9 Hz, 1H), 5.54 (s, 2H), 3.85–3.76 (m, 3H), 3.73–3.69 (m, 6H), 3.42–3.40 (m, 2H), 2.81–2.76 (m, 2H), 2.48 (s, 3H), 2.18–2.05 (m, 2H). Q-DEPT NMR (101 MHz, water-d2): δ 172.4 (np), 165.2 (np), 159.4 (np), 143.4 (np), 142.5 (np), 139.9, 111.0, 57.3 (np), 48.9, 44.6 (np), 44.2 (np), 42.1 (np), 41.3 (np), 40.7 (np), 28.4 (np), 25.3 (np), 12.5. HRMS (ESI): m/z calculated for C17H28N5O4, [M + H]+ 366.2141, found 366.2133. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity >71%.
1-(2-(4-(4,5-diaminopentanoyl)piperazin-1-yl)-2-oxoethyl)-3-hydroxypyridin-2(1H)-one dihydrochloride (Ib-2). Compound Ib-2 was obtained from compound 28a (0.3 mmol) according to the general procedure (6 h) as an off-white solid (181 mg, 100%). mp 181 °C. νmax: 3034, 2917, 2853, 1586, 1442, 1216, 1117, 1014, 870, 618 cm−1. 1H NMR (400 MHz, water-d2): δ 7.13–7.07 (m, 2H), 6.46–6.42 (m, 1H), 5.01 (s, 2H), 3.94–3.86 (m, 1H), 3.72–3.52 (m, 8H), 3.34–3.17 (m, 2H), 2.77–2.75 (m, 2H), 2.12–2.07 (m, 2H). Q-DEPT NMR (101 MHz, water-d2): δ 172.3 (np), 167.1 (np), 158.8 (np), 145.4 (np), 130.1, 119.2, 108.3, 51.1 (np), 48.9, 44.6 (np), 44.2 (np), 41.8 (np), 41.4 (np), 40.7 (np), 28.4 (np), 25.3 (np). HRMS (ESI): m/z calculated for C16H26N5O4, [M + H]+ 352,1985, found 352,1981. LCMS: tr 1.0 min (98% A/2% B), 190 nm: purity > 84%.
1-(5,6-diaminohexyl)-3-hydroxy-2-methylpyridin-4(1H)-one dihydrochloride (IIa-1). Compound IIa-1 was obtained from compound 25d (0.4 mmol) according to the general procedure (3 h) as a off-white pasty solid (110 mg, 86%). νmax: 3355, 2973, 2932, 1633, 1509, 1339, 1249, 1163, 1048 cm−1. 1H NMR (400 MHz, methanol-d4): δ 8.25 (d, 3J = 7.0 Hz, 1H), 7.13 (d, 3J = 6.9 Hz, 1H), 4.42 (t, 3J = 7.6 Hz, 2H), 3.68–3.61 (m, 1H), 3.31–3.27 (m, 2H), 2.64 (s, 3H), 1.96–1.77 (m, 4H), 1.62–1.54 (m, 2H). Q-DEPT NMR (101 MHz, methanol-d4): δ 159.5 (np), 145.0 (np), 143.1 (np), 139.3, 111.8, 57.4, 50.6, 42.1, 31.0, 30.7, 22.7, 12.9. HRMS (ESI): m/z calculated for C12H22N3O2, [M + H]+ 240.1712, found 240.1713. LCMS: tr 0.8 min (98% A/2% B), 190 nm: purity >99%.
1-(10,11-diaminoundecyl)-3-hydroxy-2-methylpyridin-4(1H)-one dihydrochloride (IIa-2). Compound IIa-2 was obtained from compound 25e (0.1 mmol) according to the general procedure (18 h) as a brown pasty solid (23 mg, 100%). νmax: 3373, 2925, 2855, 1630, 1506, 1336, 1257, 1032, 715, 635 cm−1. 1H NMR (400 MHz, water-d2): δ 8.05 (m, 1H), 7.12 (m, 1H), 4.35–4.31 (m, 2H), 3.65-3.61 (m, 1H), 3.35–3.34 (m, 2H), 2.61 (s, 3H), 1.85–1.67 (br s, 4H), 1.46–1.42 (m, 2H), 1.36–1.32 (m, 14H). 13C NMR (101 MHz, water-d2): δ 158.5, 142.2, 138.4, 120.0, 110.9, 56.8, 49.4, 40.7, 30.0, 29.4, 28.4, 28.3 (2C), 26.5, 25.3, 24.0, 12.2. HRMS (ESI): m/z calculated for C17H32N3O2, [M + H]+ 310.2495, found 310.2490. LCMS: tr 2.8 min (98% A/2% B), 190 nm: purity >99%.
1-(5,6-diaminohexyl)-3-hydroxypyridin-2(1H)-one dihydrochloride (IIa-3). Compound IIa-3 was obtained from compound 28c (0.1 mmol) according to the general procedure (18 h) as a brown pasty solid (8 mg, 60%). νmax: 3588, 3380, 1648, 1557, 1541, 673 cm−1. 1H NMR (400 MHz, methanol-d4): δ 7.30–7.23 (m, 1H), 6.97–6.93 (m, 1H), 6.46–6.36 (m, 1H), 4.19–4.14 (m, 2H), 3.63–3.59 (m, 1H), 3.30–3.24 (m, 2H), 1.99–1.76 (m, 4H), 1.57–1.45 (m, 2H). HRMS (ESI): m/z calculated for C11H20N3O2, [M + H]+ 226.1556, found 226.1557. LCMS: tr 0.8 min (98% A/2% B), 190 nm: purity >89%.
1-(10,11-diaminoundecyl)-3-hydroxypyridin-2(1H)-one dihydrochloride (IIa-4). Compound IIa-4 was obtained from compound 28d (0.1 mmol) according to the general procedure (18 h) as a brown pasty solid (28 mg, 74%). νmax: 3375, 2923, 2852, 1646, 1579, 1461, 1258, 1049, 750, 645cm−1. 1H NMR (400 MHz, water-d2): δ 7.24–7.20 (m, 1H), 7.07–7.03 (m, 1H), 6.44–6.40 (m, 1H), 4.03–3.99 (m, 2H), 3.66–3.62 (m, 1H), 3.38–3.34 (m, 2H), 1.75–1.71 (br s, 4H), 1.44–1.40 (m, 2H), 1.31–1.27 (m, 14H). 13C NMR (101 MHz, water-d2): δ 158.4, 145.3, 129.5, 118.5, 108.3, 50.4, 49.4, 40.8, 29.9, 28.5, 28.4, 28.3, 28.2, 28.1, 25.6, 24.0. HRMS (ESI): m/z calculated for C16H30N3O2, [M + H]+ 296.2338, found 296.2338. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity >91%.
1-(3-(4-(4,5-diaminopentyl)piperazin-1-yl)propyl)-3-hydroxypyridin-2(1H)-one dihydrochloride (IIb-1). Compound IIb-1 was obtained from compound 28e (0.3 mmol) according to the general procedure (2 h) as a off-white pasty solid (120 mg, 78%). νmax: 3365, 2924, 2852, 1647, 1537, 1270, 1068, 752 cm−1. 1H NMR (400 MHz, dimethylsulfoxide-d6): δ 8.77 (br s, 4H), 7.21 (d, 3J = 6.5 Hz, 1H), 6.72 (dd, 3J = 7.2, 1.7 Hz, 1H), 6.13 (t, 3J = 7.0 Hz, 1H), 4.47–4.11 (m, 2H), 4.04–4.01 (m, 2H), 3.89–3.66 (m, 4H), 3.51–3.41 (m, 4H), 3.39–3.32 (m, 1H), 3.19–3.04 (m, 4H), 2.14–2.11 (m, 2H), 1.97–1.83 (m, 2H), 1.76–1.67 (m, 2H). Q-DEPT NMR (101 MHz, dimethylsulfoxide-d6): δ 157.8 (np), 146.8 (np), 128.0, 115.0, 105.8, 50.2, 46.3 (np, 4C), 46.0 (np, 2C), 39.6 (np, 2C), 25.8 (np), 23.3 (np), 18.8 (np). HRMS (ESI): m/z calculated for C17H32N5O2, [M + H]+ 338.2556, found 338.2552. LCMS: tr 0.9 min (98% A/2% B), 190 nm: purity >86%.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









