Abstract
The chloroplast trnL-F region has been extensively utilized for evolutionary analysis in plants. In the Brassicaceae this fragment contains 1–12 tandemly repeated trnF pseudogene copies in addition to the functional trnF gene. Here we assessed the potential of these highly variable, but complexly evolving duplications, to resolve the population history of the model plant Arabidopsis lyrata. While the region 5’ of the duplications had negligible sequence diversity, extensive variation in pseudogene copy number and nucleotide composition revealed otherwise cryptic population structure in eastern North America. Thus structural changes can be phylogeographically informative when pseudogene evolutionary relationships can be resolved.
1. Introduction
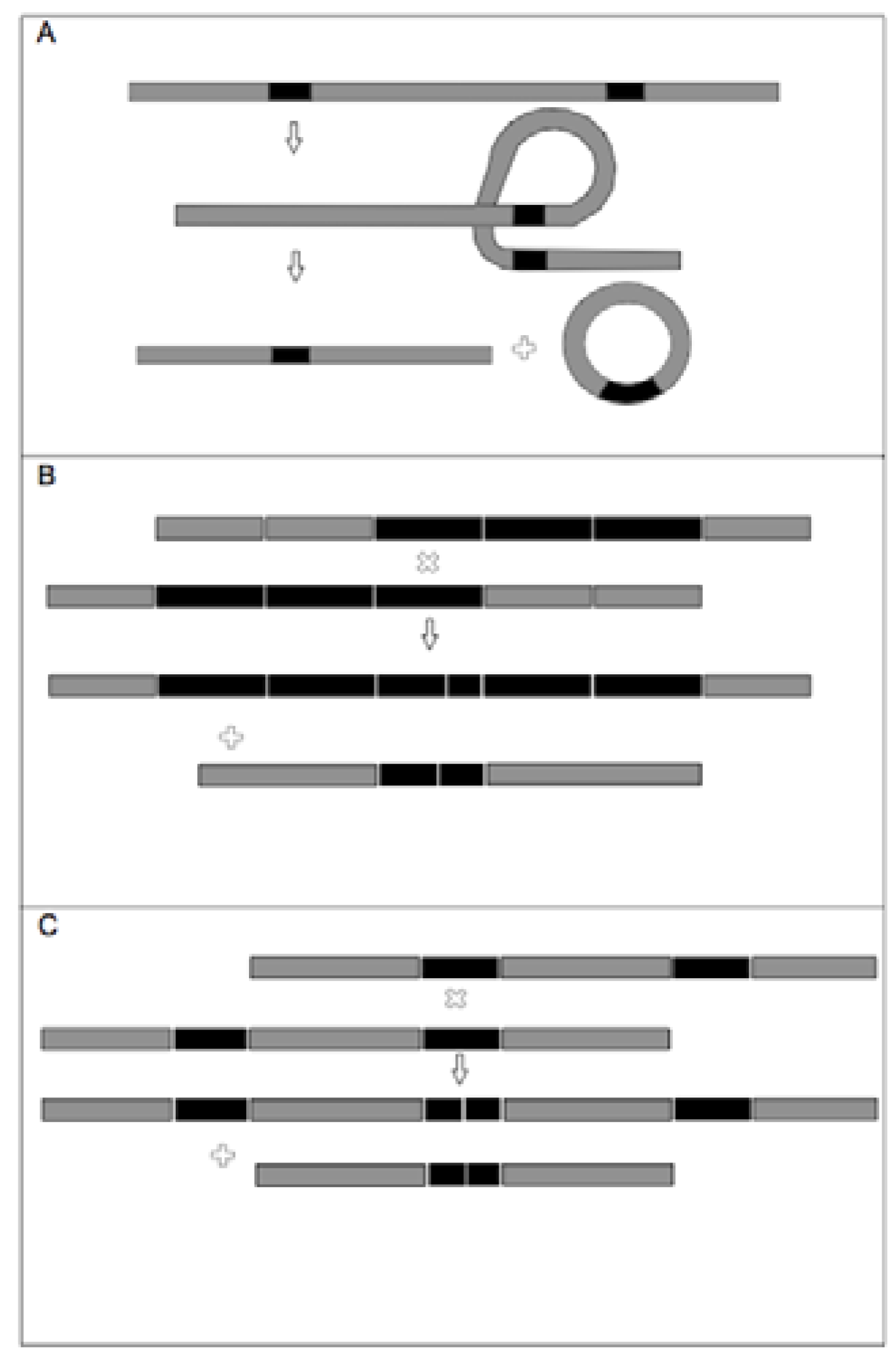
The chloroplast genome has been widely used for phylogenetic studies and its highly conserved nature allows structural changes, in the form of indels and repeat sequences, as well as base substitutions to be considered phylogenetically informative [1,2]. Low base pair substitution rates within the chloroplast genome has lead to the use of indels in population level studies, with small structural changes (< 10 bp) being useful for increasing phylogenetic resolution [3], and increasing the ability to discriminate within species variation [4]. Larger structural changes, and complex structural rearrangements, such as inversions, translocations, loss of repeats and gene duplications are much less common, but have been reported occasionally [5]. However, there is some evidence to suggest that the value of these indel-linked, structural changes is compromised for use in evolutionary studies, due to uncertainty about the underlying mechanisms by which they arise [6]. Two main mechanisms have been suggested to account for large structural changes, such as pseudogene (non functioning duplications of functional genes) formation. (1) Intramolecular recombination between two similar regions on a single double-stranded DNA (dsDNA) molecule can result in the excision of the intermediate region, yielding a shorter dsDNA molecule and a separate circularised dsDNA molecule. It has been suggested that this will be mediated by short repeats formed by slip-strand mispairing [7]. (2) Intermolecular recombination between two genome copies has also been proposed because the plastid contains a large number of copies of the chloroplast genome. Recombination by this method could act to increase pseudogene variation through uneven crossing over [8,9] (Appendix 1).
Since the development of universal primers [10] the non-coding trnL (UAA)-trnF(GAA) intergenic spacer region (trnL-F IGS) of the chloroplast genome has been extensively utilized for plant evolutionary analysis [9,11,12,13,14] (Appendix 2). In the Brassicaceae, the trnL-F region from at least 20 genera has been documented to contain multiple copies of a duplicated trnF pseudogene [11,13,14]. These copies are thought to be non-functional, and are made up of partial trnF gene fragments ranging between c. 50 bp–c. 100 bp in length. The origin of these duplications is uncertain, but Ansell et al. [11] identified two similar motifs that reside around the trnF gene in Brassica (which has no pseudogenes) and these may have functioned as sites for intermolecular recombination. There is evidence of extensive trnL-F fragment length variation in Arabidopsis [13], Boechera [9], Cardamine [15], Rorippa [16] and Lepidium [17], and that the pseudogene duplications serve as a useful lineage marker within the Brassicaceae, as they are absent from many genera, including Brassica, Draba, and Sinapis [13]. Structural mutations within the Brassicaceae trnL-F spacer region have caused the trnF gene copy number to be increased to 12 in certain genera [14], and within the Arabidopsis lyrata complex [13,18,19], the copy number varies between one and six, making this highly variable region attractive for exploring evolutionary histories.
Earlier analyses of Arabidopsis pseudogene variation detected complex patterns of parallel length change among trnF haplotypes [11,12,13] and so focused on the sequence 5’ to the pseudogene copy region (pre-tandem repeat region). This system was used to study the phylogeography of European populations of Arabidopsis lyrata [11,12], which were considered as A. lyrata ssp. petraea [19]. Taxonomy of this genus has subsequently been altered, and now both subspecies are considered as the Arabidopsis lyrata complex [19,20]. However, careful analysis based on dense sampling of related taxa within both the Arabidopsis and Boechera demonstrated that it is possible to reconstruct ancestries and pseudogene copy evolution [9,13], suggesting that the pseudogenic region can increase phylogenetic resolution. This was given further weight by subsequent studies reviewing trnF evolution in cruciferous plants [14] and evolution within the Brassicaceae [21].
Within North American populations, a variation of this approach was used by Hoebe et al. [22], which assessed trnL-F intergenic spacer region variation among 13 populations of A. lyrata sampled from the Great lakes region (which was previously considered as A. lyrata ssp. lyrata [18]). For this approach, the pseudogenic variation was recognized, but the relationship between copies was not addressed, and enabled three fragment length haplotypes to be distinguished among the populations sampled [22]. Based on these data, Hoebe et al. [22] concluded there had been at least two independent colonisations of glaciated areas around the Great Lakes. As part of a more extensive phylogeographic study comparing nuclear and cpDNA markers, the same TrnL-F region was sequenced from eight individuals from each of the populations used by Hoebe et al. [22], plus 11 additional populations sampled from more eastern and northern populations around the Great Lakes, in order to assess population structure in relation to postglacial expansion and mating system history (Foxe et al. Unpublished data). We found that population structure using this marker was congruent to structure elucidated from nuclear DNA markers and micro-satellites. However, while in this study we considered mutations within length variants to define haplotypes, inability to infer relationships between length variants reduced the potential for resolving phylogeographic scenarios based only on cpDNA. The degree of phylogenetic resolution may be improved through more explicit incorporation of pseudogene nucleotide information, as demonstrated by wider studies of the genus [13,14].
Another limitation of the previous studies was that no samples were included from outside of the last glacial maximum. The objectives of this study were to investigate the utility of using variation in pseudogene copy number, as well as point substitutions, to infer phylogeographic patterns and relative diversity in a broader sampling of North American populations of A. lyrata, with European data as a reference. Specifically, we addressed the following questions: (1) do conclusions about relationships between North American and European populations change when different methods of assessing trnF variation are employed? (2) do patterns of diversity in relation to phylogeography within North American populations change when different methods of assessing trnF variation are employed?
2. Materials and Methods
2.1. Samples
Leaf material from 72 diploid plants (six per population) of A. lyrata ssp. lyrata was collected from 12 populations, from localities in eastern North America in 2007 by Yvonne Willi (Table 1). Field-collected tissues were desiccated prior to extraction using silica gel (Drierite, 8 MESH, Acros Organics, New Jersey, USA). These samples were supplemented with sequence data from the 24 populations (eight individuals per population) reported by Foxe et al. (Unpublished data). In addition to this, 35 herbarium samples, generously provided by the Illinois Natural History Survey (INHS), Bell herbarium (University of Minnesota) and the University of Wisconsin-Madison herbarium (Table 1) were also added. Herbarium tissue collection consisted of removing one leaf from each herbarium specimen that was large enough not to destroy the collection. Latitude and longitude data for each sampling location were plotted onto a map using ArcGIS v9.0 (Environmental Systems Research Institute Inc, USA).

Table 1.
Sampled populations, geographic co-ordinates, date collected, collector and trnF haplotypes. Haplotype assignments are based on the P-TR assignment method and the full sequence haplotype method.
| Population name | Abbreviation | Latitude | Longitude | Holmgren list acronym | Number of Individuals | Date collected | Collector | Herbarium (inc. Date) | Full sequence haplotype 1 (Number) | Full sequence haplotype 2 (Number) | P-TR haplotype (Number) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Point Pelee† | PTP | 41.93 | −82.51 | - | 8 | 2003 | BM | - | L1 | - | 1 |
| Iona Marsh* | IOM | 41.30 | −73.98 | - | 8 | 2007 | YW | - | L4 | - | 1 |
| Pictured Rock† | PIR | 46.67 | −86.02 | - | 8 | 2003 | BM | - | L1 | - | 1 |
| Presque Isle† | PRI | 42.17 | −80.07 | - | 8 | 2007 | BM. AT. PH. | - | L4 | - | 1 |
| Long Point† | LPT | 42.58 | −80.39 | - | 8 | 2007 | BM. AT. PH. | - | L1 | - | 1 |
| Indiana Dunes† | IND | 41.62 | −87.21 | - | 8 | 2007 | BM. AT. PH. | - | L1 (4) | L2 (4) | 1 |
| Rondeau† | RON | 42.26 | −81.85 | - | 8 | 2007 | BM. AT. PH. | - | L1 | - | 1 |
| Sleeping Bear dunes† | SBD | 44.94 | −85.87 | - | 8 | 2007 | BM. AT. PH. | - | S3 | - | 1 |
| North Carolina, Mayodan† | NCM | 36.41 | −79.97 | - | 8 | 2007 | DM | - | S2 | - | 1 |
| Tobermoray Alvar† | TSSA | 45.19 | −81.59 | - | 8 | 2007 | BM. AT. PH. | - | S1 (4) | L1 (4) | 1 |
| Lake Superior Park† | LSP | 47.57 | −84.97 | - | 8 | 2003 | BM | - | S1 | - | 1 |
| Tobermory Cliff† | TC | 45.25 | −81.52 | - | 8 | 2007 | BM. AT. PH. | - | S1 | - | 1 |
| Old Woma Bay† | OWB | 47.79 | −84.90 | - | 8 | 2003 | BM | - | S1 | - | 1 |
| Pic River† | PIC | 48.60 | −86.30 | - | 8 | 2003 | BM | - | S1 | - | 1 |
| Pukaskwa National Park† | PUK | 48.40 | −86.19 | - | 8 | 2003 | BM | - | S1 | - | 1 |
| Manitoulin Island† | MAN | 45.67 | −82.26 | - | 8 | 2003 | BM | - | L2 | - | 1 |
| Tobermoray SS† | TSS | 45.19 | −81.58 | - | 8 | 2007 | BM. AT. PH. | - | S1 | - | 1 |
| Pinery† | PIN | 43.27 | −81.83 | - | 8 | 2007 | BM. AT. PH. | - | L1 | - | 1 |
| Beaver Island* | BEI | 45.76 | −85.51 | - | 8 | 2007 | YW | - | S1 | - | 1 |
| Headland Dunes† | HDC | 41.76 | −81.29 | - | 8 | 2007 | BM. AT. PH. | - | L4 | - | 1 |
| Kitty Todd† | KTT | 41.62 | −83.79 | - | 8 | 2007 | BM. AT. PH. | - | L3 | - | 11 |
| Port Cresent† | PCR | 44.00 | −83.07 | - | 8 | 2007 | BM. AT. PH. | - | L2 | - | 1 |
| Wasaga Beach† | WAS | 44.52 | −80.01 | - | 8 | 2003 | BM | - | L4 (6) | L1 (2) | 1 |
| White fish Dunes | WFD | 44.92 | −87.19 | - | 8 | 2007 | BM. AT. PH. | - | L4 (4) | S3 (4) | 1 |
| Indian Ladder* | INL | 42.66 | −74.02 | - | 6 | 2007 | YW | - | S4 | - | 1 |
| Dover Plains* | DOP | 41.74 | −73.58 | - | 6 | 2007 | YW | - | S1 (4) | S4 (2) | 1 |
| Fort Montgomery* | FOM | 41.33 | −73.99 | - | 6 | 2007 | YW | - | L4 | - | 1 |
| Illinois Beach* | ILB | 42.42 | −87.81 | - | 6 | 2007 | YW | - | L2 | - | 1 |
| Apostle Island* | API | 46.94 | −90.74 | - | 6 | 2007 | YW | - | L2 | - | 1 |
| Bete Grise Bay* | BGB | 47.39 | −87.96 | - | 6 | 2007 | YW | - | L1 (4) | L4 (2) | 1 |
| Isle Royal* | ISR | 48.00 | −88.83 | - | 6 | 2007 | YW | - | S1 | - | 1 |
| Ludington* | LUD | 43.96 | −86.45 | - | 6 | 2007 | YW | - | L4 | - | 1 |
| Saugatuck* | SAU | 42.68 | −86.18 | - | 6 | 2007 | YW | - | L2 | - | 1 |
| Friedensville* | FDV | 40.55 | −75.41 | - | 6 | 2007 | YW | - | L4 | - | 1 |
| Fillmore Co. MN | H-FIL | 43.67 | −92.10 | MIN | 3 | - | - | 1977/1941 | L6 (2) | L9 (1) | 1 |
| Houston Co. MN | H-HOU | 43.67 | −92.24 | MIN | 2 | - | - | 1942/1962 | L6 (1) | L8 (1) | 1(1) 12(1) |
| Wabasha Co. MN | H-WAB | 44.28 | −91.77 | MIN | 1 | - | - | 1997 | L6 | - | 1 |
| Winona Co. MN | H-WIN | 43.97 | −91.77 | MIN | 1 | - | - | 1992 | L7 | - | 1 |
| Trempealeau Co. WI | H-TRE | 44.32 | −91.35 | WIS | 2 | - | - | NR | L2 (1) | L7 (1) | 1 |
| Eau Claire, WI | H-EAU | 44.82 | −91.50 | WIS | 2 | - | - | NR | L6 | - | 1 |
| Marquette Co. WI | H-MAR | 43.82 | −89.40 | WIS | 1 | - | - | NR | L6 | - | 1 |
| Richland Co. WI | H-RIC | 43.38 | −90.43 | WIS | 1 | - | - | NR | L6 | - | 1 |
| Sauk Co. WI | H-SAU | 43.45 | −89.95 | WIS | 1 | - | - | NR | L6 | - | 1 |
| Waushara Co. WI | H-WAU | 44.12 | −89.29 | WIS | 1 | - | - | NR | L6 | - | 1 |
| Cass Co. MN | H-CAS | 46.92 | −94.28 | MIN | 6 | - | - | 1992/1997 | L6 (5) | L5 (1) | 1(5) 12(1) |
| Goodhue Co. MN | H-GOH | 44.42 | −92.72 | MIN | 2 | - | - | 1987/1940 | L6 | - | 1 |
| Crow wing Co. MN | H-CRO | 46.47 | −94.08 | MIN | 1 | - | - | 1936 | L6 | - | 1 |
| Anoka Co. MN | H-ANK | 45.25 | −93.25 | MIN | 1 | - | - | 1960 | L6 | - | 1 |
| Hennepin Co. MN | H-HEN | 43.08 | −92.24 | MIN | 1 | - | - | 1922 | L6 | - | 1 |
| Sheboygan Co. WI | H-SHE | 43.73 | −87.93 | WIS | 1 | - | - | NR | L5 | - | 12 |
| Wadena Co. MN | H-WAA | 46.58 | −94.97 | MIN | 1 | - | - | 1992 | L5 | - | 12 |
| Oconto Co. WI | H-OCO | 45.00 | −88.18 | WIS | 3 | - | - | NR | L2 | - | 1 |
| Washington Co. MN | H-WAH | 45.03 | −92.92 | MIN | 1 | - | - | 1961 | L2 | - | 1 |
| Morrison CO. MN | H-MOR | 46.02 | −94.30 | MIN | 1 | - | - | 1990 | L2 | - | 1 |
| Cook Co. MN | H-COK | 47.92 | −90.55 | MIN | 1 | - | - | 1980 | L2 | - | 1 |
| Milwaukee Co. WI | H-MIL | 43.00 | −87.97 | WIS | 1 | - | - | NR | L1 | - | 1 |
* New populations described in this paper and collected by Yvonne Willi. † Data taken from Foxe et al. (unpublished).
2.2. Sequencing
Total genomic DNA was either extracted from 100 mg of desiccated leaf tissue using the FastDNA kit (QBiogene101, MP Biomedicals) or by DNeasy kits (Qiagen Inc) and staff of the Genome laboratory at the John Innes Center (Norwich, UK). The trnL(UAA)–trnF(GAA) IGS and trnF gene region was amplified by PCR using the 'E' and 'F' primers of Taberlet et al. [10]. Amplifications were performed using HotStar Taq polymerase (Qiagen Inc.) under the following conditions: 94 °C for 2 min, followed by 28 cycles of 94 °C for 30 s, 50 °C for 30 s, 72 °C for 30 s, and one cycle of 72 °C for 5 min. PCR products were visualized with ethidium bromide under UV, and the bands were excised and purified with QiaQuick gel extraction kits (Qiagen Inc.). Products were sequenced on an ABI 3730 sequencer (by The Sequencing Service, University of Dundee, and The Gene Pool, University of Edinburgh). In order to compare diversity in North American populations to more extensively studied populations in Europe, we also utilised the sequence data published for 42 diploid European populations of Arabidopsis lyrata; Genbank numbers DQ989814–DQ989862 and GU456721-GU456722 [11] (Appendix 3).
2.3. TrnF Sequence Alignment and Pseudogene Recognition
Sequences were manually aligned using Geneious 3.7 (Biomatters Ltd). Sequences containing the maximum number of tandem repeat copies detected (six for A. lyrata) were initially aligned, allowing for shorter variants to be subsequently aligned by the addition of gaps. Manual checks of the trnF pseudogene tandem repeats showed that no additional ‘F’ primer annealing sites were present, and all sequences contained at least 40 bases of the functional trnF gene at the 3’ end of the sequence. This allowed for the identification of the complete tandem repeat region (TR), so to reliably verify the total number of pseudogene copies. Pseudogene recognition followed the protocol outlined by Ansell et al. [11]. A slightly different approach was used by Koch et al. [13] and related studies.
2.4. TrnF Sequence Variation Based Only On the P-TR Region
Pre-tandem repeat (P-TR) haplotypes (alignment positions 1–180 bp) were initially assigned according to the method described in Ansell et al. [11]. The pre-tandem repeat region is shared throughout a number of species [21], including taxa that lack the pseudogene duplications (i.e., Brassica nigra). New P-TR haplotypes not found in previous studies were assigned a haplotype number sequentially, following on from those previously described. A minimum spanning network (MSN), including both published European sequences and our North American sequences, was created using the minimum number of differences between P-TR haplotype pairs (Arlequin 3.11) [23]. A haplotype network was also constructed according to the parsimony-based algorithm developed by Templeton et al. [24], as implemented in the program TCS 1.13 [25].
2.5. TrnL-F Full Sequence Variation Based Only On Mutations within Length Variants
The diversity of length variations in European and North American samples were compared. Our previous studies of North American populations [22], did not attempted to infer evolutionary relationships between long and short fragment length haplotypes because only mutations within length variants were considered. Using this approach, full sequences (i.e., including both the P-TR and tandem repeat regions) of equal length were aligned using Sequencher 4.7 (Gene Codes, Inc.). A full sequence (FS) haplotype network was constructed according to the parsimony-based algorithm developed by Templeton et al. [24], as implemented in the program TCS 1.13 [25].
2.6. TrnL-F Full Sequence Variation Considering Evolution of Copy Number Variants
In order to infer relationships among length variants by considering pseudogene copy number variation, as well as point substitutions and to allow comparison between European and North American populations, a new method of haplotype assignment was employed. This was achieved by: (1) assigning a number to each pseudogene copy in the tandem repeat region and scoring the presence or absence of said copy in a particular sequence; (2) dissecting each pseudogene copy present from the aligned sequence (following the alignment given in Table 2); (3) scoring base pair substitutions within a copy in sequential order starting at the 5’ end; (4) assigning a unique variant to the combination of copy number and point substitutions for each pseudogene copy (Table 3); (5) combining each copy variant within a sequence, with the haplotype assignments based on the pre-tandem repeat region to define a unique haplotype for each sequence type. For example, FS haplotype L1 is comprised of six pseudogene copies with the following variants: copy 1, variant 1 (referred to as 1.1 in Table 3), along with 2.1, 6.6, 7.1, 9.1 and 10.10. This, along with its P-TR haplotype (in this case, haplotype 1; Table 3), makes up its FSPS haplotype. The probability of length changes taking place in tandem repeats increases with increasing numbers of repeats [26], and consequently copy losses are more likely than gains when long TA exist, hence we consider a six copied state as more-likely ancestral. Tandem arrays with three or six copies appear to be the most common across a variety of species [9,11,12,14,21].
Table 2.
Criteria for trnF pseudogene alignment used in this study. P-TR refers to all sequence before the tandem repeat region (pseudogene copies). Origin species is the species in which certain pseudogene copies are found.
| Psuedogene copy number | Present in A.lyrata | Species* | Size (bp) | Start position (bp) |
| P-TR | Y | Unknown | 179 | 1 |
| 1 | Y | A. lyrata | 93 | 180 |
| 2 | Y | A. lyrata | 77 | 273 |
| 3 | N | A.thaliana | 32 | 350 |
| 4 | N | A.halleri | 100 | 382 |
| 5 | N | A.halleri | 98 | 480 |
| 6 | Y | A. lyrata | 99 | 579 |
| 7 | Y | A. lyrata | 67 | 646 |
| 8 | N | A.halleri | 67 | 713 |
| 9 | Y | A. lyrata | 68 | 781 |
| 10 | Y | A. lyrata | 125 | 906 |
| trnF gene | Y | Unknown | 1,032 |
* Refers to the species this pseudogene was identified in.
Table 3.
A. thaliana, European A. lyrata (PET), North American A. lyrata (L1–L9 & S1–S4) trnL-trnF IGS sequences and their trnF pseudogene composition. Numbers refer to the pseudogene variant present at each copy, for each sequence. Variants represent variation by one or more point mutations from the common sequence (which is given the prefix ‘x.1’; i.e., in pseudogene copy 2, the common sequence type is 2.1) A single variant per pseudogene copy is given to each sequence. A. thaliana is given its own variant for each copy labelled AT.
| Species & Haplotype | P-TR Haplotype | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Arabidopsis lyrata ssp. Petraea | |||||||||||
| PET 1A | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| PET IB | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| PET 1C | 2 | 1.6 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| PET 2 | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.2 |
| PET 3 | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.3 |
| PET 4 | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| PET 5 | 2 | 1.6 | 2.1 | – | – | – | 6.3 | 7.1 | – | 9.1 | 10.1 |
| PET 6 | 2 | 1.1 | 2.1 | – | – | – | 6.1 | 7.3 | – | – | – |
| PET 7 | 2 | 1.2 | – | – | – | – | – | – | – | – | – |
| PET 8 | 2 | 1.3 | – | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| PET 9 | 2 | 1.4 | – | – | – | – | – | – | – | 9.1 | 10.1 |
| PET 10 | 2 | 1.1 | – | – | – | – | 6.1 | – | – | 9.2 | 10.1 |
| PET 11 | 2 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.7 |
| PET 12 | 2 | 1.1 | 2.1 | – | – | – | 6.2 | – | – | 9.1 | 10.3 |
| PET 13 | 2 | 1.1 | 2.1 | – | – | – | – | – | – | – | 10.5 |
| PET 14 | 2 | 1.6 | 2.1 | – | – | – | 6.1 | 7.4 | – | – | – |
| PET 15 | 2 | 1.3 | – | – | – | – | – | – | – | – | 10.7 |
| PET 16 | 2 | 1.1 | 2.1 | – | – | – | – | – | – | – | 10.7 |
| PET 17 | 1 | 1.1 | – | – | – | – | – | – | – | – | – |
| PET 18 | 3 | 1.1 | 2.3 | – | – | – | 6.3 | – | – | 9.3 | 10.4 |
| PET 19A | 3 | 1.1 | 2.3 | – | – | – | – | – | – | – | 10.6 |
| PET 19B | 6 | 1.1 | 2.3 | – | – | – | – | – | – | – | 10.8 |
| PET 20 | 5 | 1.5 | 2.1 | – | – | – | 6.1 | 7.2 | – | 9.1 | 10.4 |
| PET 21A | 6 | 1.6 | 2.1 | – | – | – | 6.4 | 7.5 | – | 9.1 | 10.4 |
| PET 21B | 6 | 1.6 | 2.1 | – | – | – | 6.4 | 7.7 | – | 9.1 | 10.4 |
| PET 21C | 6 | 1.6 | 2.1 | – | – | – | 6.5 | 7.7 | – | 9.1 | 10.4 |
| PET 21D | 7 | 1.6 | 2.1 | – | – | – | 6.4 | 7.7 | – | 9.1 | 10.4 |
| PET 21E | 10 | 1.6 | 2.1 | – | – | – | 6.5 | 7.7 | – | 9.1 | 10.4 |
| PET 22 | 6 | 1.1 | 2.1 | – | – | – | 6.4 | 7.7 | – | 9.1 | 10.4 |
| PET 23 | 6 | 1.6 | 2.4 | – | – | – | 6.4 | 7.6 | – | 9.1 | 10.4 |
| PET 24 | 6 | 1.1 | – | – | – | – | 6.4 | 7.7 | – | 9.1 | 10.4 |
| PET 25 | 6 | 1.1 | – | – | – | – | 6.4 | 7.7 | – | 9.1 | 10.4 |
| PET 26 | 6 | 1.1 | – | – | – | – | 6.4 | 7.6 | – | 9.1 | 10.4 |
| PET 27 | 6 | 1.7 | – | – | – | – | – | – | – | 9.1 | 10.4 |
| PET 28 | 6 | 1.6 | 2.1 | – | – | – | 6.4 | 7.8 | – | – | – |
| PET 29 | 6 | 1.6 | 2.4 | – | – | – | 6.4 | 7.8 | – | – | – |
| PET 30 | 6 | 1.1 | 2.1 | – | – | – | 6.4 | 7.8 | – | – | – |
| PET 31A | 6 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.8 |
| PET 31B | 8 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.8 |
| PET 31C | 4 | 1.8 | 2.1 | – | – | – | – | – | – | – | 10.7 |
| PET 31D | 9 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.8 |
| PET 31E | 6 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.9 |
| PET 31F | 6 | 1.6 | 2.1 | – | – | – | – | – | – | – | 10.8 |
| PET 32 | 6 | 1.6 | 2.4 | – | – | – | – | – | – | – | 10.9 |
| PET 33 | 6 | 1.9 | – | – | – | – | – | – | – | – | – |
| PET 34 | 6 | 1.9 | – | – | – | – | – | – | – | – | – |
| Arabidopsis lyrata ssp. lyrata | |||||||||||
| L1 | 1 | 1.1 | 2.1 | – | – | – | 6.6 | 7.1 | – | 9.1 | 10.10 |
| L2 | 1 | 1.2 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.10 |
| L3 | 11 | 1.1 | 2.1 | – | – | – | 6.6 | 7.1 | – | 9.1 | 10.10 |
| L4 | 1 | 1.1 | 2.1 | – | – | – | 6.1 | 7.1 | – | 9.1 | 10.1 |
| L5 | 12 | 1.6 | 2.1 | 6.1 | 7.1 | 9.1 | 10.10 | ||||
| L6 | 1 | 1.1 | 2.1 | 6.1 | 7.1 | 9.1 | 10.10 | ||||
| L7 | 1 | 1.11 | 2.1 | 6.1 | 7.1 | 9.1 | 10.1 | ||||
| L8 | 12 | 1.6 | 2.6 | 6.1 | 7.1 | 9.1 | 10.10 | ||||
| L9 | 1 | 1.1 | 2.1 | 6.1 | 7.1 | 9.4 | 10.10 | ||||
| S1 | 1 | 1.1 | 2.1 | – | – | – | 6.7 | – | – | – | – |
| S2 | 1 | 1.4 | – | – | – | – | – | – | – | 9.1 | 10.1 |
| S3 | 1 | 1.4 | – | – | – | – | – | – | – | 9.1 | 10.10 |
| S4 | 1 | 1.1 | 2.1 | – | – | – | 6.1 | – | – | – | – |
| Arabidopsis thaliana | |||||||||||
| A.thaliana | 0 | 1.AT | 2.AT | 3.AT | – | – | 6.AT | – | – | 9.AT | 10.AT |
Each full sequence haplotype was then superimposed onto the P-TR haplotype network described above. Each pseudogene copy a particular haplotype contains is then coloured based on its copy number variant (essentially, a pseudogene copy haplotype for each pseudogene copy), allowing similarities between full sequence haplotypes to be visually assessed. This allows a visual representation of haplotypes that have copy number variation, and the associated length variation this brings, which also allows inferences about parallel independent copy number evolution between continents.
3. Results
3.1. Sequence Variation Based On the P-TR Region
Our detailed study of eastern North American A. lyrata ssp. lyrata populations identified three variable positions in the region 5’ to the tandem repeat pseudogenes, allowing the discrimination of three distinct haplotypes (Table 4, Figure 2), two of which are newly reported here (P-TR11, P-TR12). P-TR 11 and 12 differ from the previously reported P-TR 1 [11] by two alternative single point mutations (Table 4). In total, 12 distinct P-TR haplotypes were recognised when our new data was combined with the existing European study of Ansell et al. [11,12] and the resulting minimum spanning network had two star-shaped clades (Figure 1), with all eastern North American samples restricted to clade 1. In European the P-TR 1 was found from a single population [11] at Vöslauer Hütte in Austria.
Of the three P-TR haplotypes recovered in eastern North America, P-TR 1 was the most common, detected from 96% of samples from 54/57 populations (including herbarium sample regions) (Figure 1, Figure 2a). P-TR 11 was confined to the all eight individuals sequenced in the Kitty Todd population (KTT) to the west of Lake Erie (Figure 2b), while P-TR 12 was present in four herbarium samples, including one individual in Houston county MN (H-HOU), one individual in Wadena county MN (H-WAA), one individual in Sheboygan county WI (H-SHE) and a final individual in Cass county MN (H-CAS). All other individuals contained P-TR haplotype 1. Thus, on the basis of this conservative analysis of trnL-F nucleotide variation, there is little genetic diversity to resolve the phylogeographic history of these populations (Figure 1).
Table 4.
Pre-tandem repeat (P-TR) haplotype alignment criteria.
| Position of trnL - trnF alignment | ||||||||||||
| Haplotype | 46 | 47 | Indel A | 88 | 96 | 101 | 111 | 119 | 136 | 154 | 156 | 167 |
| 0 | A | T | − | C | A | T | G | C | A | T | C | C |
| 1 | A | A | − | C | A | T | G | C | A | T | C | C |
| 2 | A | A | + | C | A | T | G | C | A | T | C | C |
| 3 | A | A | − | C | T | T | G | C | A | T | C | C |
| 4 | A | A | − | C | A | T | G | C | A | T | A | C |
| 5 | A | A | − | C | A | T | G | C | A | G | C | C |
| 6 | A | A | − | T | A | T | G | C | A | T | C | C |
| 7 | A | A | − | T | A | T | G | C | C | T | C | C |
| 8 | A | A | − | T | A | T | G | T | A | T | C | C |
| 9 | G | A | − | T | A | T | G | C | A | T | C | C |
| 10 | A | A | − | T | A | T | G | C | A | T | C | C |
| 11* | A | A | − | T | A | T | T | C | A | T | C | T |
| 12* | A | A | − | T | A | A | G | C | A | T | C | C |
* Indicates new P-TR haplotypes described in this study.
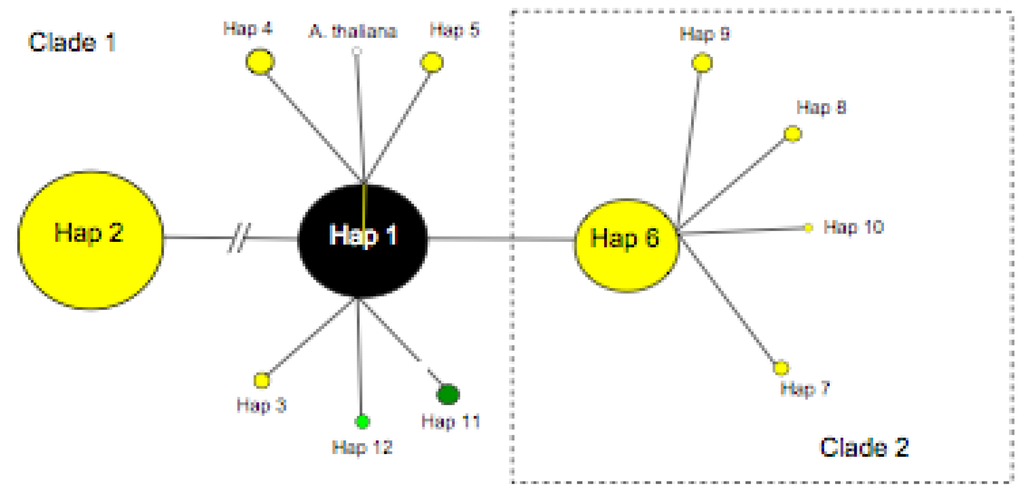
Figure 1.
Arabidopsis lyrata pre-tandem repeat (P-TR) haplotype network including all North American (coloured black, light green and green) and European variants (coloured yellow). A. thaliana P-TR haplotype (open circle) included for reference. Circle size is proportional to haplotype frequency.
Figure 1.
Arabidopsis lyrata pre-tandem repeat (P-TR) haplotype network including all North American (coloured black, light green and green) and European variants (coloured yellow). A. thaliana P-TR haplotype (open circle) included for reference. Circle size is proportional to haplotype frequency.
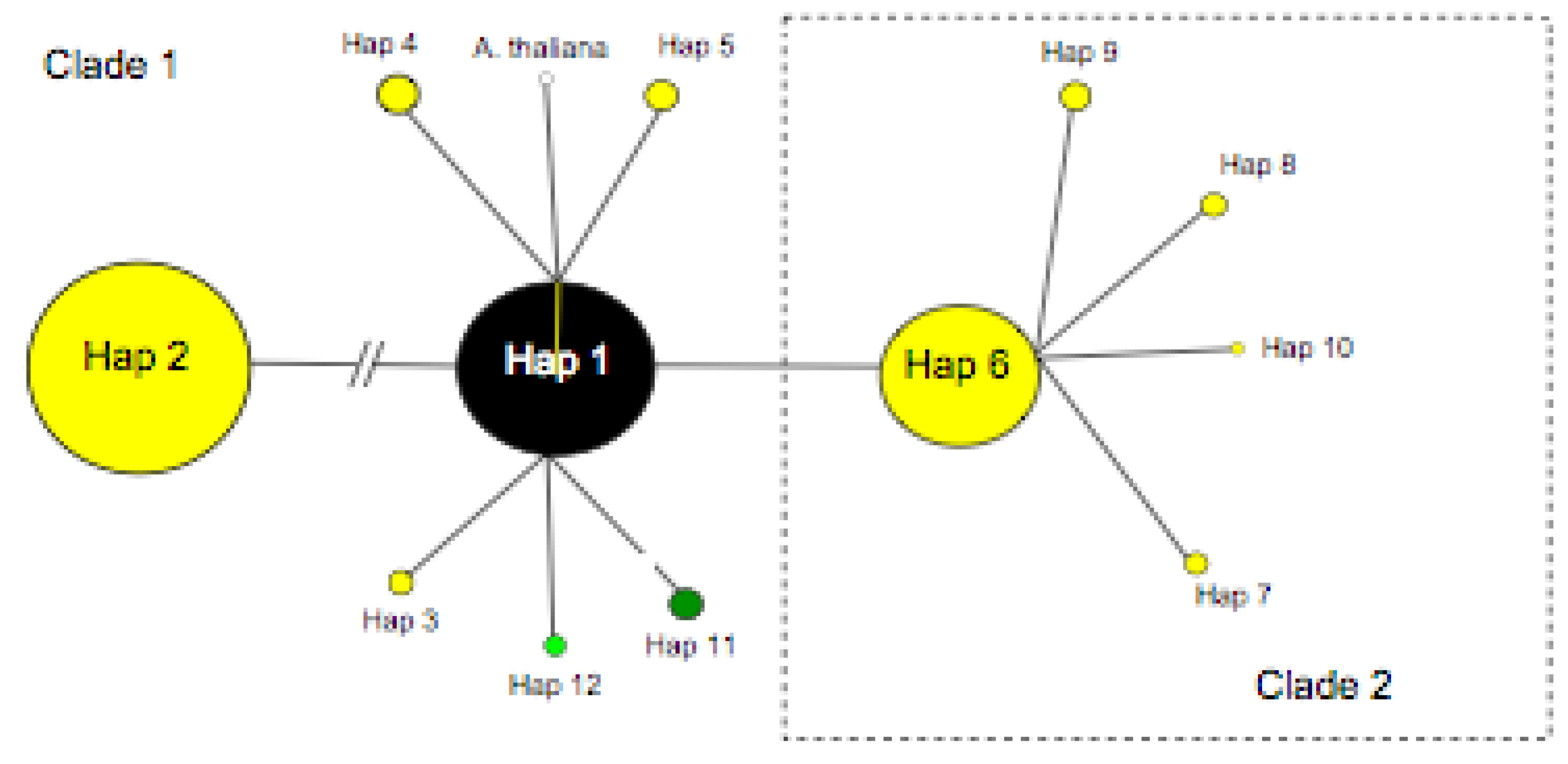
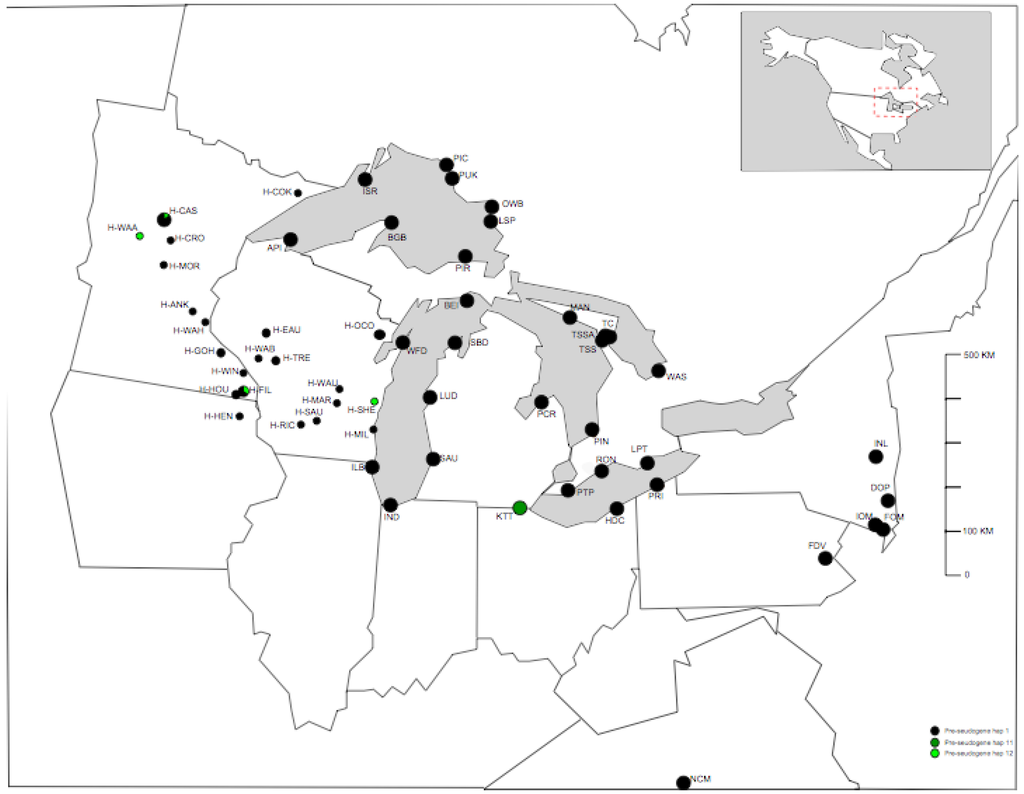
Figure 2a.
Distribution of pre-tandem repeat (P-TR) haplotypes around the Great Lakes area in North America. Full population names are given in Table 1.
Figure 2a.
Distribution of pre-tandem repeat (P-TR) haplotypes around the Great Lakes area in North America. Full population names are given in Table 1.
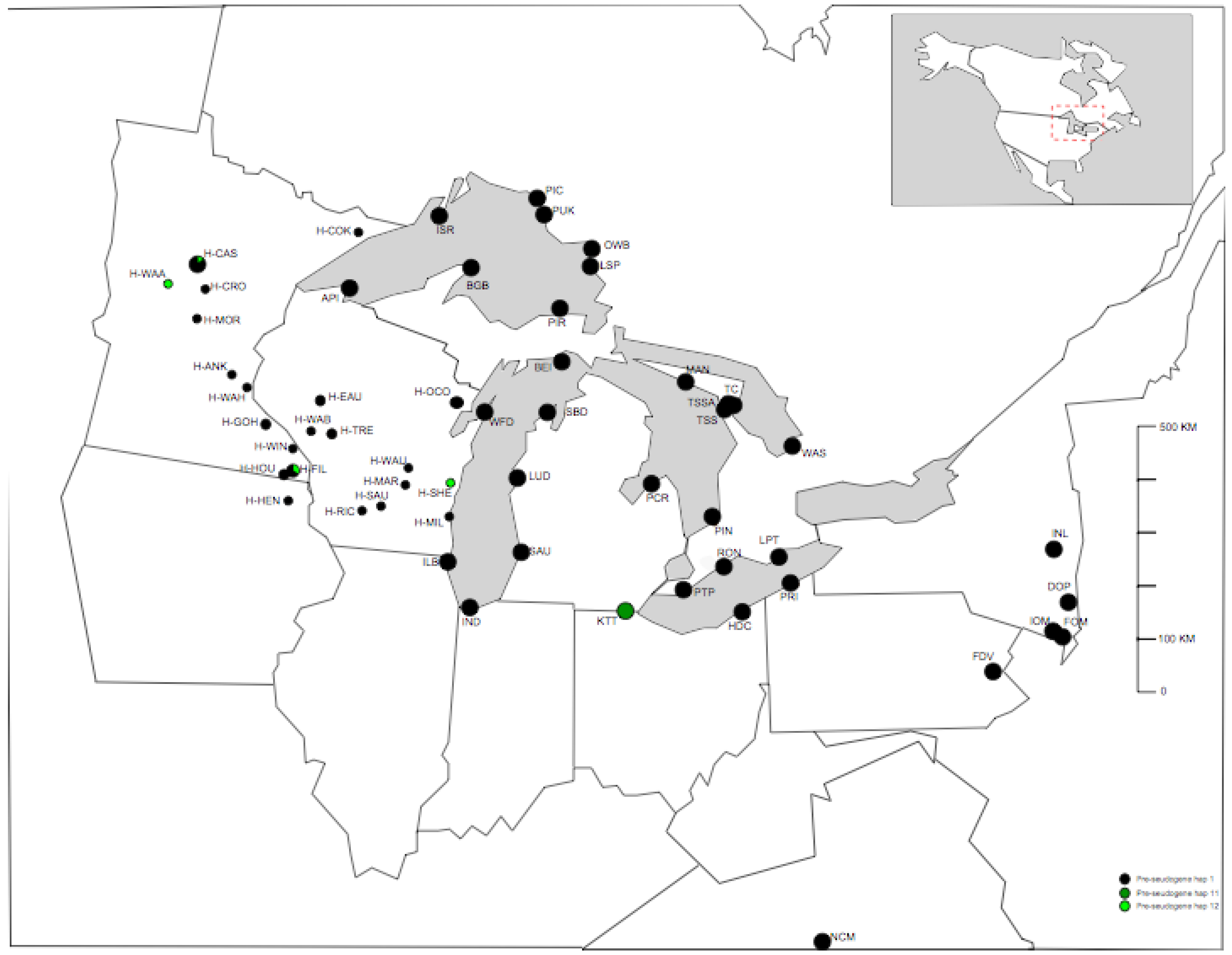
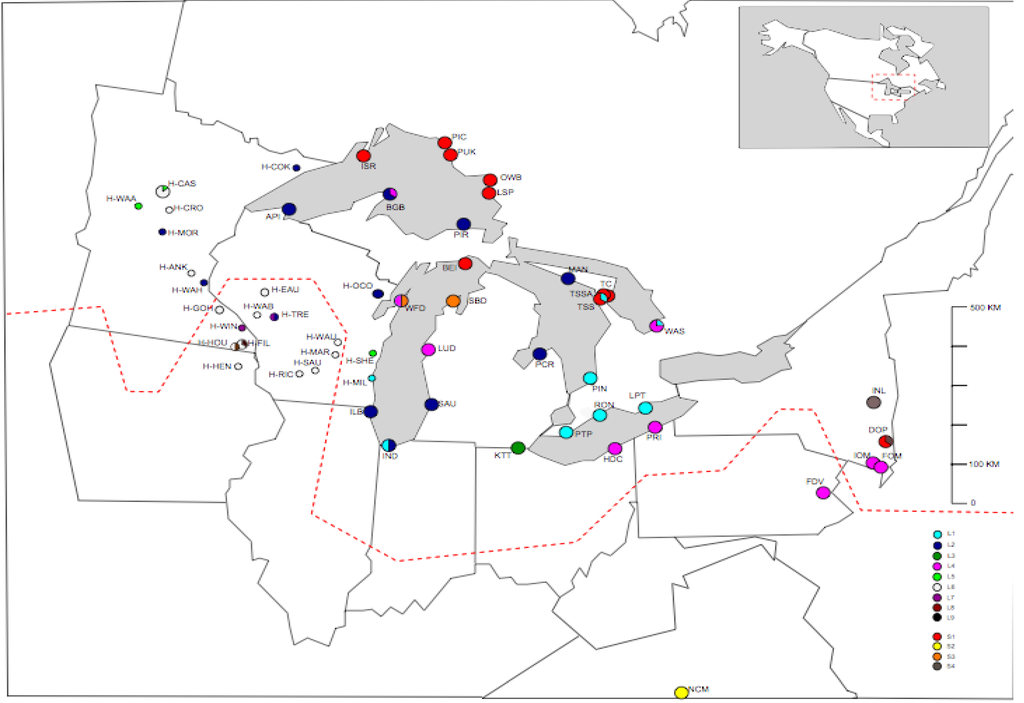
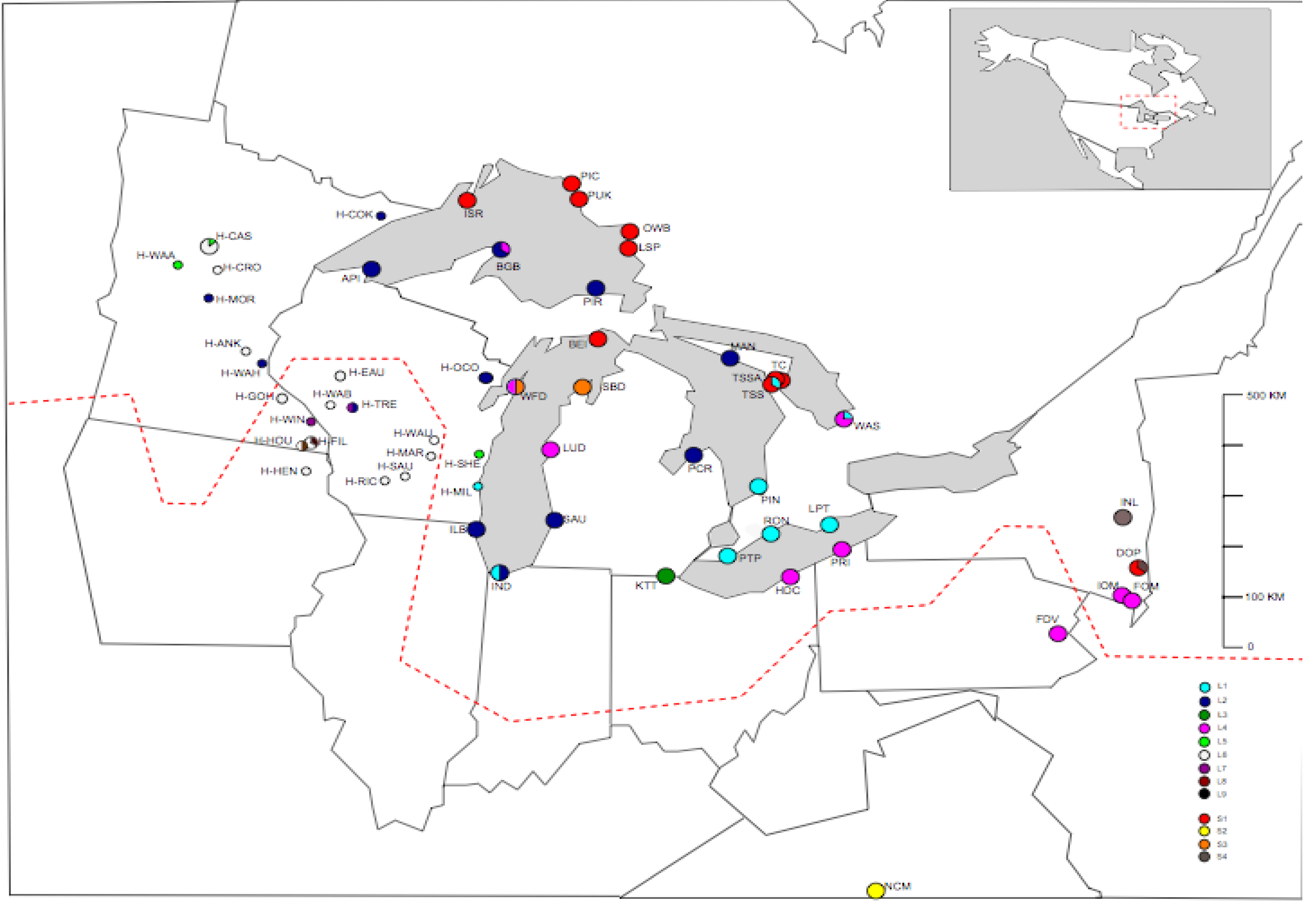
Figure 2b.
Distribution of full sequence haplotypes (L1–L9 and S1–S4) around the Great Lakes area in North America. Circles are proportional to relative frequency of haplotypes at each population. Dashed line represents the maximum southern extent of the Wisconsin ice sheets (modified from Holman, 1992).
Figure 2b.
Distribution of full sequence haplotypes (L1–L9 and S1–S4) around the Great Lakes area in North America. Circles are proportional to relative frequency of haplotypes at each population. Dashed line represents the maximum southern extent of the Wisconsin ice sheets (modified from Holman, 1992).
3.2. TrnL-F Full Sequence Variation Based Only On Mutations within Length Variants
PCR amplification of the IGS region from North American populations indicated that there are three main length variants, a ‘long’ fragment of 741 bp and two ‘short’ fragments of 515 bp and 498 bp. The European samples show eight distinct length variants ranging from 339 bp to 741 bp (haplotypes which include P-TR 2 in their full sequence haplotype will be four base pairs longer due to the insert in the pre-tandem repeat region), which include two of the three main lengths present in North America (the 515 bp variant and the 741 bp variant). There is, however, variation in base pair composition relating to mutations between European and North American sequences within these length variants so none are identical between continents. One of the two short length variants (498 bp, consisting of pseudogene copies one, two and six) is not present in Europe. Alignment of the full sequences (including both the P-TR and tandem repeat pseudogene region) from the North American sequences showed that there were 13 distinct full sequence haplotypes (Figure 3a; Figure 4) compared to 46 distinct full sequence (FS) haplotypes in Europe. Only two of the North American FS haplotypes are similar to those in Europe, L4 and S2, although both of these haplotypes are present in European haplotype P-TR2 samples (PET1A and PET9 respectively), and so they vary by the four base pair insert in the pre-tandem repeat region. Each FS haplotype is delimited from its counterparts by at least one point mutation, which can occur in the P-TR or the tandem repeated duplications.
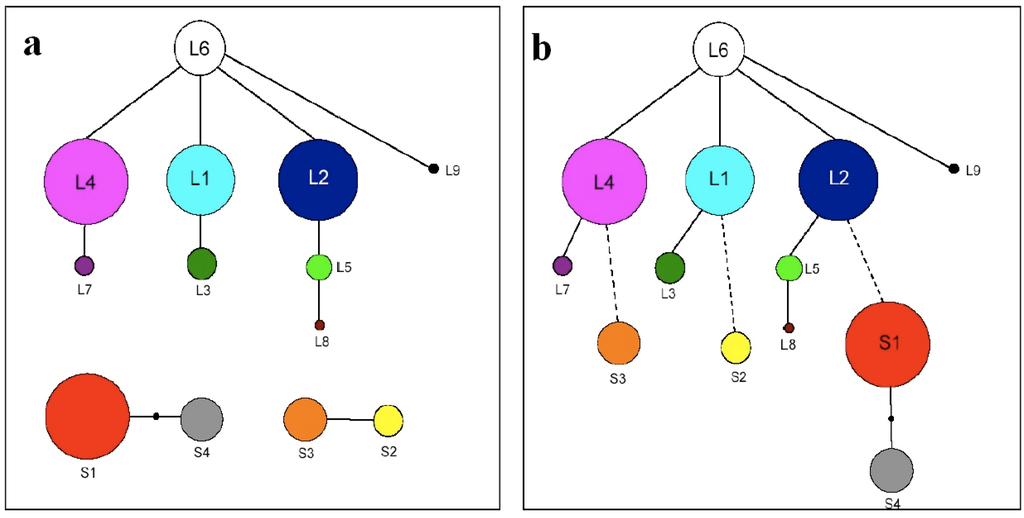
Figure 3a.
Full sequence (FS) haplotype network showing three major clades including the ‘long’ clade in which full sequence haplotype L6 is ancestral, and two ‘short’ clades (including S1 & S4 and S2 & S3 respectively) where ancestral state cannot be assigned. Circle size is proportional to frequency. 3b. Full sequence pseudogene structure (FSPS) haplotype network showing a single major clade in which haplotype L6 is assumed ancestral. Circle size is proportional to frequency. Dashed lines represent putative pseudogene loss events.
Figure 3a.
Full sequence (FS) haplotype network showing three major clades including the ‘long’ clade in which full sequence haplotype L6 is ancestral, and two ‘short’ clades (including S1 & S4 and S2 & S3 respectively) where ancestral state cannot be assigned. Circle size is proportional to frequency. 3b. Full sequence pseudogene structure (FSPS) haplotype network showing a single major clade in which haplotype L6 is assumed ancestral. Circle size is proportional to frequency. Dashed lines represent putative pseudogene loss events.
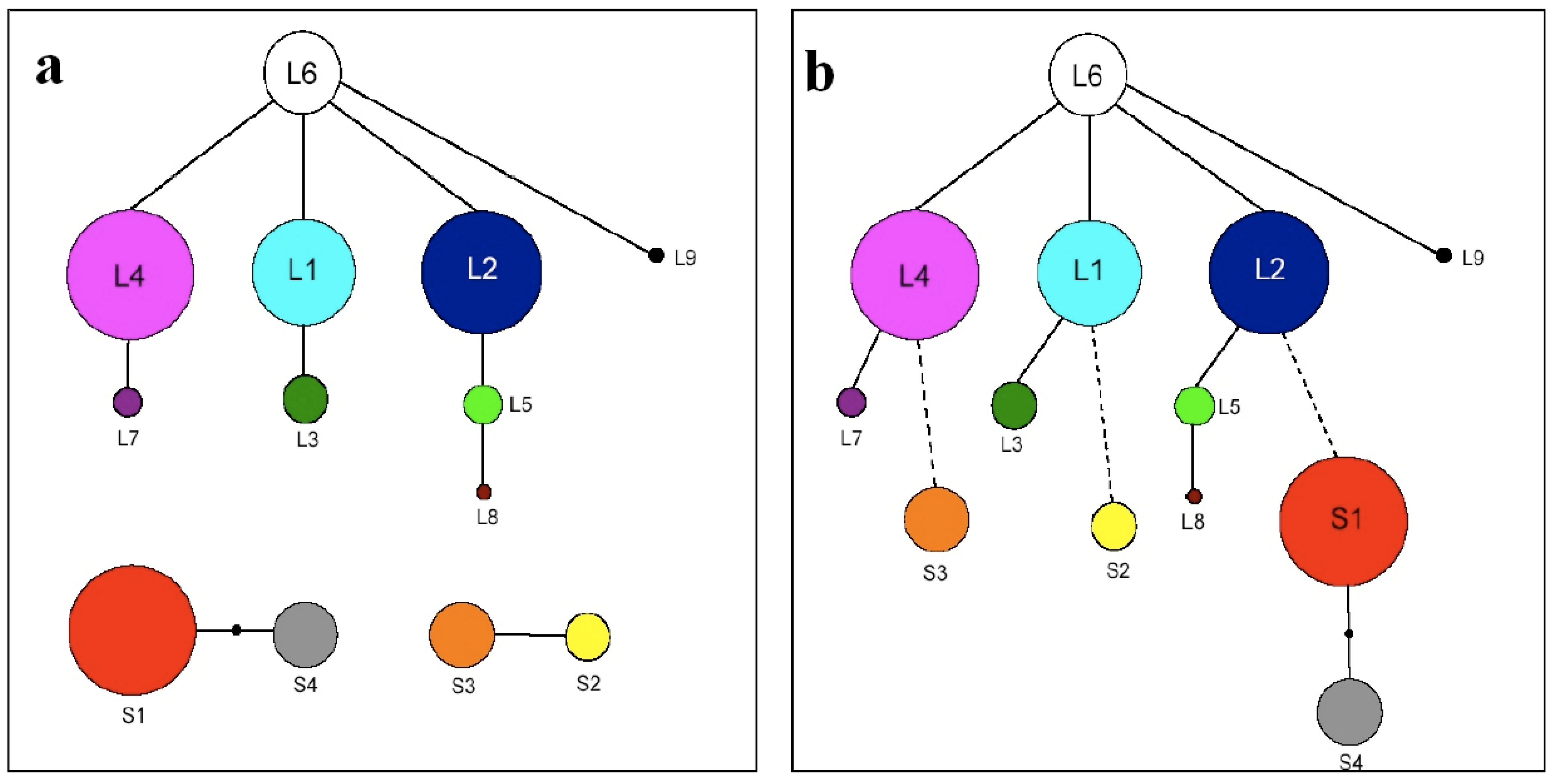
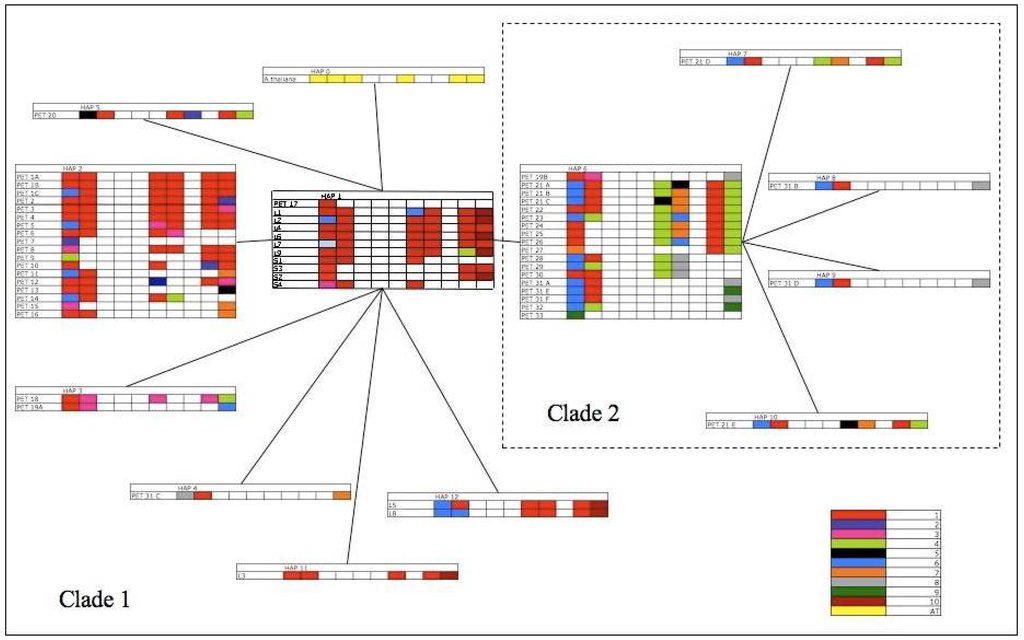
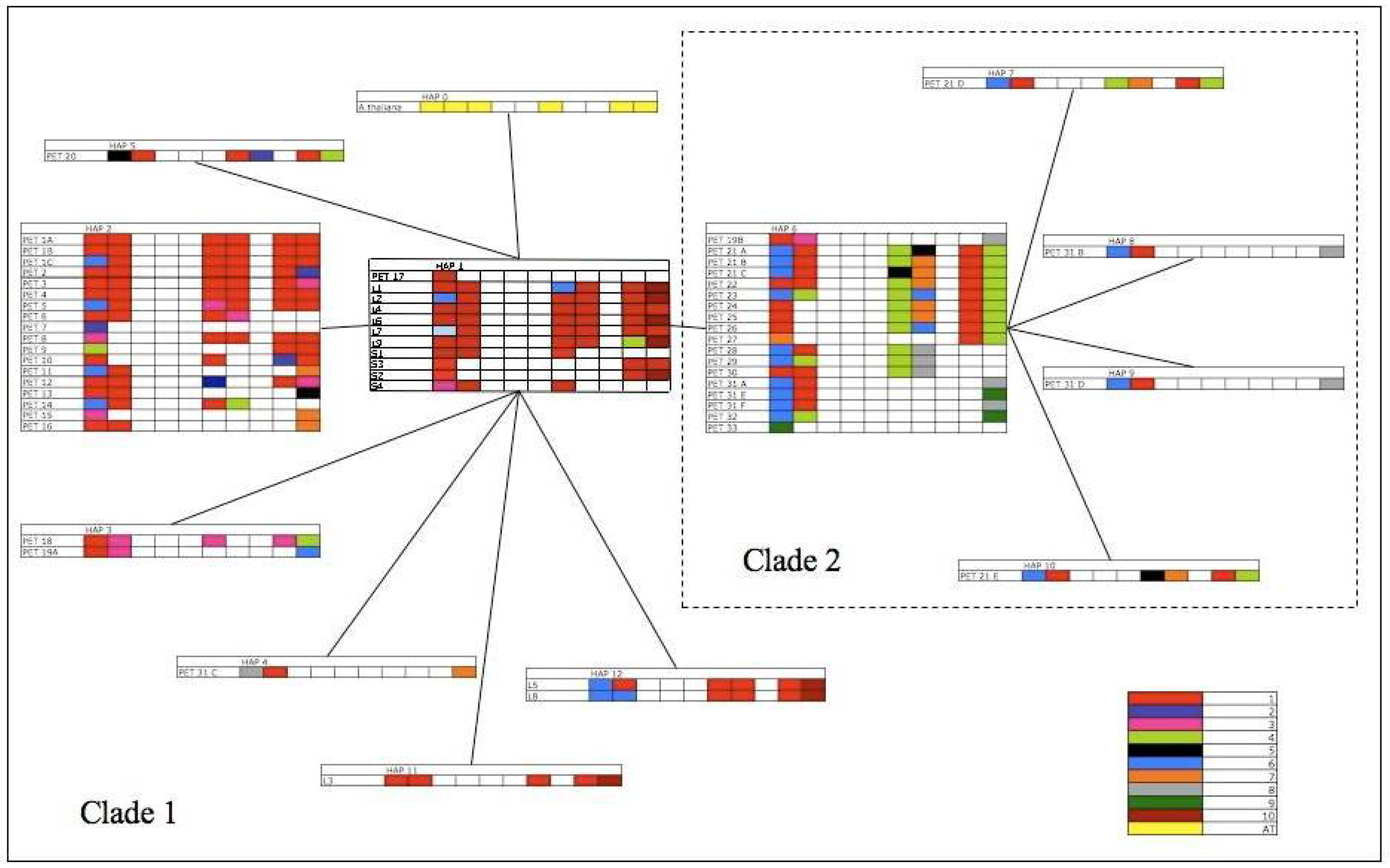
Figure 4.
Minimum spanning network of Arabidopsis trnL-trnF pre-pseudogene haplotypes. Pre-pseudogene haplotypes 1–5 and 11–12 are Clade 1, haplotypes 6–10 are clade 2. Tandem array structure is added to each Pre-pseudogene haplotype, with each copy present in a given sequence shaded. Pseudogene copy variation is represented by colour.
Figure 4.
Minimum spanning network of Arabidopsis trnL-trnF pre-pseudogene haplotypes. Pre-pseudogene haplotypes 1–5 and 11–12 are Clade 1, haplotypes 6–10 are clade 2. Tandem array structure is added to each Pre-pseudogene haplotype, with each copy present in a given sequence shaded. Pseudogene copy variation is represented by colour.
The FS haplotype network (Figure 3a) for North America shows the clustering of nine ‘long’ haplotypes in a single clade, and FS haplotype L6 is identified as ancestral by the TCS analysis, based on both number of connections and relative frequency using the principles of parsimony. Two distinct ‘short’ clades were also recovered, each containing two FS haplotypes (515 bp fragments: S1&S4 and 495 bp fragments: S2&S3). Haplotypes S1, L1 and L2 had previously been described by Hoebe et al. [22] and S2, S3, L3 and L4 by Foxe et al. (unpublished data).
S1 is principally restricted to the north (BEI, ISR, PIC, PUK, OWB and LSP) and northeast (TC, TSS and TSSA) of the Great lakes (Figure 2b), with a single population located in the extreme southeast (DOP in New York state). S3 is present in a single other population, on the opposite shore of Lake Michigan from White Fish Dunes at Sleeping Bear Dunes (SBD). Full sequence haplotype S2 is found in a single population in North Carolina (NCM), which is the only population that was sampled far to the south of the glaciated region. The final ‘short’ full sequence haplotype, S4, is found in two populations in New York state, in the south east of the Great lakes region, Indian Ladder (INL), where all six individuals share this full sequence haplotype and the mixed Dover Plains (DOP) population, where three of the eight individuals sequenced contain S4, and the remaining five have S1.
L1 is generally restricted to the tip of lake Michigan in the south and the northern shore of lake Erie south east of the Great lakes. L2 is widespread and occurs on Lake Michigan on the southeastern shore (Saugatuck; SAU) the south west shore (Illinois Beach; ILB). On Lake superiors southern shore at Pictured Rock (PIR) and Apostle Island (API), and on Lake Hurons northern shore at Manatoulin (MAN) and southern shore at Port Cresent (PCR). Further L2 haplotypes were found in herbarium specimens from Wisconsin (H-OCO & H-TRE) and from Minnesota (H-MOR, H-COK & H-WAH) (Figure 2b). Indiana dunes (IND), on the southern shore of Lake Michigan is a mixed population containing both L1 and L2. There are two further mixed populations containing L1, both on Georgian Bay in southern Ontario: Tobermory Singing Sands (TSS), where three of the eight samples sequenced were L1 and the remaining five were S1; and Wasaga beach (WAS), where two of the eight individuals sequenced were L1 and the remaining six L4. L3 is present in a single population, Kitty Todd nature reserve (KTT), which is located on the remnants of a sand flat towards the western edge of lake Erie in Toledo, Ohio. L4 has a relatively large range, spreading from the extreme south east of the Great lakes in New York state and Pennsylvania (IOM, FOM and FDV) in a north westerly direction encompassing two populations on the south shore of Lake Erie (HDC and PRI) and a population on the eastern shore of Lake Michigan (LUD). L4 is also present in White Fish Dunes (WFD), which is located on the western shore of lake Michigan and also contains a short haplotype, S3 (with four individuals of each haplotype present). The remaining full sequence haplotypes (L5–L9) are all found in herbarium samples to the west of the Great Lakes in Minnesota and Wisconsin. The most common of these is the putative ancestral sequence for the long haplotypes, L6. It is found throughout the west of the Great Lakes region in both Minnesota and Wisconsin. Full sequence haplotypes L8 and L9 are each only found in a single individual: L8 in Houston county, Minnesota (H-HOU) and L9 in Filmore County, Minnesota (H-FIL).
3.3. TrnL-F Full Sequence Variation Considering Evolution of Copy Number Variants
The 13 distinct FS haplotypes for North America, and 46 FS haplotypes from Europe, were mapped onto the haplotype network recovered from the conservative analysis of the P-TR nucleotide variation. This revealed parallel independent changes in pseudogene copy number in both Europe and North America, with tandem repeats that contain various combinations of particular pseudogene copies (one, three, four, or six) are present in both clades (e.g., PET 12 and PET 25), and also between continents (L4 and PET 5). Only a small proportion of IGS variants have undergone changes in the P-TR region, but this can also be seen on both continents (e.g., Europe—PET 31A compared to PET 31B/31C, North America—L2 compared to L3).
By comparing pseudogene copy variation, and the mutations contained within, evolutionary links between ‘short’ and ‘long’ clade haplotypes could be established. The resultant full sequence pseudogene structure (FSPS) haplotype network contains a single clade (Figure 3b). Within the “long” clade, each of the full sequence haplotypes contains the same six pseudogene copies (one, two, six, seven, nine and ten), with each haplotype being separated from one another by at least a single point mutation. These mutations equate to variation within a particular pseudogene copy or, in the case of L3, L5 and L9, in the P-TR region. Each of the four ‘short’ full sequence haplotypes contains three pseudogene copies. S1 and S4 contain copies 1, 2 and 6 whereas S2 and S3 contain copies 1, 9 and 10. S2 and S3 are separated by a single point mutation and S1 and S4 separated by two point mutations. Based on this analysis, haplotype L6 remains the most parsimonious potential ancestral haplotype based on the number of single base connections, with the structuring among ‘long’ FS haplotypes remaining unchanged. Both ‘short’ clades from the FS haplotype network are now incorporated into the ‘long’ clade, but the relationship between ‘short’ haplotypes has changed quite significantly. S2 and S3, previously linked by a single base pair substitution, are now separate. S2 can be linked to L1 based on shared pseudogene mutations with a putative pseudogene loss event, and S3 can be linked to L4 based on the same principles, with the loss of pseudogene copies six and seven. Although this still constitutes only a single base pair difference, we consider that this arrangement requires fewer mutational steps (one loss event each) than a single loss event followed by separate point mutations. The second short clade from the FS haplotype network remains together, with haplotype S1 now linked to ‘long’ haplotype L2, and haplotype S4 remains linked to S1 (separated by two base pair changes).
L1 populations, being located principally on the northern shore of Lake Erie and the southern tip of Lake Huron are in close proximity to the related ‘long’ haplotype L3, and although the suggested closely related ‘short’ haplotype S2 is some distance away, it falls well below the estimated extent of the last glacial maxima [27](Figure 2b), suggesting it is a much older population. FSPS haplotype L2’s broad distribution means it is geographically located in close proximity to its suggested ‘short’ counterpart, S1, on the shores of both Lake Superior (PIR, BGB, API and H-COK) and Lake Huron (MAN). L4’s wide, narrow distribution in a north-westerly direction (from New York/ Pennsylvania in the east to the north of Lake Michigan) culminates in its close proximity to, and mixture with S3, it’s suggested ‘short’ relation in WFD (of the eight individuals screened, four are L4 and four are S3) and SBD (non mixed S3 population). Haplotype S4 is found in two populations only, both in New York State. Indian Ladder (INL) contains all S4 individuals (six) and is in close proximity to Dover Plains (DOP), which contains two S4 individuals and four S1 individuals, supporting the evolutionary link between these two haplotypes (Figure 2b; Figure 3a).
4. Discussion
4.1. Summary of Chloroplast Variation within Arabidopsis Lyrata
It is clear from this study that there is a relatively large amount of variation within the trnL-trnF IGS region, across A. lyrata as a whole, and this pattern is also seen between related species and genera [9,14]. Much more variation was found in European samples relative to eastern North America, but this is consistent with earlier nucleotide studies on A. lyrata [28,29,30], and is concordant with the suggestion that the origins of North American populations are firmly centred in Europe [29,30,31], with lower levels of genetic diversity found in North American (with respect to European A. lyrata) being suggested to demonstrate a possible long-term population bottleneck associated with the colonization of North America from European populations [30].
While variation appears to be maintained in both the P-TR (with three distinct variants in NA and 10 variants in Europe) and the pseudogene tandem array portions of the trnL-F fragment (with 13 distinct haplotypes in NA and 46 distinct variants in Europe) (Figure 4), variation within pseudogene copies globally was higher, with up to 10 distinct variants for each copy. How this variation is considered has important implications for use of the trnF region for phylogeographic studies. A recent study by Schmickl et al. [32] assessed the global phylogeography of the Arabidopsis lyrata complex using numerous molecular markers including the trnL-F region of the chloroplast genome. Their method involved classifying variation in this region into haplotypes, which include pseudogenes, but then classifying these haplotypes into suprahaplotypes (which do not contain the pseudogene portions) to infer phylogeographic relationships. Their findings support our own by suggesting that North American populations contain much lower genetic diversity than that seen in Europe, and that North American populations are likely derived from a long-term split from a European lineage. Interestingly, their findings also include the newly incorporated A. lyrata complex species Arabidopsis arenicola into the same suprahaplotype as North American A. lyrata. However, this method was employed to look at global variation within the A. lyrata species complex but also other species with the genus Arabidopsis (including A. halleri), and not population level variation, which is the goal of our study.
4.2. TrnF Sequence Variation Based Only On the P-TR Region
If we assign haplotypes based only on variation in the P-TR region, as has been suggested previously [11], then populations in Europe show a high degree of variation, and North American populations are almost completely devoid of variation. North American populations appear to be more closely related to European haplotypes in Clade 1 than Clade 2 (Figure 4), which is consistent with Europe being the centre of genus diversity and source for the North American populations, as has been suggested previously [13,29,30]. However, in North America, despite sampling similar numbers of “populations” as in Europe, we sampled only a very small part of the range, whereas in Europe the sampling has been more comprehensive; this could partly explain the increased diversity in the latter. Nevertheless, the comparatively low amounts of variation in North America compared to Europe could be related to their different histories of post-glacial expansion. Glacial events in both regions are relatively well understood. However, post-glacial expansion in plants has not been as well studied in North America as it has in Europe. It is possible, therefore, that, differences in glacial maxima, glacial retreat times or the suspected bottleneck event associated with colonization of North America [33], gives rise to the disparity between the two continents. Although there is also a shift towards inbreeding in some of the North American populations sampled, our previous work has demonstrated that there is not a resulting difference in chloroplast diversity [22].
Based only on the P-TR haplotype allocations, the trnL-trnF IGS region would not be a good candidate for studying phylogeography in North America. It shows very little variation, with only three pre-tandem repeat haplotypes present, only one of which occurs at high frequency. Each is comprised of a single base pair mutation from the common P-TR haplotype 1. However, from the analysis in this study, we consider that using only the P-TR region to assess phlyogeograpy does not represent all the available variation found in the Great Lakes region. This is given extra weight by the findings of Foxe et al. (unpublished data), which found clear phylogeographic structuring based on microsatelite and nuclear data in this species; although cpDNA data did not conflict with conclusions based on the nuclear loci, we were unable to draw strong conclusions about phylogeography based on the distribution of length variants in the cpDNA data alone.
4.3. TrnF Full Sequence Variation Based only on Mutations within Length Variants
Considering variation across the tandem repeat region, as well as the P-TR region allows the assignment of more phylogeographically informative haplotypes. Both North America and Europe exhibit variation within full sequence haplotypes. European haplotypes form eight different length variations due to varying pseudogene copy numbers present in the tandem repeat region. Tandem repeat composition varied from a single pseudogene copy, to six pseudogene copies, with variation in pseudogene position being common also (e.g., PET 14 contains four pseudogene copies in positions one, two, six and seven; PET 10 also contains four pseudogene copies but in positions one, six, nine and ten). This variation in length variants in Europe represents greater diversity than is present in North America, where only three length variants are found, two of which are present in the European samples and a third length variant which is not found in Europe. When we consider all unique haplotypes, which fall into these length variants, then European samples form 46 full sequence haplotypes. Again this level of variation represents much greater diversity than present in North America, whereas only 13 full sequence haplotypes have been described. When considering FS haplotypes like this, no North American haplotypes are identical to those in Europe, and so suggesting potential origins of North American populations becomes very difficult with this method.
When we consider the full sequence haplotypes for North America but without considering shared copy variants the FS haplotype network separates sequences into three distinct clades: A “long” clade containing L1–L9, and two distinct “short” clades containing S1 & S4 and S2 & S3, respectively. Both ‘short’ clades contain two full sequence haplotypes, with S2 and S3 separated by a single point mutation and S1 and S4 separated by two point mutations. Neither haplotype can be considered ancestral based on the number of connections, unless haplotype frequency data are added (TCS assumes a haplotype to be ancestral by calculating both the number of individuals which contain it and the number of connections it has, based on the principles of parsimony) but our focus on sampling single herbarium specimens does not allow this. Whilst this network appears justified at the sequence level based on shared pseudogene copies, there are aspects of it that do not make strict geographical sense. One example of this is that full sequence haplotypes S2 and S3, which are considered closely related in this network, are actually spatially separated (Figure 2b), with no evidence to support a shared colonization route history.
4.4. Trn-F Full Sequence Variation Considering Evolution of Copy Number Variants
When considering the FSPS haplotype assignment method, we find that globally, variation within a pseudogene copy is large, with up to 10 variants being found at any given position. Unique variants are found in both Europe and North America; however, as for all of the other analyses, diversity (in this case the number of unique within copy variants present on either continent) is much higher in Europe. The complexity of variation in copy number within Europe still makes it difficult to infer which full sequence variants are more closely related to North American haplotypes.
However, within North America, when shared pseudogene copy variation between full sequence haplotypes is considered to be evidence of shared ancestry, full sequence haplotypes previously included in either of the ‘short’ clades can be included in a single network with the ‘long’ sequences (FSPS haplotype network; Figure 4). The loss of pseudogenes from the tandem array is considered a single evolutionary step, and is indicated with a dashed line (Figure 3b). Using this approach, the S2–S3 ‘short’ clade is separated, with S2 being linked to L1 and S3 being linked to L4. S1 and S4 are still linked together, and now join the single clade network at L1. This single clade full sequence haplotype network represents a better geographical fit, with closely related sequences separated by a pseudogene loss event being spatially closer than in the FS haplotype network (Figure 2b).
Although limited variation within populations restricts the utility of formalized phylogeographic tests for the North American samples, we can draw some conclusions about historical patterns of colonization of the Great Lakes region following the last glacial maximum. Following the retreat of the Wisconsin glaciation 10,000 bp, various scenarios of post-glacial colonization have been suggested. For reptiles and amphibians in particular, it has been suggested that at least two routes of colonization is most parsimonious with molecular and geographic data [34,35,36], with likely routes being to the east, and the west of Lake Michigan. Hoebe et al. [22] agreed that two colonization routes also were likely for A. lyrata, suggesting that individuals with the S1 full sequence haplotype moved northwards along the west side of Lake Michigan, colonizing the area to the north of Lake Superior and those with full sequence haplotype L1 was suggested to have moved northwards on the west side of Lake Michigan. In this study, we have found that the divide between ‘short’ and ‘long’ haplotypes presented by Hoebe et al. [22] is much less clear when further haplotypes are added. Foxe et al. (unpublished data) found that FS haplotype distributions for 24 of the 33 populations used here (Table 1) were consistent with Bayesian clustering patterns based on combined nuclear DNA markers and micro-satellites, which supported the findings of Hoebe et al. [22] suggesting at least two routes of colonization. In that study, we only considered mutations within length variants, and by using this method we were unable to link ‘short’ and ‘long’ clade haplotypes together. However, if we consider that full sequence haplotype S1 is linked to full sequence haplotype L2 by a single pseudogene loss event, this suggests a northern post-glacial colonisation on either the east or west side of Lake Michigan, with S1 principally clustering at the north of Lake Superior, and L2 showing a more scattered distribution (Figure 2b). It is possible that there may be a third colonization route not seen in previous studies, moving westwards from the New York state area towards Wisconsin. This is supported by current distributions for full sequence haplotype L4, and its probable derivatives S3 and L7. This colonization route may be supported by the fact that the L4 population at Friedensville (FDV) is below the estimated position of the Wisconsin glacial maxima [27].
It is possible that the Friedsville population (FDV) in Pennsylvania (Figure 2b) and a number of populations in Minnesota and Wisconsin (H-EAU, H-WAB, H-WAU, H-MAR, H-SAU, H-RIC, H-TRE, H-WIN, H-GOH, H-HOU, H-FIL and H-HEN) are much older than the other populations, as they are currently present in what should have been non-glaciated areas. This might support the view that full sequence haplotype L6 is ancestral.
4.5. Pseudogenes in a Population Level Study
Earlier phylogeographic studies on Arabidopsis based on the trnL-F IGS employed conservative approaches to analysing sequence variation, due to structurally mediated parallel changes in pseudogene copies, and elected to base their interpretations on the pre-tandem array portion of the fragment [11,20,32]. This approach was appropriate for Europe, where there is more extensive variation in the pre-tandem array region, and correspondingly, strong evidence for multiple changes in pseudogene copy number [11,14]. In the relatively genetically depauperate eastern North American populations of A. lyrata there is limited nucleotide diversity in the pre-tandem array region, allowing for greater certainty when assessing relationships between sequences that differ by pseudogene content. As a consequence, by following a simple set of assumptions, we show that the nucleotide informative available within the tandem array can be used to make inferences about evolutionary links between populations that have varying sequence length, and to recover otherwise cryptic population structure.
Acknowledgements
We thank Aileen Adam for technical assistance; Yvonne Willi for providing seeds from 13 populations; Marc Stift for comments on the manuscript. We thank Parks Canada, Ontario Parks, Michigan State Parks Authority, U.S. National Park Service, Ohio Department of Natural Resources, and the Ohio Nature Conservancy for access to protected park areas and advice on plant locations. AT was supported on a Natural Environment Research Council (NERC) PhD studentship tied to a grant awarded to BKM (NE/D013461/1), SA was supported by the Department of Botany at the NHM in London, PH was supported on a University of Glasgow Institute of Biomedical and Life Sciences PhD studentship, and BKM on a NERC Advanced Research Fellowship (NE/B500094X/1).
References
- Palmer, J.D. Plastid chromosomes: structure and evolution. In Cell Culture and Somatic Cell Genetics in Plants:The Molecular Biology of Plastids; Bogorad, L., Vasil, I.K., Eds.; Acedemic Press: San Deigo, CA, USA, 1991; Volumn 7, pp. 5–53. [Google Scholar]
- Raubson, L.A.; Jansen, R.K. Chloroplast Genomes of Plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Muse, S.V. Examining rates and patterns of nucleotide substitutions in plants. Pl. Mol. Biol. Rep. 2000, 42, 481–490. [Google Scholar]
- Mitchell-Olds, T.; Al.-Shehbaz, I.; Koch, M.; Sharbel, T. Crucifer evolution in the post-genomic era. In Plant Diversity and Evolution:Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Publishing: Oxon, UK, 2005; pp. 119–136. [Google Scholar]
- Hiratsuka, J.; Shimada, H.; Whittier, R.; Ishibashi, T.; Sakamoto, M.; Mori, M.; Kondo, C.; Honji, Y.; Sun, C.R.; Meng, B.Y.; Li, Y.Q.; Kanno, A.; Nishizawa, Y.; Hirai, A.; Shinozaki, K.; Sugiura, M. The Complete Sequence of the Rice (Oryza-Sativa) Chloroplast Genome—Intermolecular Recombination between Distinct Transfer-RNA Genes Accounts for a Major Plastid DNA Inversion during the Evolution of the Cereals. Mol. Gen. Genet. 1989, 217, 185–194. [Google Scholar] [CrossRef]
- Ingvarsson, P.K.; Ribstein, S.; Taylor, D.R. Molecular Evolution of Insertions and Deletion in the Chloroplast Genome of Silene. Mol. Biol. Evol. 2003, 20, 1737–1740. [Google Scholar] [CrossRef]
- Ogihara, Y.; Terachi, T.; Sasakuma, T. Intramolecular Recombination of Chloroplast Genome Mediated by Short Direct-Repeat Sequences in Wheat Species. Proc. Nat. Acad. Sci.USA 1988, 85, 8573–8577. [Google Scholar] [CrossRef]
- Pyke, K.A. Plastid Division and Development. Plant Cell 1999, 11, 549–556. [Google Scholar] [CrossRef]
- Dobeš, C.; Kiefer, C.; Kiefer, M.; Koch, M.A. Plastidic trnF(UUC) pseudogenes in North American genus Boechera (Brassicaceae): Mechanistic aspects of evolution. Plant Biol. 2007, 9, 502–515. [Google Scholar] [CrossRef]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal Primers for Amplification of 3 Noncoding Regions of Chloroplast DNA. Pl. Mol. Biol. Rep. 1991, 17, 1105–1109. [Google Scholar] [CrossRef]
- Ansell, S.W.; Schneider, H.; Pedersen, N.; Grundmann, M.; Russell, S.J.; Vogel, J.C. Recombination diversifies chloroplast trnF pseudogenes in Arabidopsis lyrata. J. Evol. Biol. 2007, 20, 2400–2411. [Google Scholar] [CrossRef]
- Ansell, S.W.; Stenoien, H.K.; Grundmann, M.; Schneider, H.; Hemp, A.; Bauer, N.; Russell, S.J.; Vogel, J. Population structure and historical biogeography of European Arabidopsis lyrata. Heredity 2010, in press. [Google Scholar] [CrossRef]
- Koch, M.A.; Dobeš, C.; Matschinger, M.; Bleeker, W.; Vogel, J.; Kiefer, M.; Mitchell-Olds, T. Evolution of the trnF(GAA) gene in Arabidopsis relatives and the Brassicaceae family: Monophyletic origin and subsequent diversification of a plastidic pseudogene. Mol. Biol. Evol. 2005, 22, 1032–1043. [Google Scholar] [CrossRef]
- Schmickl, R.; Kiefer, C.; Dobeš, C.; Koch, M. Evolution of trn F(GAA) pseudogenes in cruciferous plants. Plant Syst. Evol. 2008, 282, 229–240. [Google Scholar] [CrossRef]
- Lihova, J.; Fuertes Aguilar, J.; Marhold, K.; Nieto Feliner, G. Origin of the disjunct tetraploid Cardamine amporitana (Brassicaceae) assessed with nuclear and chloroplast DNA sequence data. Am. J. Bot. 2004, 91, 1231–1242. [Google Scholar] [CrossRef]
- Bleeker, W.; Hurka, H. Introgressive hybridization in Rorippa (Brassicaceae): gene flow and its consequences in natural and anthropogenic habitats. Mol. Ecol. 2001, 10, 2013–2022. [Google Scholar] [CrossRef]
- Mummenhoff, K.; Bruggemann, H.; Bowman, J.L. Chloroplast DNA phylogeny and biogeography of Lepidium (Brassicaceae). Amer. J. Bot. 2001, 88, 2051–2063. [Google Scholar] [CrossRef]
- Al-Shehbaz, I.A.; O'Kane, S.L. Taxonomy and phylogeny of Arabidopsis (Brassicaceae). In The Arabidopsis Book; Somerville, C.R., Meyerowitz, E.M., Eds.; American Society of Plant Biologist: Rockville, MD, USA, 2002; pp. 1–22. [Google Scholar]
- Schmickl, R.; Jorgensen, M.; Brysting, A.; Koch, M. Phylogeographic implications for the North American boreal-arctic Arabidopsis lyrata complex. Pl. Ecol. Divers. 2008, 1, 245–254. [Google Scholar] [CrossRef]
- Koch, M.; Matschinger, M. Evolution and genetic differentiation among relatives of Arabidopsis thaliana. Proc. Nat. Acad. Sci. USA 2007, 104, 6272–6277. [Google Scholar] [CrossRef]
- Koch, M.A.; Dobeš, C.; Kiefer, C.; Schmickl, R.; Klimes, L.; Lysak, M.A. Supernetwork identifies multiple events of plastid trnF(GAA) pseudogene evolution in the Brassicaceae. Mol. Biol. Evol. 2007, 24, 63–73. [Google Scholar]
- Hoebe, P.N.; Stift, M.; Tedder, A.; Mable, B.K. Multiple losses of self-incompatibility in North-American Arabidopsis lyrata: Phylogeographic context and population genetic consequences. Mol. Ecol. 2009, 18, 4924–4939. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinformatics 2005, 1, 47–50. [Google Scholar]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A Cladistic-Analysis of Phenotypic Associations with Haplotypes Inferred from Restriction Endonuclease Mapping and DNA-Sequence Data . 3. Cladogram Estimation. Genetics 1992, 132, 619–633. [Google Scholar]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef]
- Kelchner, S.A. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann. Mo. Bot. Gard. 2000, 87, 482–498. [Google Scholar] [CrossRef]
- Holman, J.A. Late Quaternary Herpetofauna of the Central Great-Lakes Region, USA Zoogeographical and Paleoecological Implications. Quaternary Sci. Rev. 1992, 11, 345–351. [Google Scholar] [CrossRef]
- Balañá-Alcaide, D.; Ramos-Onsins, S.E.; Boone, Q.; Aguade, M. Highly structured nucleotide variation within and among Arabidopsis lyrata populations at the FAH1 and DFR gene regions. Mol. Ecol. 2006, 15, 2059–2068. [Google Scholar] [CrossRef]
- Ross-Ibarra, J.; Wright, S.I.; Foxe, J.P.; Kawabe, A.; DeRose-Wilson, L.; Gos, G.; Charlesworth, D.; Gaut, B.S. Patterns of Polymorphism and Demographic History in Natural Populations of Arabidopsis lyrata. Plos. One. 2008, 3, e2411. [Google Scholar] [CrossRef]
- Clauss, M.J.; Mitchell-Olds, T. Population genetic structure of Arabidopsis lyrata in Europe. Mol. Ecol. 2006, 15, 2753–2766. [Google Scholar] [CrossRef]
- Wright, S.I.; Lauga, B.; Charlesworth, D. Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol. Ecol. 2003, 12, 1247–1263. [Google Scholar] [CrossRef]
- Schmickl, R.; Jorgensen, M.; Brysting, A.; Koch, M. The evolutionary history of the Arabidopsis lyrata complex: A hybrid in the amphi-Beringian area closes a large distribution gap and builds up a genetic barrier. BMC Evol. Biol. 2010. In press. [Google Scholar] [CrossRef]
- Clauss, M.J.; Cobban, H.; Mitchell-Olds, T. Cross-species microsatellite markers for elucidating population genetic structure in Arabidopsis and Arabis (Brassicaeae). Mol. Ecol. 2002, 11, 591–601. [Google Scholar] [CrossRef]
- Holman, J.A. Pleistocene amphibians and reptiles in North America. Oxford Monogr. Geol. Geop. 1995, 32, 1–243. [Google Scholar]
- Placyk, J.S.; Burghardt, G.M.; Small, R.L.; King, R.B.; Casper, G.S.; Robinson, J.W. Post-glacial recolonization of the Great Lakes region by the common gartersnake (Thamnophis sirtalis) inferred from mtDNA sequences. Mol. Phylogenet. Evol. 2007, 43, 452–467. [Google Scholar] [CrossRef]
- Soltis, D.E.; Morris, A.B.; McLachlan, J.S.; Manos, P.S.; Soltis, P.S. Comparative phylogeography of unglaciated eastern North America. Mol. Ecol. 2006, 15, 4261–4293. [Google Scholar] [CrossRef]
Appendixes
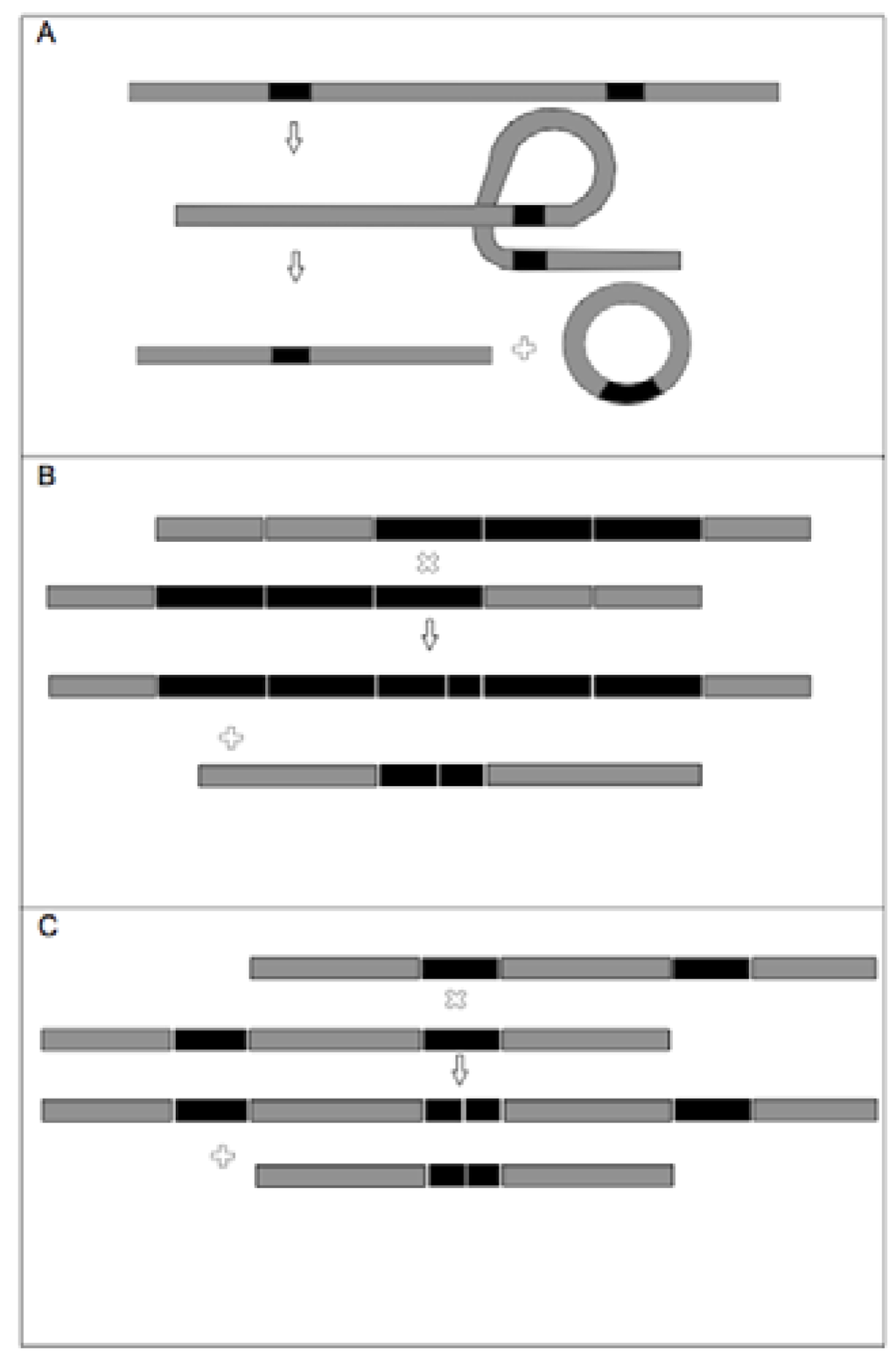
Appendix 1.
(Adapted from Dobeš et al. 2007). A graphical representation illustrating the main types of mutation mechanisms, potentially responsible for DNA length variation among pseudogenes, as discussed in the text. Grey and black lines represent double stranded DNA. Direct (oriented in the same direction) DNA repeats are given in solid black. The ‘x’ symbol marks crossing over points. A) Intramolecular recombination between two similar regions on a single double-stranded DNA (dsDNA) molecule can result in the excision of the intermediate region, yielding a shorter dsDNA molecule and a separate circularised dsDNA molecule. B & C) Unequal intermolecular recombination: Recombination by crossing over between repeats of different sequential position within tandem repeat, occurring: B) within a series of direct repeats separated by intervening DNA fragments, or C) the increase in size of one DNA fragment at the expense of the other.
Appendix 1.
(Adapted from Dobeš et al. 2007). A graphical representation illustrating the main types of mutation mechanisms, potentially responsible for DNA length variation among pseudogenes, as discussed in the text. Grey and black lines represent double stranded DNA. Direct (oriented in the same direction) DNA repeats are given in solid black. The ‘x’ symbol marks crossing over points. A) Intramolecular recombination between two similar regions on a single double-stranded DNA (dsDNA) molecule can result in the excision of the intermediate region, yielding a shorter dsDNA molecule and a separate circularised dsDNA molecule. B & C) Unequal intermolecular recombination: Recombination by crossing over between repeats of different sequential position within tandem repeat, occurring: B) within a series of direct repeats separated by intervening DNA fragments, or C) the increase in size of one DNA fragment at the expense of the other.
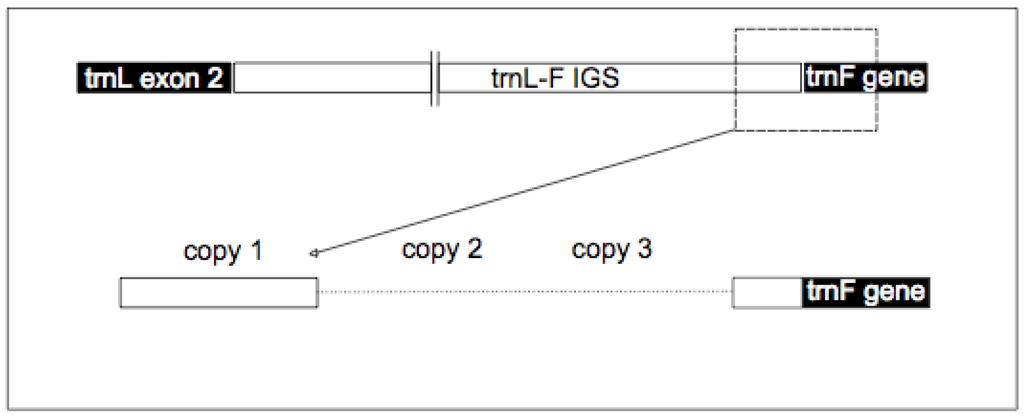
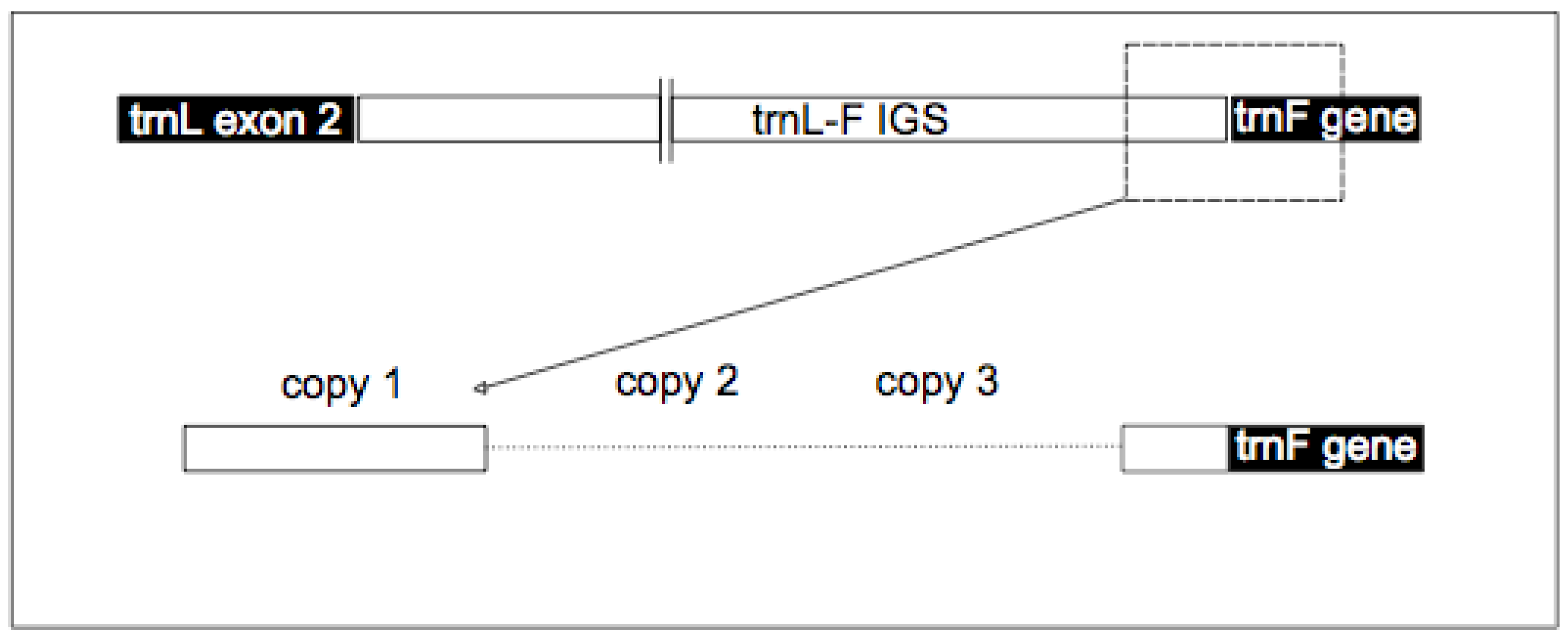
Appendix 2.
(Adapted from Dobeš et al. 2007). A graphical representation of the trnL-F IGS and the trnF (UAA) gene, detailing the region sequenced in this study.
Appendix 2.
(Adapted from Dobeš et al. 2007). A graphical representation of the trnL-F IGS and the trnF (UAA) gene, detailing the region sequenced in this study.
Appendix 3.
Genbank accession numbers for all European samples used in this study.
| Sequence Name | Accession number | Author |
| PET 1A | DQ989814 | Ansell et al. 2007 |
| PET IB | DQ989815 | Ansell et al. 2007 |
| PET 1C | DQ989816 | Ansell et al. 2007 |
| PET 2 | DQ989817 | Ansell et al. 2007 |
| PET 3 | DQ989818 | Ansell et al. 2007 |
| PET 4 | DQ989819 | Ansell et al. 2007 |
| PET 5 | DQ989820 | Ansell et al. 2007 |
| PET 6 | DQ989821 | Ansell et al. 2007 |
| PET 1 | DQ989822 | Ansell et al. 2007 |
| PET 8 | DQ989823 | Ansell et al. 2007 |
| PET 9 | DQ989824 | Ansell et al. 2007 |
| PET 10 | DQ989825 | Ansell et al. 2007 |
| PET 11 | DQ989826 | Ansell et al. 2007 |
| PET 12 | DQ989827 | Ansell et al. 2007 |
| PET 13 | DQ989828 | Ansell et al. 2007 |
| PET 14 | DQ989829 | Ansell et al. 2007 |
| PET 15 | DQ989830 | Ansell et al. 2007 |
| PET 16 | DQ989831 | Ansell et al. 2007 |
| PET 17 | DQ989832 | Ansell et al. 2007 |
| PET 18 | DQ989833 | Ansell et al. 2007 |
| PET 19A | DQ989834 | Ansell et al. 2007 |
| PET 19B | DQ989835 | Ansell et al. 2007 |
| PET 20 | DQ989836 | Ansell et al. 2007 |
| PET 21A | DQ989837 | Ansell et al. 2007 |
| PET 21B | DQ989838 | Ansell et al. 2007 |
| PET 21C | DQ989839 | Ansell et al. 2007 |
| PET 21D | DQ989840 | Ansell et al. 2007 |
| PET 21E | DQ989841 | Ansell et al. 2007 |
| PET 22 | DQ989842 | Ansell et al. 2007 |
| PET 23 | DQ989843 | Ansell et al. 2007 |
| PET 24 | DQ989844 | Ansell et al. 2007 |
| PET 25 | DQ989845 | Ansell et al. 2007 |
| PET 26 | DQ989846 | Ansell et al. 2007 |
| PET 27 | DQ989847 | Ansell et al. 2007 |
| PET 28 | DQ989848 | Ansell et al. 2007 |
| PET 29 | DQ989849 | Ansell et al. 2007 |
| PET 30 | DQ989850 | Ansell et al. 2007 |
| PET 31A | DQ989851 | Ansell et al. 2007 |
| PET 31B | DQ989852 | Ansell et al. 2007 |
| PET 31C | DQ989853 | Ansell et al. 2007 |
| PET 31D | DQ989854 | Ansell et al. 2007 |
| PET 31E | DQ989855 | Ansell et al. 2007 |
| PET 31F | DQ989856 | Ansell et al. 2007 |
| PET 32 | DQ989857 | Ansell et al. 2007 |
| PET 33 | DQ989858 | Ansell et al. 2007 |
| PET 34 | DQ989859 | Ansell et al. 2007 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).