
(E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole

Abstract
1. Introduction
2. Results and Discussion
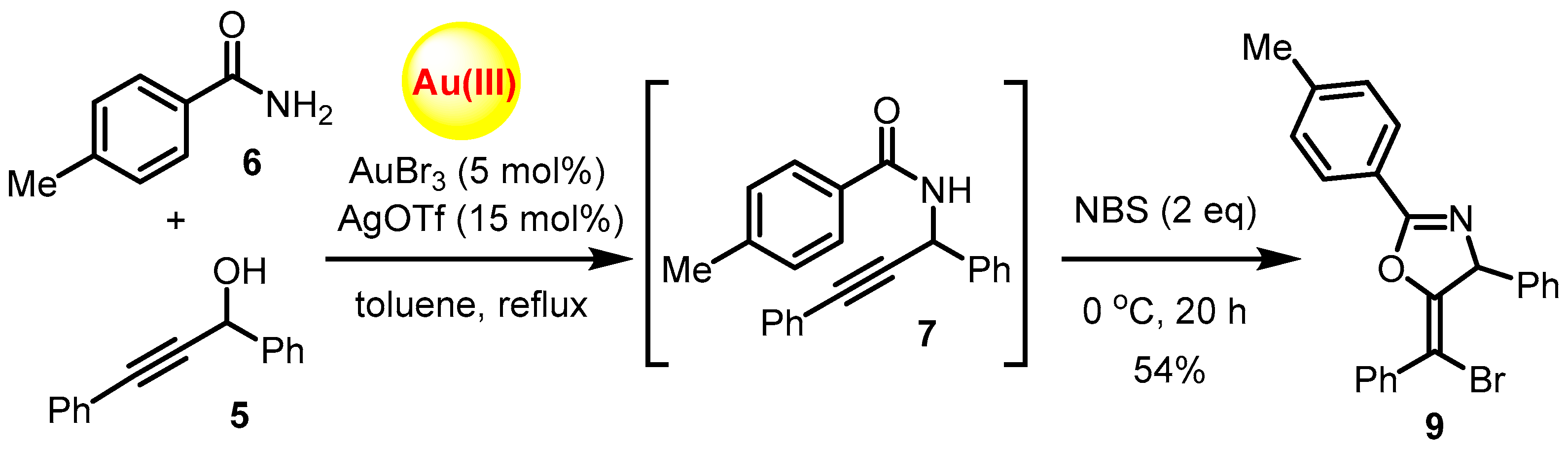
2.1. Chemistry
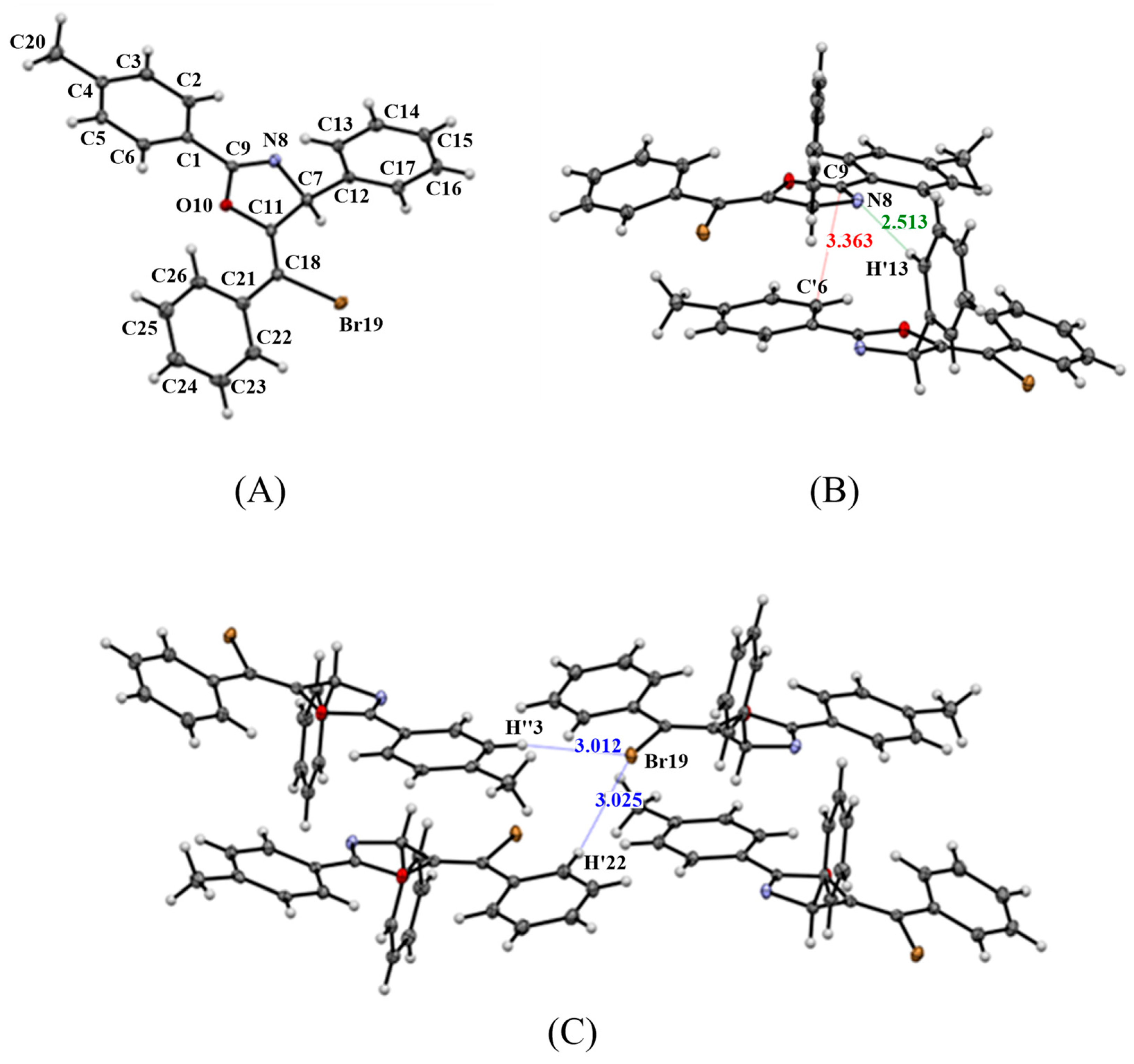
2.2. X-ray Structure Analysis
3. Materials and Methods
3.1. General Information
3.2. Synthesis of (E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole (9)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yeh, V.S.C. Recent advances in the total synthesis of oxazole-containing natural products. Tetrahedron 2004, 60, 11995–12042. [Google Scholar] [CrossRef]
- Wipf, P. Synthetic studies of biologically active marine cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Lipshutz, B.H. Five-membered heteroaromatic rings as intermediates in organic synthesis. Chem. Rev. 1986, 86, 795–819. [Google Scholar] [CrossRef]
- Wipf, P.; Venkatraman, S. From aziridines to oxazolines and thiazolines: The heterocyclic route to thiangazole. Synlett 1997, 1997, 1–10. [Google Scholar] [CrossRef]
- Dong, K.; Gurung, R.; Xu, X.; Doyle, M.P. Enantioselective catalytic cyclopropanation-rearrangement approach to chiral spiroketals. Org. Lett. 2021, 23, 3955–3959. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, S.; Tomkinson, N.C.O. Transition metal-mediated synthesis of oxazoles. Heterocycles 2014, 89, 2479–2543. [Google Scholar]
- Senadi, G.C.; Hu, W.-P.; Hsiao, J.-S.; Vandavasi, J.K.; Chen, C.-Y.; Wang, J.-J. Facile, selective, and regiocontrolled synthesis of oxazolines and oxazoles mediated by ZnI2 and FeCl3. Org. Lett. 2012, 14, 4478–4481. [Google Scholar] [CrossRef] [PubMed]
- Okamura, Y.; Sato, D.; Yoshimura, A.; Zhdankin, V.V.; Saito, A. Iodine(III)-mediated/catalyzed cycloisomerization-amination sequence of N-propargyl carboxamides. Adv. Synth. Catal. 2017, 359, 3243–3247. [Google Scholar] [CrossRef]
- Yu, X.; Xin, X.; Wan, B.; Li, X. Base-catalyzed cyclization of N-sulfonyl propargylamides to sulfonylmethyl-substituted oxazoles via sulfonyl migration. J. Org. Chem. 2013, 78, 4895–4904. [Google Scholar] [CrossRef]
- Morita, N.; Chiaki, H.; Aonuma, S.; Tanaka, K., III; Hashimoto, Y.; Tamura, O. (Z)-5-Benzylidene-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole. Molbank 2023, 2023, M1600. [Google Scholar] [CrossRef]
- Weyrauch, J.P.; Hashmi, A.S.K.; Schuster, A.; Hengst, T.; Schetter, S.; Littmann, A.; Rudolph, M.; Hamzic, M.; Visus, J.; Rominger, F.; et al. Cyclization of propargylic amides: Mild access to oxazole derivatives. Chem. Eur. J. 2010, 16, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Urbanaitė, A.; Jonušis, M.; Bukšnaitienė, R.; Balkaitis, S.; Čikotienė, I. Electrophile-mediated reactions of functionalized propargylic substrates. Eur. J. Org. Chem. 2015, 2015, 7091–7113. [Google Scholar] [CrossRef]
- Hu, Y.; Yi, R.; Wang, C.; Xin, X.; Wu, F.; Wan, B. From propargylamides to oxazole derivatives: NIS-mediated cyclization and further oxidation by dioxygen. J. Org. Chem. 2014, 79, 3052–3059. [Google Scholar] [CrossRef] [PubMed]
- Paradise, C.L.; Sarkar, P.R.; Razzak, M.; De Brabander, J.K. Gold-catalyzed synthesis of amino acid-derived 2,5-disubstituted oxazoles. Org. Biomol. Chem. 2011, 9, 4017–4020. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhang, M.; Ciccarelli, S.; Lee, J.; Catano, B. AuIII-Catalyzed Formation of α-Halomethyl Ketones from Terminal Alkynes. Eur. J. Org. Chem. 2017, 2017, 781–785. [Google Scholar] [CrossRef]
- CCDC-2321720. Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 25 December 2023).
- Weiss, H.-C.; Boese, R.; Smith, H.L.; Haley, M.M. ≡CH⋯p versus ≡CH⋯halogen interactions–The crystal structures of the 4-halogenoethynylbenzenes. Chem. Commun. 1997, 2403–2404. [Google Scholar] [CrossRef]
- Podsiadto, M.; Olejniczak, A.; Katrusiak, A. Halogen⋯halogen contra C–H⋯halogen interactions. CrystEngComm 2014, 16, 8279–8285. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Data |
| Identification | C23H18BrNO |
| Formula weight | 404.29 |
| Temperature/K | 293(2) |
| Crystal system | monoclinic |
| Space group | P21/c |
| Unit cell dimensions | a/Å 10.0295(2) α/° 90 |
| b/Å 22.6360(5) β/° 99.839(2) | |
| c/Å 8.09410(10) γ/° 90 | |
| Volume/Å3 | 1810.56(6) |
| Z | 8 |
| Dcalc./g cm−3 | 1.483 |
| μ/mm−1 | 3.172 |
| F(000) | 824.0 |
| Crystal size/mm−1 | 0.22 × 0.15 × 0.12 |
| Radiation | CuKα (λ = 1.54184) |
| 2Θ range for data collection/° | 3.906 to 77.142 |
| Index range | −12 ≤ h ≤ 12, −26 ≤ k ≤ 27, −7 ≤ l ≤ 9 |
| Reflections collected | 13,128 |
| Independent reflections | 3560 [Rint = 0.0265, Rsigma = 0.0242] |
| Data/restrains/parameters | 3560/0/236 |
| Goodness-of-fit on F2 | 1.179 |
| Final R indexes (I) | R1 = 0.0380, wR2 = 0.1053 |
| Final R indexes (all data) | R1 = 0.0385, wR2 = 0.1056 |
| Largest diff. peak/hole/e Å−3 | 1.309/−0.460 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morita, N.; Kurami, S.; Ishii, N.; Tanaka, K., III; Hashimoto, Y.; Tamura, O. (E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole. Molbank 2024, 2024, M1769. https://doi.org/10.3390/M1769
Morita N, Kurami S, Ishii N, Tanaka K III, Hashimoto Y, Tamura O. (E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole. Molbank. 2024; 2024(1):M1769. https://doi.org/10.3390/M1769
Chicago/Turabian StyleMorita, Nobuyoshi, Saki Kurami, Naho Ishii, Kosaku Tanaka, III, Yoshimitsu Hashimoto, and Osamu Tamura. 2024. "(E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole" Molbank 2024, no. 1: M1769. https://doi.org/10.3390/M1769
APA StyleMorita, N., Kurami, S., Ishii, N., Tanaka, K., III, Hashimoto, Y., & Tamura, O. (2024). (E)-5-[Bromo(phenyl)methylene]-4-phenyl-2-(p-tolyl)-4,5-dihydrooxazole. Molbank, 2024(1), M1769. https://doi.org/10.3390/M1769