1. Introduction
Figure 1 and
Figure 2 show two previous compounds reported related to this project [
1,
2,
3]. The compounds are
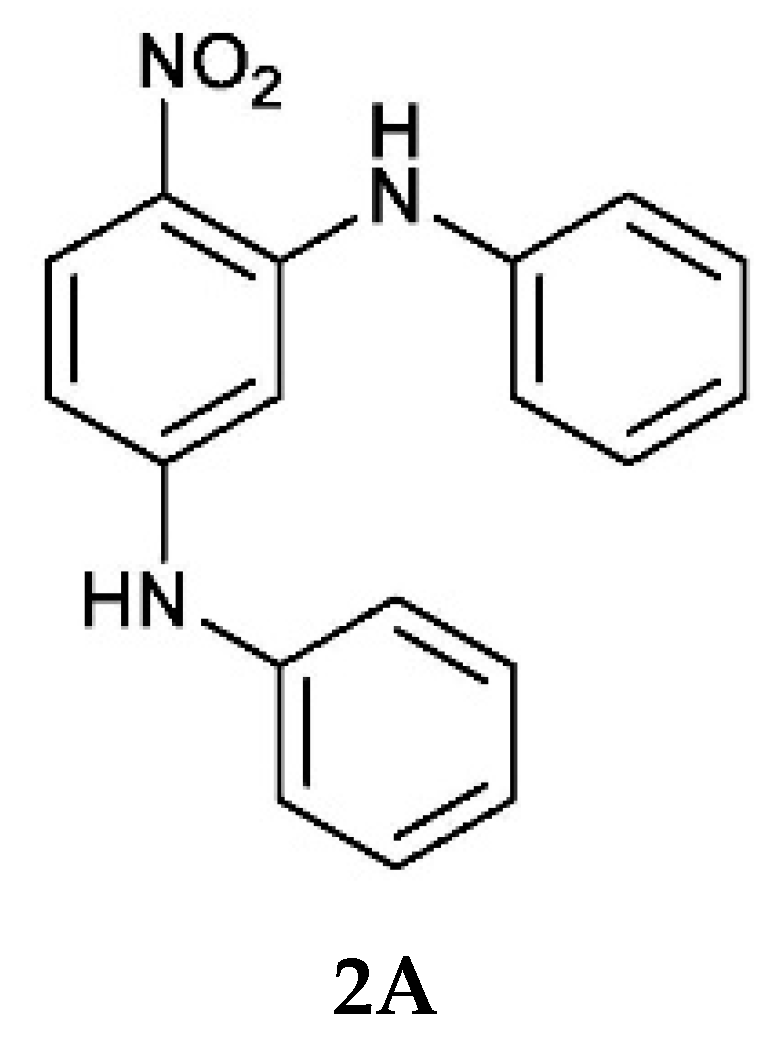
1A and
2A. Drawings
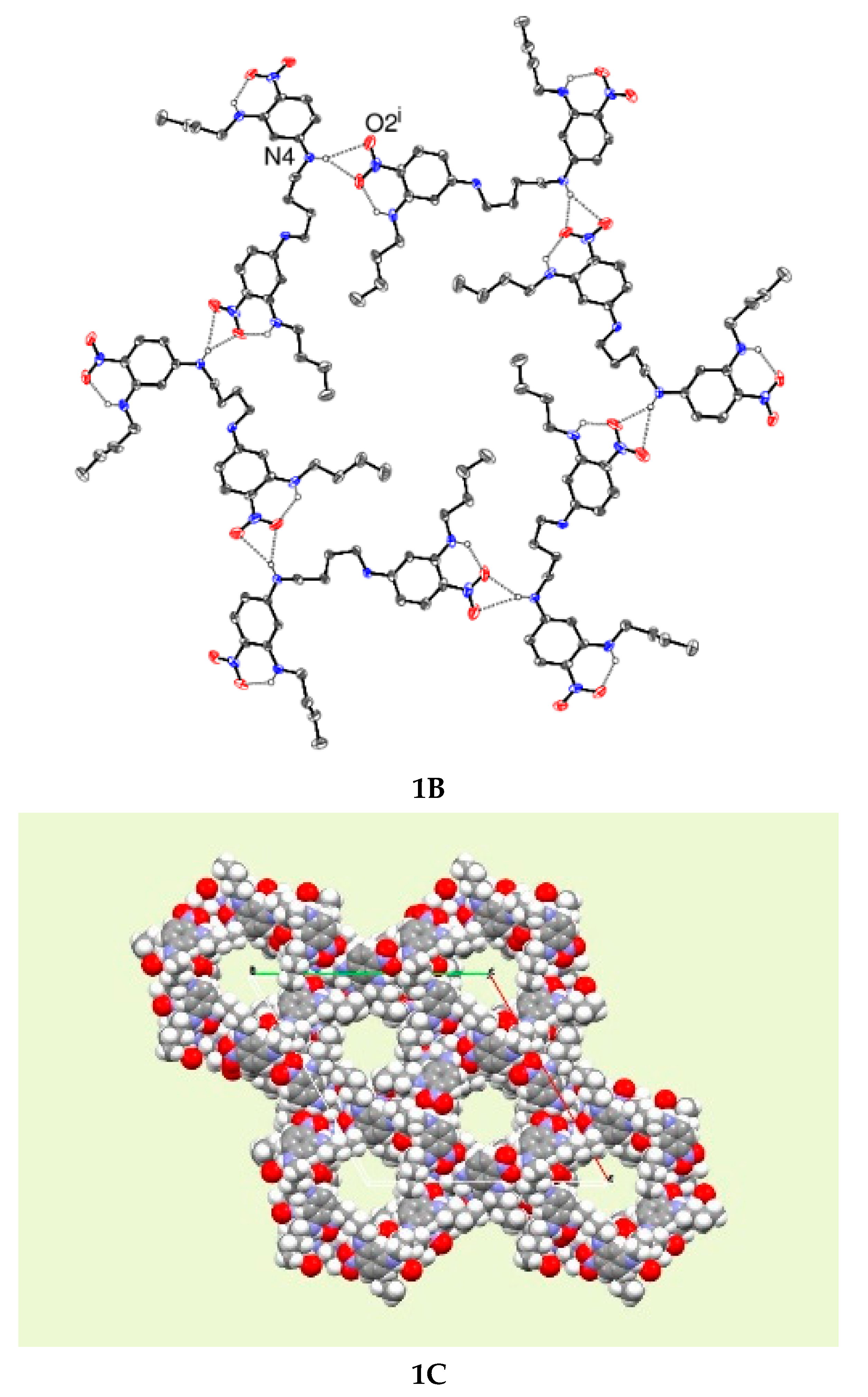
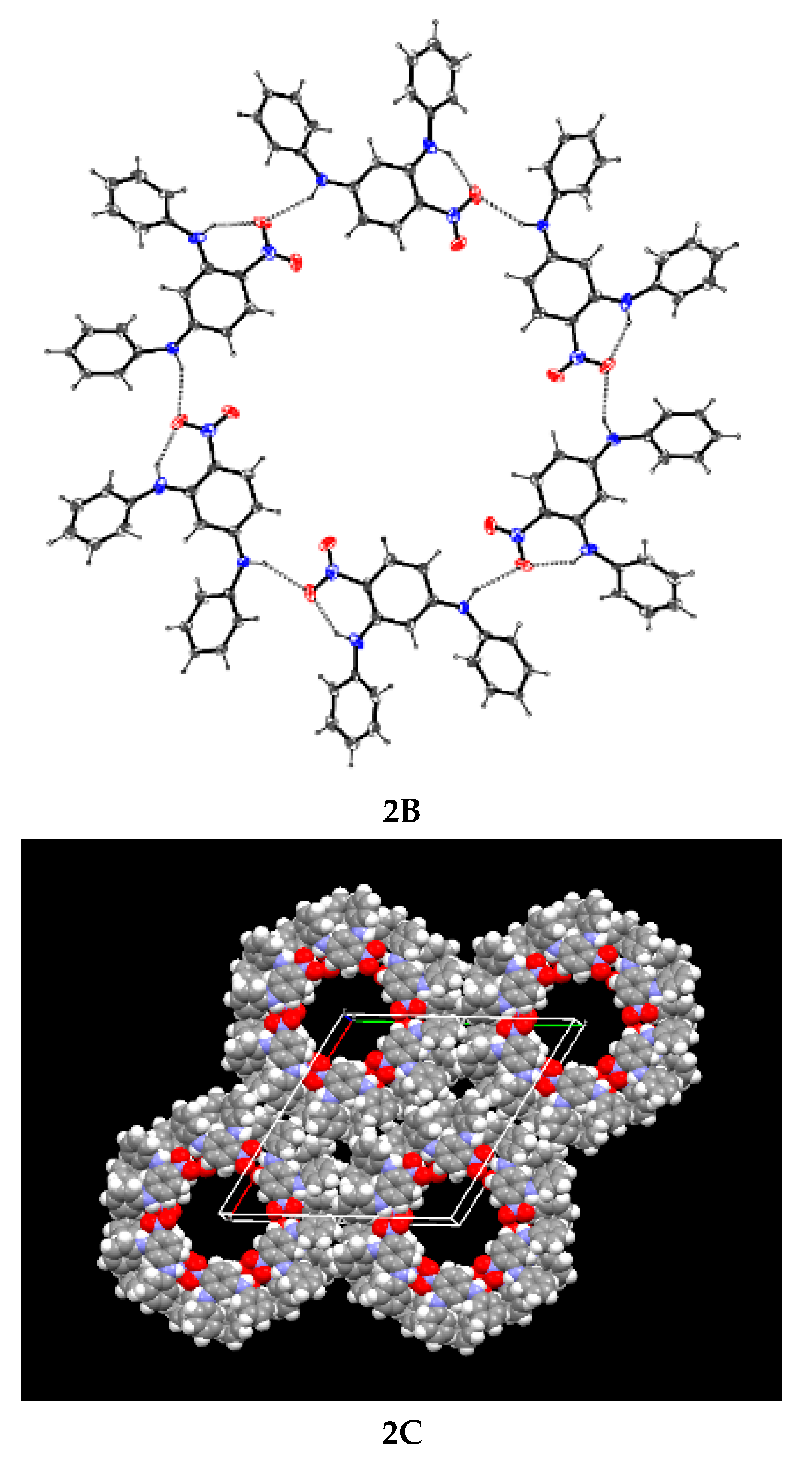
1B and
2B are packing diagrams showing how channels form and drawings
1C and
2C show the open framework channels in the lattices.
In compound 1, the butyl side chains pack forming a channel stabilised by two different hydrogen bonds. One intramolecular bifurcated hydrogen bond forms from the butylamino group to the nitro group and an intermolecular bifurcated hydrogen bond forms to the nitro group from the NH hydrogen atom of the diaminobutane side chain. These hydrogen bonds stabilise the packing.
Compound 2 is stabilised by bifurcated hydrogen bonds formed between the nitro group and two PhNH hydrogen atoms. The structure is unusual because, instead of forming infinite chains, it has a hexameric motif. The work reported in this paper enhances our knowledge and understanding of how different, but related, compounds crystallise.
2. Discussion
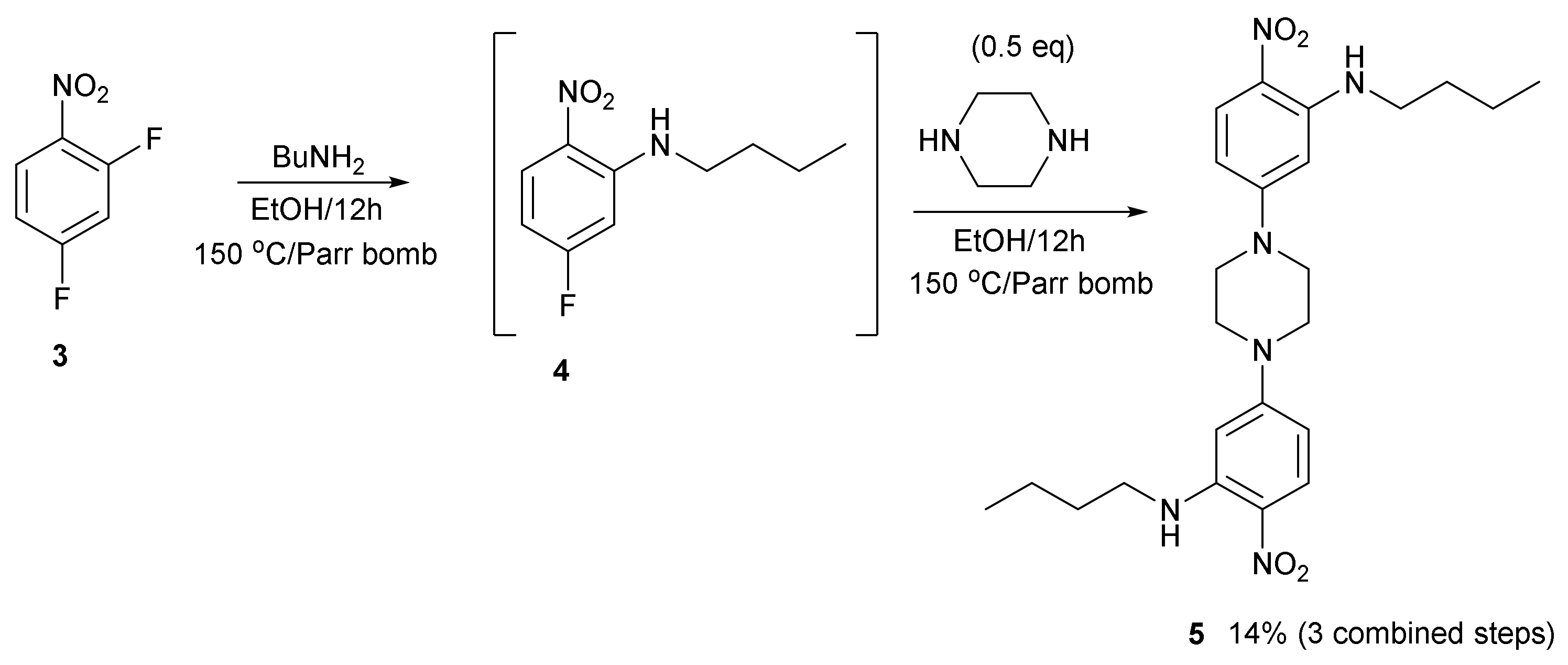
Compound
5 is reported in this paper (
Figure 3) and the spectroscopic data is in the
Supplementary Materials. It was made by the sequential treatment of 2,4-difluoronitrobenzene
3 with butylamine and then piperazine in a digestion bomb at 150 °C. Three steps were combined into one step. The fluorine atom
ortho to the nitro group displaces first with good selectivity. Unlike other similar building blocks, the crystal structure of compound
5 is closely packed, rather than porous (
Figure 4). However, the knowledge gained helped us to construct a picture of what types of molecule to make using this approach, which is likely to lead to open framework porous materials. Firstly, diversity is possible here as there are two sites, two amines, where the molecular structure can be varied. Secondly, compound
5 is a more rigid molecule, which packs closely compared to the more flexible framework of compound
1, which packs forming channels. Further studies will continue to investigate flexibility versus rigidity as a predictive way of uncovering open framework channels in crystal structures.
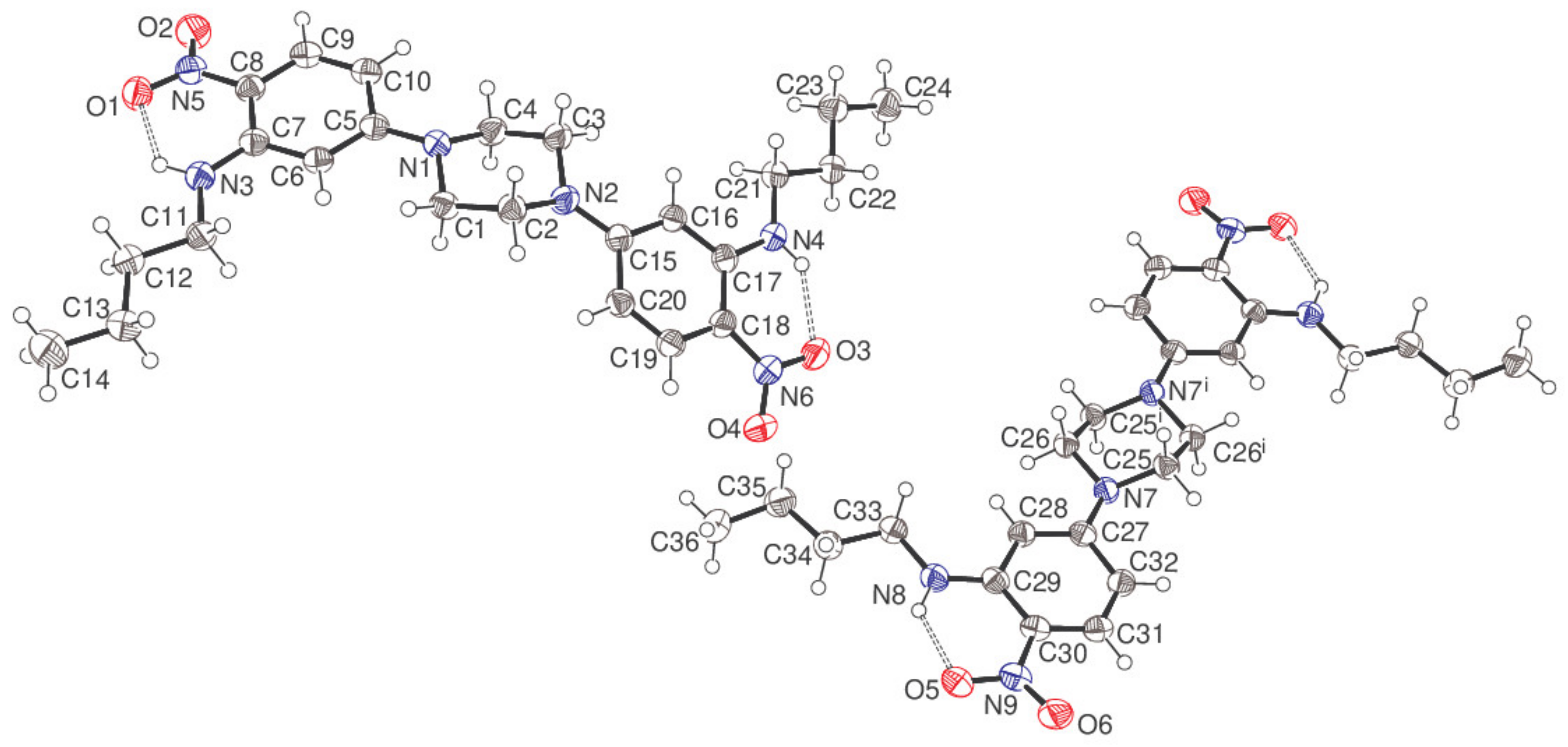
The asymmetric unit of compound 5 consists of a complete C24H34N6O4 molecule (containing C1) and a half-molecule (containing C25), with the latter completed by crystallographic inversion symmetry at the centre of the piperazine ring, which results in the uncommon situation of Z = 12 in the orthorhombic space group Pbca.
The piperazine ring of the C1 molecule adopts a typical chair conformation with the exocyclic N1—C5 and N2—C15 bonds in equatorial orientations. The dihedral angles between the central C1–C4/N1/N2 ring (all atoms) and the adjacent C5–C11 and C15–C20 aromatic rings are 11.95 (7) and 34.16 (7)°, respectively. The N5/O1/O2 and N6/O3/O4 nitro groups are close to co-planar with their attached rings (dihedral angles = 11.72 (16)° and 7.51 (15)°, respectively), with these conformations being reinforced by intramolecular N3—H1n
…O1 [H
…O = 1.933 (19) Å, N—H
…O = 130.7 (15)°] and N4—H2n
…O3 [1.95 (2) Å, 134.2 (16)°] hydrogen bonds, both of which close
S(6) rings, using the topological graph-set classification scheme of Bernstein [
4]. Both
N-butyl chains adopt extended conformations with all torsion angles within 4° of ±180°. Otherwise, all the bond lengths and angles may be regarded as normal.
In the C25 molecule, the dihedral angle between the piperazine ring and the pendant C27–C32 benzene ring attached via the exocyclic N7—C27 bond is 22.45 (7)° and an intramolecular N8—H3n…O5 hydrogen bond [1.877 (19) Å, 131.8 (15)°] is observed, which presumably establishes a near coplanar conformation for the C27–C32 ring and the N9/O5/O6 nitro group (dihedral angle = 9.37 (17)°). The conformation of the N-butyl chain deviates slightly further from fully extended than in the C1 molecule, with the smallest torsion angle being C33—C34—C35—C36 at 169.24 (15)°.
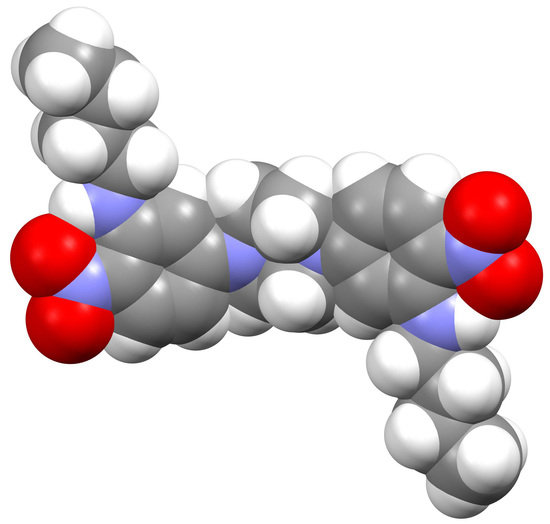
In the extended structure of compound
5, some weak C—H
…O interactions with H
…O separations in the range 2.45–2.60 Å and very weak aromatic π–π stacking contacts (shortest centroid–centroid separation = 3.9458 (9) Å) may help to consolidate the packing, which is otherwise presumably dominated by Van der Waals’ interactions. A PLATON [
5] analysis of the packing indicated that there was no free space and the structure may, therefore, be regarded as ‘closely packed’.
3. Experimental Procedure
IR spectra were recorded on a diamond Attenuated Total Reflection (ATR) Fourier transform infrared (FTIR) spectrometer (ThermoScientific, Hemel Hempstead, UK); Ultraviolet (UV) spectra were recorded using a PerkinElmer Lambda 25 UV-Vis spectrometer (PerkinElmer, Coventry, UK) with EtOH as the solvent. The term sh means shoulder. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 and 100.5 MHz, respectively, using a Bruker 400 spectrometer (Bruker, Oxford, UK). Chemical shifts, δ, are given in ppm and measured by comparison with the residual solvent. Coupling constants, J, are given in Hz. High-resolution mass spectra were obtained at the University of Wales, Swansea, using an Atmospheric Solids Analysis Probe (ASAP) (Positive mode) Instrument: Xevo G2-S ASAP (SpectraLab, Markham, ON, Canada). Melting points were determined with a Dynalon (Raptor Supplies, London, UK).
3.1. Synthesis of Bis(3-butylamino-4-nitro)piperazine 5
2,4-Difluoronitrobenzene 3 (500 mg, 3.15 mmol) in EtOH (20 mL) was treated with butylamine (230 mg, 3.15 mmol) and triethylamine (635 mg, 6.3 mmol) and heated at 150 °C for 12 h in a 45 mL PTFE lined Parr digestion bomb. After cooling, the mixture was treated with piperazine (135 mg, 1.57 mmol), sealed, and heated again for 12 h. After cooling, maroon needle-like crystals of the title compound (350 mg, 0.74 mmol, 47%), mp 197–198 °C, were harvested. These were washed with water and then with a small amount of 10% EtOH/H2O and allowed to dry. 1H NMR (400 MHz; CDCl3) 1.02 (6H, t, J = 7.0), 1.51 (4H, m), 1.77 (4H, m), 3.28 (4H, t, J = 7.0), 3.68 (8H, s), 5.87 (2H, d, J = 2.5), 6.22 (2H, d, J = 8.0), 8.14 (2H, d, J = 8.0) and 8.41 (2H, s, NH, br); 13C NMR (100.1 MHz; CDCl3) 13.8, 20.4, 30.9, 42.7, 45.9, 92.9, 103.3, 124.5, 129.3, 147.8 and 155.1; (ATR Diamond) (cm−1) 3345 (w), 2957 (w), 2928 (w), 2859 (w), 1612 (vs), 1574 (vs), 1519 (s), 1489 (s), 1470 (w), 1411 (w), 1386 (w), 1323 (s), 1265 (s), 1194 (vs), 1175 (vs), 1026 (s), 987 (s), 913 (w), 819 (w), 803 (w), 790 (w), 749 (w), 668 (w), 617 (w), 591 (w), 542 (s) and 468 (w); UV/Vis (EtOH): λmax (ε) = 414 (7943), 270 nm (10,000) and 240 (9150) mol−1dm3cm−1); HRMS (ASAP Orbitrap) m/z calcd for C24H36N6O4 + H+: 471.2720; found: 471.2720 100%.
3.2. Single-Crystal Diffraction
The crystal structure of compound 5 (yellow block, 0.14 × 0.09 × 0.04 mm, recrystallized from dichloromethane/light petroleum ether) was established using intensity data collected on a Rigaku AFC11 CCD diffractometer (Cu Kα radiation, λ = 1.54178 Å) at 100 K. An analytical absorption correction was applied, and the structure was routinely solved by dual-space methods using SHELXT and the structural model completed and optimized by refinement against |F|2 with SHELXL-2018. The N-bound H atoms were located in difference maps and their positions were freely refined. The C-bound H atoms were placed geometrically (C—H = 0.95–0.99 Å) and refined as riding atoms: the methyl groups were allowed to rotate, but not to tip, to best fit the electron density. The constraint Uiso(H) = 1.2Ueq(carrier) or 1.5Ueq(methyl carrier) was applied in all cases. Full details of the structure and refinement are available in the deposited cif.
Crystal data for compound 5 (C24H34N6O4): Mr = 470.57, orthorhombic, space group Pbca (No. 61), a = 22.9241 (2) Å, b = 7.32050 (10) Å, c = 42.5522 (4) Å, V = 7140.93 (13) Å3, Z = 12, T = 100 K, μ = 0.745 mm−1, ρcalc = 1.313 g cm−3, 31,395 reflections measured (4.2 ≤ 2θ ≤ 136.5°), 6453 unique (Rint = 0.039), R(F) = 0.043 (5944 reflections with I > 2σ(I)), wR(F2) = 0.113 (all data), Δρmin,max (e Å−3) = −0.18, +0.20, CCDC deposition number 2174311.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}