5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one

Abstract
:1. Introduction
2. Results
3. Materials and Methods
3.1. General
3.2. Synthesis of Chalcone Derivative
3.3. Synthesis of the Target Compound
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kappe, C.O.; Stadler, A. The Biginelli dihydropyrimidine synthesis. Org. React. 2004, 63, 1–116. [Google Scholar]
- Jadhav, V.B.; Holla, H.V.; Tekale, S.U.; Pawar, R.P. Bioactive dihydropyrimidines: An overview. Chem. Sin. 2012, 3, 1213–1228. [Google Scholar]
- Ozair, A.; Khan, S.A.; Siddiqui, N.; Ahsan, W.; Verma, S.P.; Gilani, S.J. Antihypertensive activity of newer 1,4-dihydro-5-pyrimidine carboxamides: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2010, 45, 5113–5119. [Google Scholar]
- Lloyd, J.; Finlay, H.J.; Atwal, K.; Kover, A.; Prol, J.; Yan, L.; Rao, B.; Vaccaro, W.; Huynh, T.; Huang, C.S.; et al. Dihydropyrazolopyrimidines containing benzimidazoles as K(V)1.5 potassium channel antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 5469–5473. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Mishra, M.; Saxena, N.; Yadav, G.P.; Maulk, P.R.; Sahoo, M.K.; Gaur, R.L.; Murthy, P.K.; Tripathi, R.P. Synthesis of 2-sulfanyl-6-methyl-1,4-dihydropyrimidines as a new class of antifilarial agents. Eur. J. Med. Chem. 2008, 43, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, C.; Ok, T.; So, W.; Jo, M.; Seo, M.; Kim, Y.; Sohn, J.H.; Park, Y.; Moon, K.J.; et al. A novel 3,4-dihydropyrimidin-2(1H)-one: HIV-1 replication inhibitors with improved metabolic stability. Bioorg. Med. Chem. Lett. 2012, 22, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, F.E.; De Clerq, E. Synthesis and anti-HIV-1 activity evaluation of 5-alkyl-2-alkylthio-6-(arylcarbonyl or alpha-cyanoarylmethyl)-3,4-dihydropyrimidin-4(3H)-ones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 2007, 50, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Ulaganathan, V.; Rath, O.; Prokopcov, H.; Dallinger, D.; Kappe, C.O.; Kozielski, F. Structural basis for inhibition of Eg5 by dihydropyrimidines: Stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J. Med. Chem. 2010, 53, 5676–5683. [Google Scholar] [CrossRef] [PubMed]
- Suwito, H.; Jumina; Mustofa; Pudjiastuti, P.; Fanani, M.Z.; Kimata-Ariga, Y.; Katahira, R.; Kawakami, T.; Fujiwara, T.; Hase, T.; et al. Design and synthesis of chalcone derivatives as inhibitors of the ferredoxin—Ferredoxin-NADP+ reductase interaction of Plasmodium falciparum: Pursuing new antimalarial agents. Molecules 2014, 19, 21473–21488. [Google Scholar] [CrossRef] [PubMed]
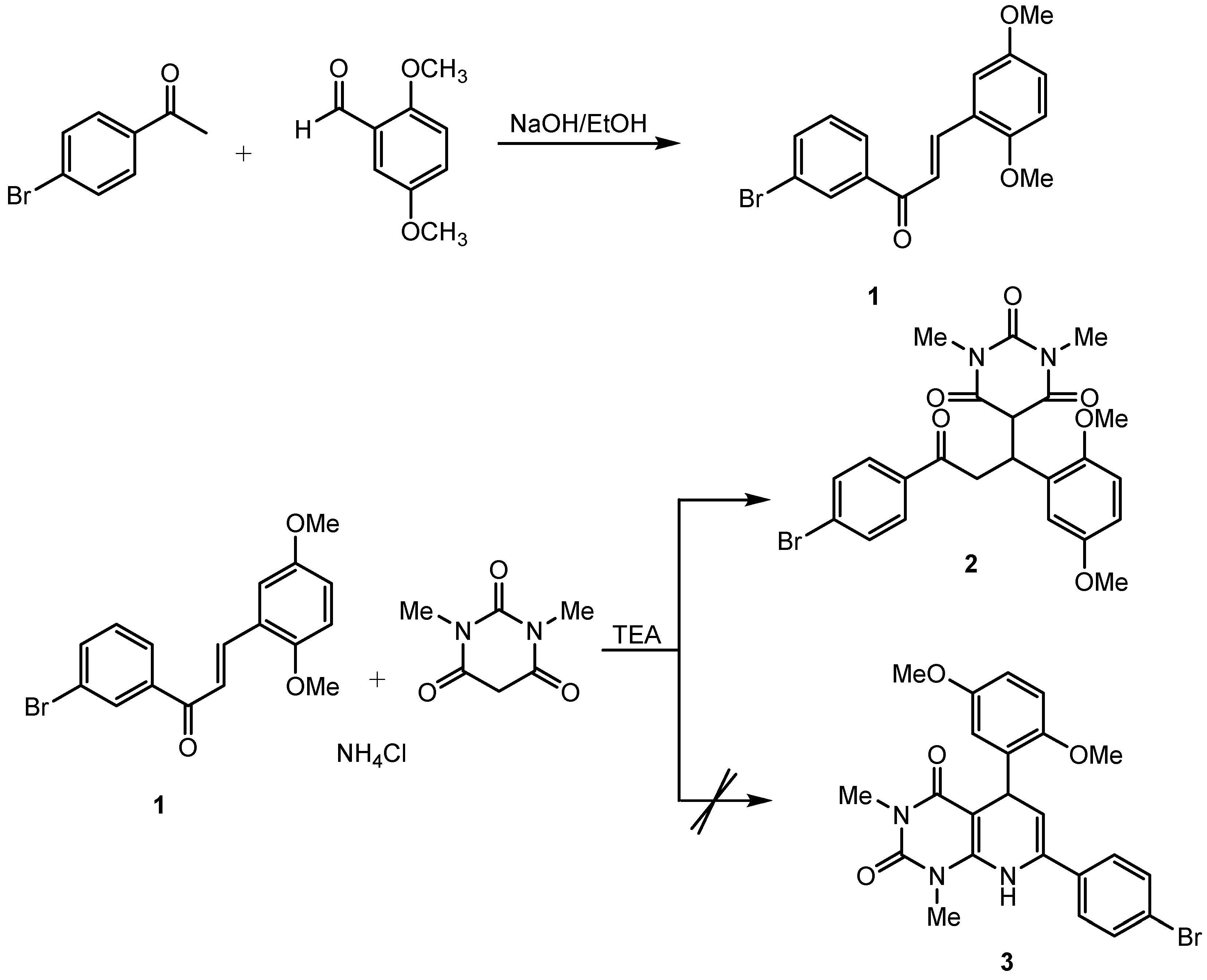

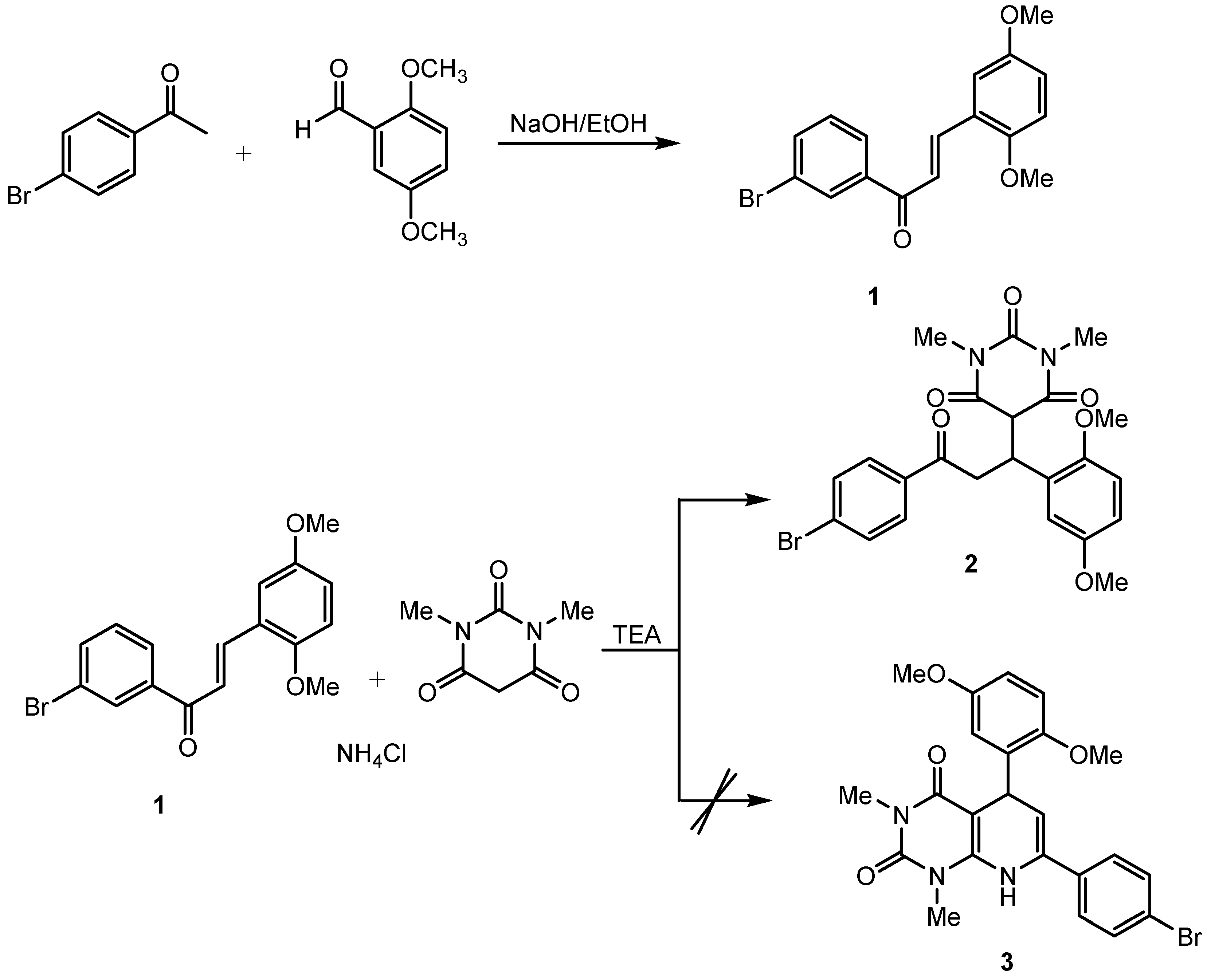{kind=link}
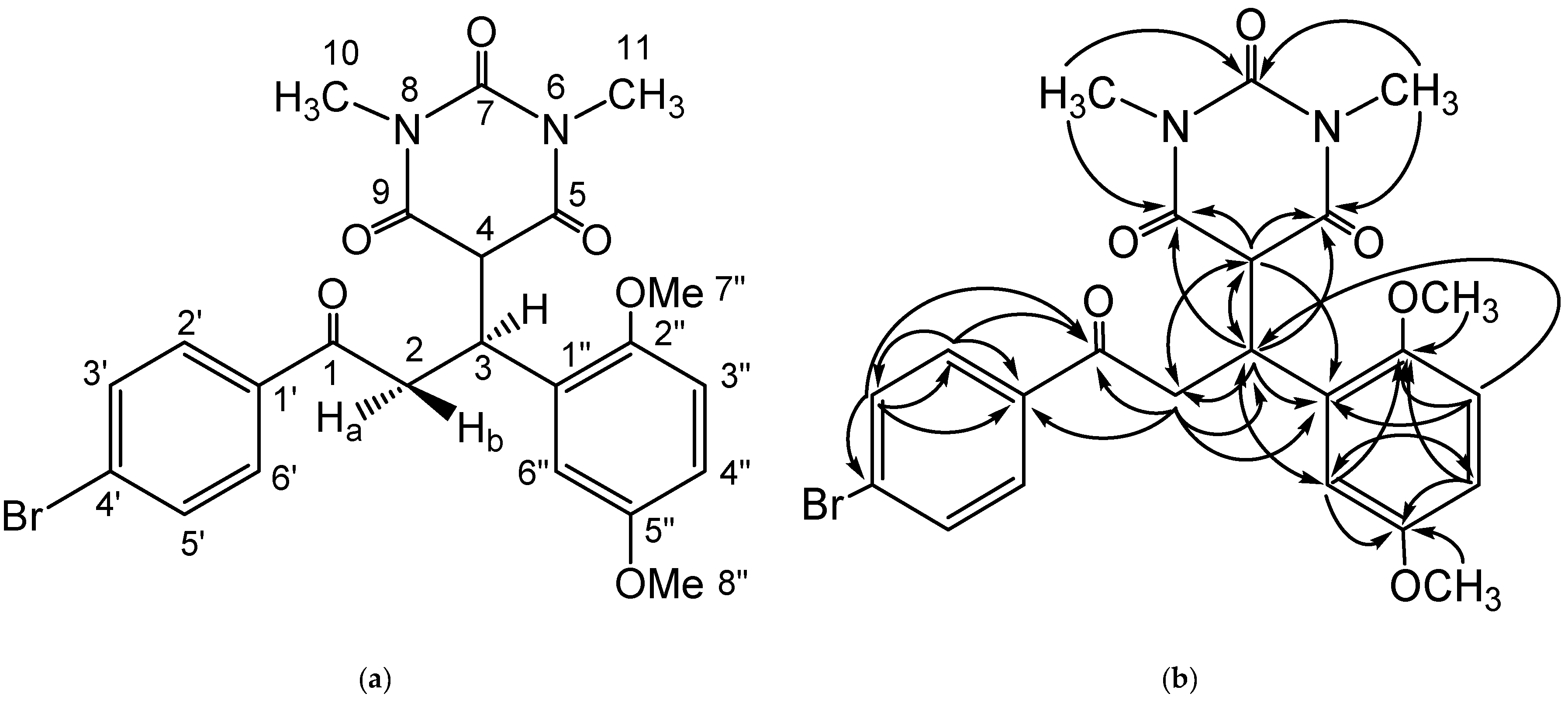
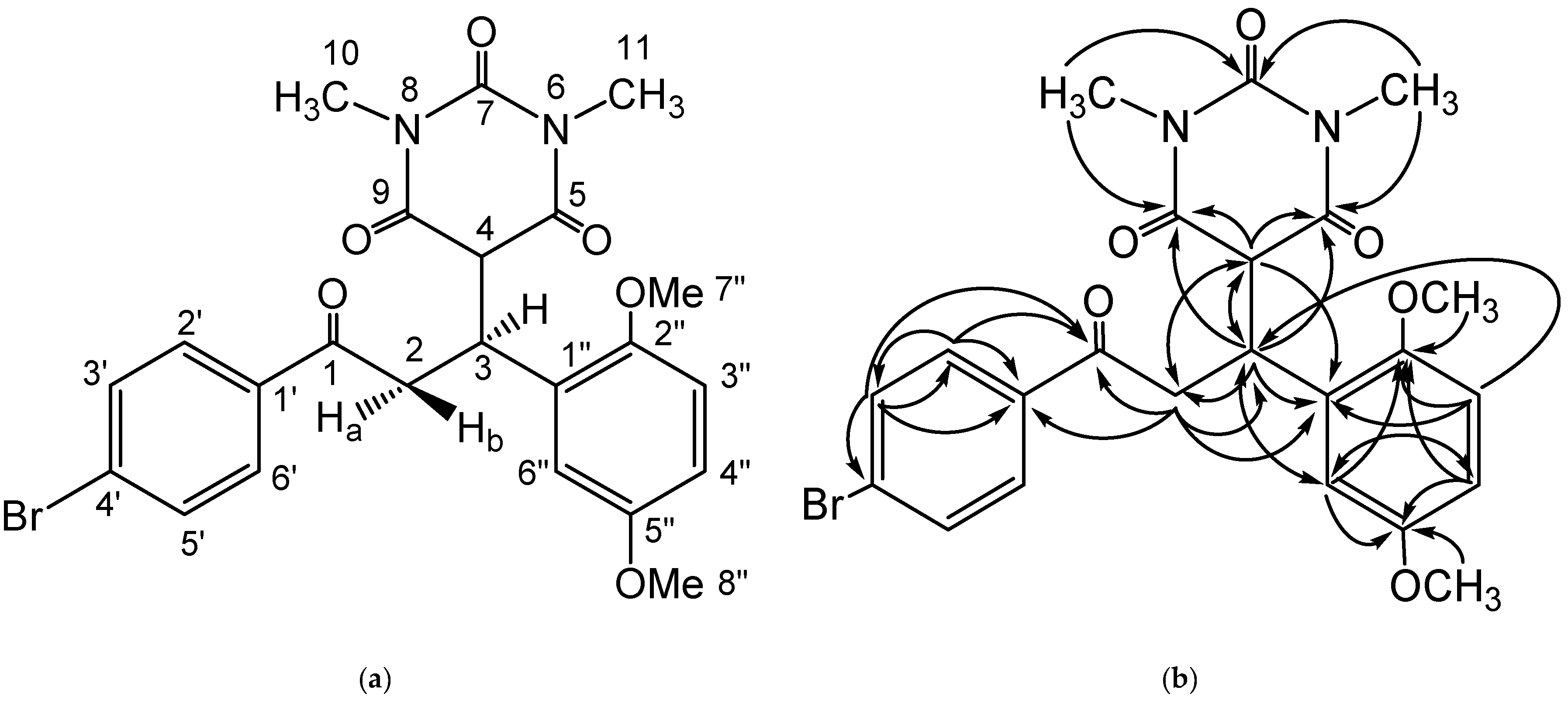{kind=link}
| No. Atom | δH (Mult, J Hz) | δC (ppm) | HMBC |
|---|---|---|---|
| 1 | - | 197.9 | |
| 2 | Ha = 3.90 (dd, 1H, J = 18.3 Hz, J = 8.3 Hz) | 40.5 | C-1, C-3, C-4, C-1′, C-1″ |
| Hb = 3.49 (dd, 1H, J = 18.3 Hz, J = 6.2 Hz) | |||
| 3 | 4.43 (ddd, 1H, J = 8.3 Hz, J = 6.2 Hz, J = 4.4 Hz) | 37.1 | C-2, C-4, C-5, C-9, C-1″, C-6″ |
| 4 | 3.79 (d, 1H, J = 4.4 Hz) | 53.2 | C-2, C-3, C-5, C-9, C-1″ |
| 5 | - | 168.7 | |
| 6 | - | - | |
| 7 | - | 151.3 | |
| 8 | - | - | |
| 9 | - | 168.3 | |
| 10 | 2.93 (s, 3H) | 28.21 | C-9, C-7 |
| 11 | 2.92 (s, 3H) | 28.31 | C-5, C-7 |
| 1′ | - | 127.9 | |
| 2′, 6′ | 7.92 (d, 2H, J = 8.5 Hz) | 132.3 | C-1, C-1′, C-3′ |
| 3′, 5′ | 7.75 (d, 2H, J = 8.4 Hz) | 130.5 | C-1, C-1′, C-2′, C-4′ |
| 4′ | - | 136.1 | |
| 1″ | - | 128.3 | |
| 2″ | - | 153.4 | |
| 3″ | 6.85 (d, 1H, J = 8.9 Hz) | 112.1 | C-3, C-1″, C-2″ |
| 4″ | 6.77 (dd, 1H, J = 8.9 Hz, J = 2.9 Hz) | 113.3 | C-2″, C-5″, C-6″ |
| 5″ | - | 151.9 | |
| 6″ | 6.61 (d, 1H, J = 2.9 Hz) | 115.2 | C-3, C-2″, C-4″, C-5″ |
| 7″ | 3.64 (s, 3H) | 56.3 | C-2″ |
| 8″ | 3.65 (s, 3H) | 55.9 | C-5″ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suwito, H.; Sari, R.H.P.; Ul Haq, K.; Kristanti, A.N. 5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one. Molbank 2018, 2018, M1013. https://doi.org/10.3390/M1013
Suwito H, Sari RHP, Ul Haq K, Kristanti AN. 5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one. Molbank. 2018; 2018(3):M1013. https://doi.org/10.3390/M1013
Chicago/Turabian StyleSuwito, Hery, Ria Hesty Purnama Sari, Kautsar Ul Haq, and Alfinda Novi Kristanti. 2018. "5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one" Molbank 2018, no. 3: M1013. https://doi.org/10.3390/M1013
APA StyleSuwito, H., Sari, R. H. P., Ul Haq, K., & Kristanti, A. N. (2018). 5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one. Molbank, 2018(3), M1013. https://doi.org/10.3390/M1013