Abstract
TP53-mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) comprise a distinct subgroup of myeloid neoplasms with unique biological and clinical features. The molecular alterations linked to TP53 mutations drive genomic instability and treatment resistance and ultimately lead to poor survival outcomes. The disease biology is further shaped by alterations in immune response within the bone marrow microenvironment and significant changes in cellular metabolism. Conventional treatments, including chemotherapy and hypomethylating agents +/− venetoclax, offer limited benefit, with high relapse rates and short remissions. Allogeneic bone marrow transplantation is the only curative approach, but the vast majority of patients relapse. Novel therapeutic approaches—ranging from p53 reactivation strategies to immunotherapy and targeted inhibition of specific signaling pathways—are under active investigation. Our review summarizes current knowledge on the molecular pathogenesis, prognostic implications, and therapeutic landscape of TP53-mutated MDS/AML and discusses ongoing challenges and opportunities for improving patient outcomes.
1. Introduction
P53 is a well-known and broadly investigated tumor suppressor protein with a crucial role in cell cycle regulation, response to DNA damage, and apoptosis [1]. As such, alterations in the TP53 gene are common across all cancer types and are typically associated with resistance to treatment and adverse prognosis [1,2]. Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are myeloid neoplasms (MNs) caused by somatic mutations in stem and progenitor hematopoietic cells. MDS is thought to be the result of evolving clonal hemopoiesis (CH), and high-risk disease exhibits a high frequency of transformation to AML (secondary AML) [3]. AML is generally characterized by a blast percentage > 20%, but new classification systems propose the MDS/AML entity, with blast count range of 10–20%, which represents a disease continuum rather than two distinct malignancy states [4,5].
TP53 mutations are present in approximately 10% of patients with MDS and AML and are even more prevalent (70%) in MNs with complex karyotype (CK) [6,7]. The TP53 gene and its locus on chromosome 17p13.1 can undergo a variety of alternations, but the first event is usually a missense mutation, followed by a bi-allelic loss due to loss of heterozygosity (LOH) and other structural variations. Loss of p53 function promotes genomic instability, resulting in complex chromosomal aberrations and, ultimately, chemoresistance and dismal outcomes for patients [7,8].The underlying mechanism of leukemogenesis is not fully understood, but it has been shown that TP53 mutations exist in founding clones and are selected under cytotoxic pressure. This also explains the higher prevalence of TP53 mutations in older patients and in therapy-related myeloid neoplasms (t-MNs) [6,9].
Additionally, in terms of treatment options, intensive chemotherapy is very rarely used, and the addition of Bcl-2 inhibition by venetoclax to hypomethylating agents (HMAs), a combination widely used in older patients ineligible for transplantation, has not demonstrated prominent responses. Allogeneic bone marrow transplantation (Allo-BMT) is the only potentially curative option, yet relapse rates remain high [8]. Moreover, a very small subset of patients achieve the depth of remission required for Allo-BMT. P53-targeting strategies are promising, but developed agents did not exhibit a substantial effect in late-phase clinical trials [3]. Because of this distinctive genomic and clinical profile, TP53-mutated MDS/AML has been recently classified separately as a unique disease entity in the World Health Organization (WHO-5) and International Classification Consortium (ICC) systems [4,5].
In this narrative review, we summarize the published data on the molecular profile of TP53-mutated MDS/AML and its correlation with clinical outcomes in patients. We also aim to describe current therapeutic strategies and novel agents under investigation in preclinical studies and clinical trials, highlighting the unmet needs in this patient subset.
2. Molecular Events and Related Changes in TP53-Mutated MDS/AML
2.1. P53 as “The Guardian of the Genome”
P53 tumor protein is a 53-kilodalton protein encoded by the TP53 gene on the locus of chromosome 17p13.1. Its tertiary structure consists of two N-terminal transactivation functional domains, a central DNA-binding domain (DBD), and a C-terminus encoding its nuclear localization signals and an oligomerization domain for transcriptional activity. P53 plays a critical role in a variety of cellular processes—cell cycle arrest, inflammation, apoptosis, autophagy, metabolism, cell senescence, and genomic stability—through cell growth inhibition in response to DNA damage (“the guardian of the genome”) [1,3]. Recent studies also suggest that abnormal p53 protein can alternate the tumor immune microenvironment, proposing a role in tumor–immune cell interactions [10,11,12].
P53 levels are kept under strict control and regulated by post-transcriptional modifications including ubiquitination, acetylation, and phosphorylation. Mouse double minute-2 homolog (MDM2) is a ubiquitin E3 ligase which drives p53 ubiquitination, leading to proteasomal degradation in the absence of genomic or cellular stress. While MDM2 protein controls p53 stability, MDM4 protein (also called MDMX) inactivates p53 by occluding its transactivation domain. Cellular stress due to hypoxia or DNA damage inhibits MDM2 function, leading to inhibition of p53 ubiquitination and stabilization in its tetrameric form [13]. Activated p53 protein serves primarily as a transcriptional factor with a broad spectrum of target genes involved in almost all cellular processes [1,3].
2.2. Alternations of TP53 Gene Locus and p53 Function in Myeloid Neoplasms
The TP53 gene and its chromosomal locus exhibit substantial heterogeneity of alternations. Most mutations are missense mutations present inside the DNA-binding domain of the protein, followed by insertions/deletions, splice-site mutations, and truncating mutations. Missense mutations affecting the DNA-binding domain result in p53’s misfolding and, thus, an impaired ability to bind to transcriptional targets. P53 can also be inactivated by TP53 gene deletions, alternative splicing, post-transcriptional modifications, or MDM2-MDM4 upregulation [6]. While TP53-mutated MNs share some common hotspot mutations with other cancers (R175, R248, R273, and R282), these neoplasms are also characterized by unique hotspots (Y220 and M237) [14,15]. In some cases, TP53 mutations have a dominant-negative effect due to the ability of mutated p53 to form tetramers with the wild-type protein, leading to its inactivation [16]. Nevertheless, missense variants may lead to gain-of-function effects by enabling p53-mutated protein to engage in neomorphic interactions with other transcriptional factors. Besides mutations, loss-of-function can also occur from loss of heterozygosity (LOH) or copy-neutral loss of heterozygosity (cnLOH) [6,7].
In most patients with TP53-mutated MDS/AML (76%), there is bi-allelic inactivation of the TP53 gene. Mechanistically, this is primarily a consequence of ≥2 mutations in 18.4–28.8% of patients, a single mutation with deletion of the trans wild-type allele in 22.5–42.2% of patients, and a single mutation with concomitant cnLOH in 12.2–20.6% of patients. Bi-allelic TP53 mutations are associated with CK, higher blast count, and higher leukemic transformation [6,17,18].
2.3. Concurrent Mutations and Chromosomal Aberrations
In a recent genomic characterization of TP53-mutated MNs, whole-genome sequencing (WGS) was performed by analyzing tumor/normal paired samples from 42 patients. In this study, it was shown that mutations in common signaling genes, such as KIT, FLT3, and WT1 are very rarely detected, while mutations in epigenetic modifiers (DNMT3, TET2, or ASXL1) or spliceosome genes (SF3B1, SRSF2) are relatively infrequent (14% and 4.8% cumulative frequency, respectively) [7]. Similar results were published in a previous study including 2200 AML/MDS-with excess blasts (MDS-EB) samples [6]. Indeed, compared with TP53 wild-type, TP53-mutated MNs are characterized by a paucity of concurrent mutations [15,18].
In the latter study, the majority of patients (84%) with TP53-mutated AML/MDS-EB had complex karyotype (i.e., ≥3 concurrent cytogenetic abnormalities) and CK was even more commonly observed in patients with bi-allelic TP53 mutations (97%), multiple TP53 mutations (94%), and in patients with larger TP53-mutated clones (94%, defined by variant allele frequency of >40%) [6]. There is also a reverse relation as follows: TP53 mutations are observed in 4.5% of cases with normal karyotype, increasing to 17.3% with two chromosomal aberrations and, ultimately, to 76.8% in CK cases. This interrelation is repeated within CK cohorts. In parallel, the paucity of concurrent mutations is more prevalent in multi-hit TP53-mutated MNs compared with single-hit [15,18]. These data suggest that even among TP53-mutated MNs, multi-hit TP53 inactivation is a molecularly distinct entity compared with single-hit.
Some additional interesting findings include reduced ETV6 gene expression in patients with TP53-mutated MDS/AML. Possible etiologies include copy-number losses of its chromosomal locus and deletions of the gene, but also unknown mechanisms. Moreover, in TP53-mutated AML, NF1 gene mutations are highly overrepresented, with deletions of one copy of the gene in 45% of cases and bi-allelic mutations in 17%. Furthermore, a surprising finding was that in TP53-mutated AML, telomere content was found to be amplified compared to other AML types, with abnormal sequences detected in interstitial chromosomal regions [7].
Regarding chromosomal aberrations, bi-allelic TP53 mutations are strongly linked to complex karyotype (Table 1). In a pool of 42 patient samples analyzed by WGS, recurrent copy-number losses and cytogenetic changes were detected, such as loss of chromosomes 5, 7, 12, 16, 17, 18, and 20q, and gains of chromosomes 21, 22, 1p, and 8 [7]. Furthermore, in myeloid malignancies with TP53 mutations, chromothripsis—a phenomenon of extensive, randomly oriented chromosomal rearrangements resulting from faulty DNA double-strand break repair—occurs in ~35% of cases [19,20]. This event most frequently involves chromosomes 5, 17, and 21 and ultimately contributes to the formation of complex karyotype [7]. TP53-mutated MDS/AML patients with complex cytogenetics have a very poor prognosis, as discussed later in this review.

Table 1.
Common cytogenetic abnormalities in TP53-mutated MDS/AML.
2.4. TP53 Mutations and Changes in Cellular Functions
P53 regulates a broad spectrum of cellular functions including apoptosis, senescence, glucose and fatty acid metabolism; thus, p53 alternations affect homeostasis beyond genomic instability (Figure 1). In order to assess these effects in cell metabolism, one group performed a multi-omics analysis after developing cell lines with TP53 mutations (R175H, R273H, and R248Q variants). Elevated expression of gene signatures related to inflammatory responses (CXCL8, HSP8) and metabolic processes (ANXA1, DUSP1) was reported in TP53 R175H-mutated cells, along with enrichment of amino acid and RNA metabolism pathways [21]. Interestingly, our laboratory recently reported that loss of p53 induces a direct, cell-autonomous upregulation of interferon gamma signaling via upregulation of C-C motif chemokine receptor-like 2 (CCRL2)/JAK2/STAT1, which is associated with venetoclax resistance [22]. These findings are consistent with other studies highlighting an association between TP53 mutations and activation of inflammatory pathways [23].
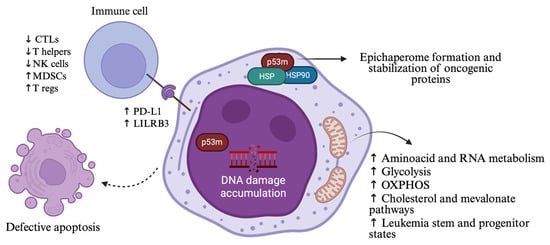
Figure 1.
Alternations in cellular functions and immune microenvironment resulting from TP53 mutations in MDS/AML. Mutant p53 induces genomic instability, impaired apoptosis, immunosuppressive signaling, metabolic rewiring, and formation of epichaperomes that stabilize oncogenic proteins, ultimately contributing to treatment resistance. P53m: mutant p53; HSP: heat shock protein; OXPHOS: oxidative phosphorylation; CTLs: cytotoxic T lymphocytes; NK cells: natural killer cells; MDSCs: myeloid-derived suppressor cells; T regs: T regulatory cells. Created in https://BioRender.com (assessed on 25 September 2025).
In the multi-omics analysis, AML cells harboring TP53 R175H and R273H mutations displayed enhanced oxidative phosphorylation (OXPHOS) and enrichment of leukemia stem cell populations. Some of their findings, including high senescence and OXPHOS states, were associated with poor clinical outcomes [21]. Besides MDS/AML, a pan-cancer analysis of TP53 mutations and related metabolic pathways showed that TP53 plays a key role in glycolysis regulation by suppressing the AKT/mTOR and NF-kB signaling pathways and the expression of related genes, such as PFKP and SLC16A3 [24]. Additionally, in a very recent study investigating metabolic drivers of chemoresistance in TP53-mutated AML, it has been shown that TP53-mutated AML cells resist cytarabine-induced death through upregulation of one-carbon metabolism, that provides greater antioxidant capacity and better cytarabine-induced reactive oxygen species (ROS) management. Remarkably, while stable TP53-mutated AML cells had decreased OXPHOS in this study, cytarabine induced an increase in OXPHOS, and the mevalonate pathway was found to play a significant role in these changes [25]. Upregulation of cholesterol pathway has also been linked to CAR-T cell therapy resistance in TP53-mutated AML cells [26].
Moreover, heat shock proteins (HSPs), such as HSP90 and other cochaperones and associated proteins that regulate proteostasis, can form epichaperomes in malignant cells. Epichaperomes are complexes of these proteins that stabilize oncogenic kinases, including FLT3, and transcription factors, such as mutant p53, further contributing to signaling aberrations [27]. These complexes have been found to promote cell growth and survival in TP53-mutated AML and AML stem/progenitor cells. PU-H71, an epichaperome-specific inhibitor that has been tested in this poor-risk disease, suppresses baseline cellular stress responses and induces apoptosis. Preclinical studies revealed that PU-H71 exhibits a synergistic apoptotic effect with the Bcl-2 inhibitor venetoclax, potentially through Mcl-1 downregulation among other mechanisms. In vivo experiments also revealed promising anti-leukemic activity, raising the hope that malignant cells could be targeted independently of specific mutations [28].
Lastly, while p53 is known to regulate cell cycle arrest and apoptosis through a variety of mechanisms, it was recently found that p53 exhibits its role in cell fate also through ferroptosis, a unique form of iron-dependent cell death mediated by lipid peroxidation. Although most data support that wild-type p53 induce ferroptosis, more research can unveil the effects of mutant p53 and how this can be further exploited in cancer treatment [29].
2.5. TP53-Mutated Clones and the Driving Force of Cancer Treatments
Clonal hemopoiesis (CH) and, especially, clonal hemopoiesis of indeterminate potential (CHIP) become more prevalent with advancing age and have been linked to an increased risk of hematologic malignancies. The TP53 gene is among the top five most frequently mutated genes in CHIP, and the affected hematopoietic stem cells (HSCs) may gain a survival and expansion advantage under certain conditions [30]. Inflammation, bone marrow microenvironment, and prior exposure to chemotherapy, radiation, and immunotherapy can facilitate clonal expansion [3]. TP53-mutated clones are more commonly found in patients presenting with t-MNs (23–37%) [9,15] and in patients with cancer predisposition syndromes, such as Li–Fraumeni and Schwachman syndromes (mutations in TP53 and SBDS genes, respectively) [31]. t-MNs have worse prognosis than de novo MDS and AML [9].
The improved outcomes of cancer survivors have brought light to late complications of these therapies. Cancer survivors have a 4.7-fold-higher risk of leukemia [9] and the relative risk for t-MDS/AML ranges from 1.5 to over 10 after chemotherapy for solid tumors [32]. CH present at the time of primary cancer treatment is associated with a 10-fold increased risk of developing t-MNs [31]. Among chemotherapeutic agents, alkylating agents and topoisomerase II inhibitors are the main promoters of t-MNs, compared with anti-metabolites and taxanes [33]. Poly(ADP-ribose) polymerase inhibitors (PARPis) have been associated with an increased incidence of MDS/AML, particularly in patients harboring TP53 CH mutations and often with a short latency after initial exposure [34]. TP53 mutations were detected in 50–75% of t-MN cases following PARPi, as described in small cohorts [35,36]. t-MNs can also emerge after chimeric antigen receptor (CAR)-T cell therapies with a cumulative incidence of 7.5–9% at three years. Remarkably, TP53 mutations are present in 44.4–50% of t-MN cases, and most patients present with t-MN in less than one year after CAR-T therapy [37,38]. Potential risk factors include older age, higher MCV, and higher ICANS grade [38]. All data support that clones harboring TP53 mutations are present prior to CAR-T therapy [37,38]. In AML patients, pre-leukemic stem cells (preLSCs) harboring somatic TP53 mutations have been identified, proposing that these mutations constitute early leukemogenic events [39].
2.6. The Effect of TP53 Mutations in Immune Microenvironment
Within the bone marrow microenvironment, multiple cell types interact to regulate hemopoiesis in response to external stimuli. Inflammation, and specifically chronic inflammation, plays a key role in MDS pathogenesis and leukemic progression [40,41]. Mutations in RNA splicing and epigenetic regulators, common in MDS, trigger a reciprocal inflammatory cycle through innate immunity signaling pathways and NRP3 inflammasome activation [42,43]. HSCs can respond to cytokines, pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs), pushing their differentiation pathway toward myeloid lineage [42].
A single-cell multi-omic analysis of hematopoietic stem/progenitor cells (HSPCs) from patients with myeloproliferative neoplasms (MPNs) showed that, under chronic inflammation, TP53-mutated HSPCs exhibit a fitness advantage, driving clonal expansion [41]. Another study compared immune features between TP53-mutated and TP53 wild-type MDS/AML. PDL1, an immune checkpoint protein, was found to be overexpressed on the surface of HSCs in TP53-mutant patients, while PD-1 expression was reduced. In addition, there was a substantially decreased number of cytotoxic T cells, T helper cells, and NK cells in the bone marrow of TP53-mutated patients. In contrast, a highly immunosuppressive microenvironment was identified, enriched in regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) (Figure 1) [10].
These findings were corroborated in a study of 61 TP53-mutated MDS/AML patients treated with the HMA azacitidine +/− the anti-PDL1 antibody durvalumab in the FUSION clinical trial. T-cell exhaustion with associated immunophenotypes [CD3+CD8+PD1+TIM3− and CD3+CD8+PD1−TIM3+] was observed in AML patients harboring TP53 mutations. Remarkably, after one treatment cycle, DNA methylation patterns at specific loci in PD-1, PD-L1, and PD-L2 genes were almost completely reversed between TP53-mutated and wild-type samples compared with baseline [12]. Further research is required to determine whether these epigenetic changes upregulate immune checkpoint expression and contribute to treatment resistance.
Lastly, leukocyte immunoglobulin-like receptor B3 (LILRB3), a myeloid immune checkpoint, shows increased expression in the myeloid cell compartment of TP53-mutated MDS/AML. LILRB3 overexpression has been linked to endured leukemic cell survival and impaired T-cell cytotoxicity, posing myeloid immune checkpoints as possible therapeutic targets [44]. These special immunological features of TP53-mutated MDS/AML have been a fertile ground, but also an obstacle in the usage of immunotherapy for this specific type of disease, as described later.
3. From Classification to Clinical Outcomes: Understanding TP53-Mutated MDS/AML
3.1. Classification Systems
As outlined in the previous section, MDS, MDS/AML, and AML with TP53 mutations share unique genetic and clinical features. The latest classification systems for myeloid neoplasms reflect these findings to improve prognosis assessment, clinical trial design, and drug discovery. In the WHO-5 classification of hematolymphoid disorders (2022), TP53-mutated MDS (0–19% blast count) requires bi-allelic inactivation—defined as a TP53 mutation accompanied by either a second mutation or a copy-number loss/cnLOH) [5,8]. However, TP53-mutated AML (>20% blasts) is not distinguished from the other types of AML, despite the substantially poorer prognosis compared to the wild-type [45,46]. The International Consensus of Classification of myelodysplastic syndromes and related entities (2022) introduces the multi-hit status, defined by either two TP53 mutations (each with VAF > 10%) or one mutation (VAF > 10%) accompanied by the following: (1) a deletion involving the TP53 locus at chromosome 17p.31, (2) VAF > 50%, or (3) copy-neutral LOH at 17p. Notably, a single TP53 mutation with complex karyotype is regarded as a multi-hit equivalent. The multi-hit status is essential for TP53-mutated MDS categorization (0–9% blasts), but for MDS/AML (10–19%) and AML (>20%) a single mutation with VAF > 10% is sufficient. Both systems require a multi-hit TP53 status for MDS, excluding MDS patients with mono-allelic inactivation or single-hit status [4]. Further analysis of differences and discrepancies between the two classification systems is beyond the purpose of this review. Finally, the European LeukemiaNet (ELN) 2022 recommendations for diagnosis and management of AML in adults classify TP53-mutated AML (VAF > 10% irrespective of allelic status) within the adverse-risk group. A hierarchical classification is proposed as follows: AML with mutated TP53 precedes AML with myelodysplasia-related gene mutations which, in turn, takes precedence over AML with myelodysplasia-related cytogenetic abnormalities [47].
3.2. Role of Allelic Status and Variant Allele Frequency of TP53 Mutations in Patient Outcomes
The impact of allelic status (mono-allelic vs. bi-allelic inactivation) on the prognosis of patients harboring TP53 mutations is still equivocal. In a large study of 3324 MDS patients, 378 carried at least one putative oncogenic TP53 mutation (VAF ≥ 2%). Patients were grouped into those with mono-allelic TP53 state and a residual TP53 wild-type allele, and those with multi-hit state and probably no residual TP53. The multi-hit group exhibited worse overall survival (OS) and higher AML transformation rates compared to the mono-allelic group. In addition, these patients were more cytopenic and had higher percentages of bone marrow blasts (median 9% vs. 4%). Median OS was 8.7 months in multi-hit state vs. 2.5 years in the mono-allelic state. Remarkably, mono-allelic patients had outcomes and responses to therapy comparable to wild-type cases (OS of wild-type patients: 3.5 years). TP53 allelic state predicted outcomes independently from the Revised International Prognostic Scoring System (R-IPSS) [18]. Additionally, although less common in AML (29% single-hit vs. 71% multi-hit), patients with TP53 single-hit disease exhibited longer OS (8 months vs. 1 month, respectively) [17]. TP53 mutations also predicted dismal outcomes in t-MDS patients irrespective of bone marrow blast percentage, VAF range, allelic status, or prior treatment type [48]. In the study encompassing 2200 AML/MDS-EB patients, this association between TP53 allelic status and survival was not confirmed [6]. Nevertheless, strong evidence suggests that multi-hit TP53 status profoundly worsens survival in AML and MDS with elevated blasts (>5%), although multi-hit cases predominate in this patient group [17]. Interestingly, a study has shown that female patients are characterized by higher CR rates possibly due to the lower percentage of the multi-hit state [49]. These findings are consistent with previous data showing that women with AML more often had normal karyotype and lower-risk mutations [50].
To simplify the categorization progress and facilitate risk stratification, a new model was proposed in 2025. The risk stratification was based on a retrospective analysis of 580 patients with MN harboring TP53 mutations (VAF > 2%) from Mayo Clinic (USA) and the South Australia Health Network between 2002 and 2023. The WHO-4 revised classification system was used. In MDS-LB (<5% blasts) with mono-allelic TP53 inactivation, the presence of CK worsened prognosis, approximating outcomes of bi-allelic disease. MDS-EB1 (5–9%), MDS-EB2 (10–19%), and AML (>20%) have been shown to have dismal outcomes regardless of allelic status. MDS-EB1/-EB2/AML cases with VAF < 10% (excluded from ICC 2022 classification) and CK exhibited similar survival rates to those with VAF > 10%. Nonetheless, cases with VAF < 10% and without CK are relatively uncommon. In this study and contrary to the previously presented data, allelic status affected outcomes only in MDS-LB (<5% blasts) group, with no impact in high-blast MDS or AML. TP53-mutated AML had the worst outcomes. An online risk calculator derived from this model estimates OS at 6, 12, and 24 months based on patient-specific WHO classification, TP53 mutation VAF, TP53 allelic status, and the presence of complex karyotype [51].
Regarding mutation types, although available data are limited, prognostic variability among TP53 variants has been observed across different myeloid neoplasms (MDS, AML, AML with myeloid-related changes, and t-AML). Interestingly, missense mutations, particularly those retaining transcriptional activity, appear to initiate MDS formation, while truncating mutations may drive leukemic transformation and disease progression [52]. TP53 splice junction mutations are also referred to predict very poor survival [49]. Several models have been proposed for functional classification of TP53 variants, including the Evolutionary Action score (EAp53)—developed in head and neck cancer—and the Relative Fitness Score (RFS), which assesses the selective advantage of each mutation in cancer evolution. In AML, low-risk TP53 mutations (RFS > −1) were associated with median OS of 12.9 months, versus 5.5 months for high-risk (RFS ≤ −1) variants, further indicating patients who would benefit from intensive treatment approaches [53]. With regard to co-mutations and their role, mutations in CUX1, U2AF1, EZH2, TET2, CBL, and KRAS genes, which comprise the ‘EPI6 signature’, are associated with inferior OS at 24 months [49].
To conclude, TP53 allelic status, mutation VAF, complex karyotype, mutation type, and co-mutational background collectively influence prognosis. The multi-hit state strongly correlates with CK, reflecting underlying genomic instability. Although various VAF cutoffs have been proposed [49,54,55], VAF does not independently affect outcomes in multi-hit disease, indicating that bi-allelic TP53-inactivated clones dominate disease biology and drive poor prognosis regardless of clone size. Similarly, co-mutational status predicts survival only in single-hit patients [18]. Lastly, although allogeneic bone marrow transplantation (allo-BMT) remains the only putative curative treatment option for TP53-mutated MDS/AML patients, early post-transplant relapses are frequent and survival remains poor. In line with previous studies [49,56,57], a very recent MD Anderson Cancer Center analysis reported that patients with lower TP53 mutation VAF (<50%) and without complex cytogenetics benefited most from allo-SCT, achieving a 2-year progression-free survival of 60% [55].
4. Treatment Armamentarium and Novel Strategies for TP53-Mutated MDS/AML
4.1. Hypomethylating Agents (HMAs) and Their Combination with Venetoclax Take Precedence over Intensive Chemotherapy
TP53 mutations are more common among elderly AML patients and those with prior exposure to various treatments for malignancies, while these patients often present with low performance status or comorbidities which preclude them from undergoing intensive chemotherapy. Although HMAs (azacitidine and decitabine) are considered the preferred frontline regimen for elderly/unfit patients with AML [58], the presence of TP53 mutations deprives this group of patients of substantial survival benefit [59]. Interestingly, a 10-day decitabine therapeutic regimen showed overall survival rates that were not significantly different between TP53-mutated and TP53 wild-type patients [60]. However, despite that decitabine is considered a mainstay therapy for TP53-mutated myeloid neoplasms, it remains a palliative approach for the majority of patients.
Venetoclax is an orally administered FDA-approved selective Bcl-2 inhibitor which is widely used in everyday clinical practice for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) and AML treatment. Venetoclax is used in combination with HMAs (particularly azacitidine) in AML patients, based also upon the results of the pivotal study VIALE-A, where the combination of two exhibited higher composite response rates [CR + complete remission with incomplete hematologic recovery (CRi)] compared to azacitidine alone (55.3% vs. 0%) [61]. Nonetheless, a post hoc analysis of the VIALE-A trial revealed that the addition of venetoclax to azacitidine do not benefit patients with TP53 mutations (median OS: 5.5 months vs. 5.4 months with azacitidine and placebo) [62]. Additionally, in a 2021 post hoc analysis of a phase 2 trial, 10-day decitabine plus venetoclax regimen produced lower response rates in patients harboring TP53 mutations (ORR 66% vs. 89%, TP53-mutated vs. TP53 wild-type disease) and shorter survival (median OS 5.2 vs. 19.4 months, respectively) [63]. Notably, in contrast to the commonly used cytotoxic doses of HMAs and venetoclax, weekly administered low-dose combination of decitabine and venetoclax can provide a comparable median OS of 11.3 months with less myelotoxicity [64].
Furthermore, the absence of intact p53 protein provides these myeloid malignancies with innate chemorefractoriness and, combined with the high-risk profile of patients, it results in limited usage of intensive chemotherapy. This is also due to the low response rates (20–40%) and median survival (5–11 months) that have been reported [6,15,65]. TP53 mutation VAF has also been reported to potentially affect survival in patients receiving intermediate- or high-dose cytarabine-based regimens. A VAF > 40% reduced median survival to 5 months compared to 18.1 months with VAF < 40% [66]. Liposomal daunorubicin and cytarabine (CPX-351) showed improved OS compared to HMA + venetoclax as a frontline regimen for AML [67], but no improvement was found when compared to classic 7 + 3 chemotherapy in a cohort of younger patients with adverse cytogenetics AML or high-risk MDS [68].
Lastly, HMA and venetoclax combinations have also been studied as bridging therapies for allo-SCT in patients with high-risk MDS or CMML. In this recent study, six patients harbored TP53 mutations and all of them had complex karyotypes. Among these patients, an ORR of 100% was achieved, but the two-year post-transplant OS was 50% with a higher two-year post-transplant cumulative incidence of relapse (CIR, 75% for TP53-mutated patients vs. 15.4% for TP53 wild-type) [69].
4.2. Allogeneic Stem Cell Transplantation (Allo-BMT)
Allo-BMT is generally considered for all patients with MNs harboring TP53 mutations (Table 2). Median post-transplant survival is reported to be 1.03 years and OS at 1 and 2 years is 51.4% and 35.1%, respectively. OS was comparable between MDS and AML patients. These are data reported in an international study from seven institutions across the United States and Australia, published in 2025, encompassing 134 patients with TP53-mutated MNs [70]. These findings are consistent with that from a previous systematic review and meta-analysis including 297 patients with TP53-mutated AML from eight studies [71]. TP53 mutations are predictors of poor prognosis following allo-BMT in patients with MDS [72]. Our group has also reported that TP53 mutations strongly predict worse outcomes after allo-BMT in MNs [73], and the presence of CK further decreases survival rates [74]. On the other side, CR at day 100 post-allo-BMT and the occurrence of chronic GvHD can improve event-free survival (EFS) and OS [75]. The role of pre-transplant TP53 mutation clearance by NGS still remains equivocal [76,77]. Taken together, although advanced age, poor PS, infections, and other factors can be hindrances for transplantation, allo-BMT remains the only putatively curative option, and risk models based on all these genetic and clinical parameters could facilitate better selection of eligible patients [78].
Table 2.
Clinical outcomes of Allo-BMT in TP53-mutated myeloid neoplasms.
Regarding conditioning regimens, there are conflicting data about post-transplant relapse rates between myeloablative (MAC) and reduced-intensity conditioning (RIC) [79,80]. Nonetheless, more recent data show that MAC and RIC regimens exhibit comparable relapse and survival rates [75]. Interestingly, in the aforementioned international study, it has been shown that melphalan-based conditioning is associated with superior relapse-free survival (RFS), but this finding might be restricted in patients with a low blast percentage of <5% prior to allo-BMT [70]. In order to reduce relapse and improve survival, different post-transplant maintenance strategies have been used, but with limited effectiveness. There are several agents investigated in clinical trials, such as azacitidine (NCT04173533, results to be posted), azacitidine and venetoclax (NCT04161885, results to be posted), decitabine and cedazuridine (NCT04980404, results to be posted), and venetoclax added to fludarabine and busulfan (NCT03613532, ongoing trial) [81,82,83,84].
4.3. The Role of Immunotherapy in TP53-Mutated MDS and AML
4.3.1. Immune Checkpoint Inhibitors (ICI)
As mentioned above, in TP53-mutated MDS and AML, PD-L1 expression is elevated in HSCs, as opposed to PD-1 expression that is reduced, and the cellular microenvironment is highly immunosuppressive. These findings have led to the investigation of immune checkpoint inhibitors, which are commonly used in solid tumors, as potential additional treatments. Nivolumab, a PD-1 inhibitor, has been tested as a combination therapy with azacitidine in a phase II study of 70 relapsed/refractory AML patients and exhibited modest efficacy, with a median OS of six months in the TP53-mutated cohort (16/70 patients) [85]. Nivolumab has also been investigated as induction therapy with cytarabine and idarubicin in patients with newly diagnosed AML or high-risk MDS (eight patients with TP53 mutations), and a median OS of 18.54 months was reported [86]. Ipilimumab, a cytotoxic T-lymphocyte-associated antigen-4 or CTLA-4 inhibitor, has been investigated in combination with decitabine in relapsed/refractory (R/R) or treatment-naïve MDS and AML. Although there was no separate analysis for TP53 mutations, ORR was 52% in the transplant-naïve group [87]. Dual blockade of PD-1 and CTLA-4 was also tested in previously untreated MDS in a clinical trial with recently published results [88]. The investigated combinations were azacitidine–nivolumab, azacitidine–ipilimumab, and azacitidine–ipilimumab–nivolumab. Azacitidine–nivolumab exhibited the best efficacy, with an ORR of 55%, while the triplet-based regimen had the highest toxicity [88]. In addition, durvalumab, a PD-L1 inhibitor, had an acceptable safety profile but did not improve outcomes in combination with azacitidine in a randomized phase II clinical trial [89]. Lastly, there is an ongoing phase I/II clinical trial investigating the combination of the hypomethylating agent guadecitabine with the anti-PD-L1 antibody atezolizumab for R/R MDS or CMML (NCT02935361) [90].
4.3.2. Anti-CD47 Targeting Strategies
CD47 is widely expressed on the surface of various malignant cells and interacts with the signal-regulatory protein (SIRP)-a on phagocytic cells, initiating an antiphagocytic “don’t-eat-me” signal. Magrolimab was the first-in-class monoclonal antibody targeting this molecule. Magrolimab showed promising results combined with azacitidine in early-phase clinical studies, achieving a 40.3% CR/CRi in patients with TP53-mutated AML [91]. In a phase Ib/II clinical trial, magrolimab was combined with azacitidine and venetoclax in patients with newly diagnosed or R/R AML ineligible for intensive chemotherapy, and while response rates were high (ORR 74% for TP53-mutated AML and 93% for wild-type), 1-year survival for TP53-mutated AML was 53% [92]. Despite these encouraging results, phase III clinical studies ENHANCE-2 [93,94] and ENHANCE-3 [95,96] were discontinued due to futility and increased mortality.
Next-generation CD47 blockers have also been developed, such as evorpacept (ALX148). Although preliminary clinical data of evorpacept in combination with azacitidine and venetoclax showed favorable tolerability and potential anti-leukemic activity, the ASPEN-05 phase 1a study never moved forward to phase 2 [97,98]. The ASPEN-02 phase I/II trial, which tested evorpacept with azacitidine in patients with high-risk MDS, was also discontinued [99]. Finally, lemzoparlimab and ligufalimab (AK117) are two novel CD47-targeting antibodies which are red blood cell-sparing. Lemzoparlimab was tested for safety in a phase 1b study with azacitidine and venetoclax, but the study was terminated due to strategic considerations [100,101]. Ligufalimab is being tested with azacitidine in an ongoing phase 2, randomized, double-blind, placebo-controlled, multicenter study for patients with newly diagnosed higher-risk MDS (NCT06196203) [102]. Phase 1 study of this drug revealed CR rate of 48.1% and limited toxicity [103].
4.3.3. Sabatolimab—A TIM-3 Inhibitor
T-cell immunoglobulin and mucin domain 3 (TIM-3) is an immune regulator expressed on immune cells and myeloid leukemic progenitors. By inhibiting TIM-3, sabatolimab facilitates T-cell activation and induction of phagocytosis. This agent was tested in a phase 1b study in combination with HMAs in patients with very high/high-risk MDS (vHR/HR-MDS) and AML. Results reported acceptable safety profile and durable responses even in patients with adverse-risk mutations, including TP53 mutations [vHR/HR-MDS: ORR: 71.4%, median duration of response (mDOR): 21.5 months, AML: ORR: 53.8%, mDOR: 12.6 months)] [104,105]. Unfortunately, subsequent phase II trials STIMULUS-MDS1 and STIMULUS-MDS2 were discontinued due to failure to reach their primary endpoints [106,107]. The STIMULUS-AML1 study, which investigated sabatolimab combined with azacitidine and venetoclax in patients with newly diagnosed AML, was also discontinued [108]. The STIMULUS-MDS3 study investigating the same combination in 20 vHR/HR-MDS patients reported an ORR of 86.7% in the 800 mg sabatolimab arm [109].
4.3.4. CD123 × CD3 Bi-Specific Antibodies
Flotetuzumab is a CD123 × CD3 dual affinity antibody that works by enhancing the formation of an immunologic synapse between cytotoxic T cells and AML cells, independently of the major histocompatibility complex (MHC) pathway. Based on data showing that TP53 mutations induce an immune-filtrated tumor microenvironment that promotes resistance to chemotherapy, flotetuzumab was tested in a phase I/II study of 88 adult patients with R/R AML as salvage immunotherapy. The drug exhibited favorable tolerability and a CR rate of 47% among 15 patients with TP53-mutated R/R AML. Median OS was 10.3 months among those who achieved a complete response [23,110]. APVO-436, another novel CD123 × CD3 bi-specific antibody, was well tolerated and exhibited potential anti-leukemic activity alone and in combination with standard-of-care regimens in a phase 1 study [111]. There is an ongoing phase 1b/2 open-label study of this agent in combination with venetoclax and azacitidine in patients with newly diagnosed AML (NCT06634394) [112].
Besides bi-specific antibodies, CD123 is also targeted by other agents, such as tagraxofusp, which is an FDA-approved drug for treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). Tagraxofusp (TAG) is a recombinant interleukin-3 protein fused to a truncated diphtheria toxin payload and preclinical data have shown that AML cells resistant to TAG were re-sensitized by azacitidine and became more dependent on Bcl-2. A phase 1b study tested TAG and azacitidine with or without venetoclax in AML patients and the triplet combination was tolerable, without further increasing the risk for infections or capillary leak syndrome. Seven out of thirteen patients (54%) with TP53 mutations achieved CR/CRi or morphologic leukemia-free state (MLFS), while median OS was reported as 14 months in the entire cohort [113]. Tagraxofusp is currently investigated in a phase I trial in combination with azacitidine and/or venetoclax for patients with AML, high-risk MDS, or BPDCN (NCT03113643) [114].
4.3.5. Chimeric Antigen Receptor (CAR)-T Cell Therapies
While CAR-T cell therapies have proven highly effective for lymphoid neoplasms such as B-cell lymphomas, they demonstrate limited applicability to AML and myeloid neoplasms because of the lack of tumor-specific targets, high off-target toxicity, and profound myelosuppression. The hostile tumor microenvironment with highly immunosuppressive features is also a strong caveat. CD33, CD123, CLL1, and IL-1RAP are antigens that CAR-T cell therapies have been developed against for treatment of AML patients in various stages and they are being tested in early-phase clinical trials [8,115]. Another novel strategy uses compound CAR (cCAR)-T cells targeting both CD33 and CLL-1 AML antigens, and initial results from a phase 1 clinical trial have shown high efficacy and manageable toxicity [116,117]. Anti-Tim-3/CD123 CAR-T cell therapy is also a compound strategy currently investigated in patients with R/R AML (NCT06125652) [118].
He et al. [119] developed bi-specific and split CAR (BissCAR) T cells targeting CD13 and TIM3 by isolating multiple nanobodies and using a sequentially tumor-selected antibody and antigen retrieval (STAR) system. In preclinical studies, it has been shown that this platform eradicated patient-derived AML cells with limited toxicity to normal HSCs [119]. Lastly, another group removed CD33 from normal HSCs by using CRISPR-Cas9 gene editing to perform CD33 targeting CAR-T cell therapy while preventing myelotoxicity [120]. Nevertheless, none of the current studies are specifically designed for TP53-mutated MDS/AML yet. An overview of different types of immunotherapies currently under investigation for this poor-risk disease is given in Figure 2.
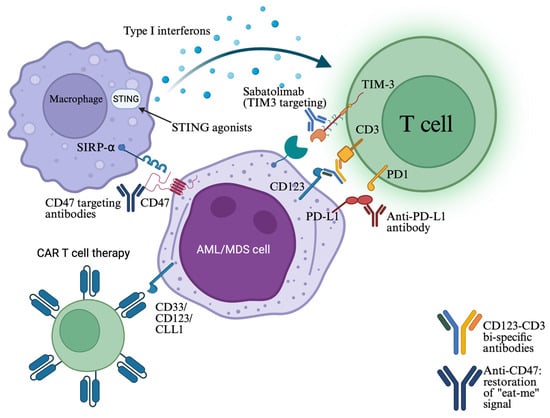
Figure 2.
Cell–cell interactions between AML/MDS cells and immune cells, such as T cells and macrophages (phagocytic cells), are crucial in immune escape. Immune checkpoint inhibitors, bi-specific antibodies facilitating T-cell engagement, as well as STING agonists that stimulate innate immune cells, have been developed for restoring immune surveillance and function. CAR-T cells targeting a broad spectrum of antigens are also under investigation (see text). Created in https://BioRender.com (assessed on 25 September 2025).
4.4. p53 Targeting Strategies
Missense TP53 mutations cause p53 protein misfolding and loss of function, and these missense mutant proteins are usually overexpressed in malignant cells as a response to oncogenic signals. As a result, refolding the missense mutant protein could induce massive apoptosis to malignant cells with mutated p53 while sparing cells with wild-type protein [121]. Eprenetapopt (APR-246) is the most investigated drug in this category. In two phase II clinical trials testing eprenetapopt in combination with azacitidine, it has been shown that higher ORR (52% and 71%) and CR rates (37% and 44%) can be reached compared to azacitidine alone, and the combination is well tolerated (≥3 cycles given). Additionally, median OS was also extended (12.1 and 10.8 months) and higher CR was associated with TP53 mutation clearance as detected by NGG with VAF < 5% [122,123]. Unfortunately, in the phase III trial, eprenetapopt plus azacitidine did not reach statistical significance in the primary endpoint (12-month CR rate 35 vs. 22% with azacitidine alone) [124]. Furthermore, eprenetapopt was tested with azacitidine and venetoclax in a phase I trial exhibiting an acceptable safety profile and encouraging activity (ORR: 64%, CR: 38%) [125]. The combination of eprenetapopt plus azacitidine was also evaluated as a post-transplant maintenance therapy in a phase II trial enrolling 33 patients with TP53-mutated AML/MDS. The results were promising, with a median RFS of 14.5 months and a median OS of 20.6 months reported [126,127]. Eprenetapopt can also induce cell death via a p53-independent redox effect by inhibiting thioredoxin reductase and thus depleting cellular glutathione levels. Chemoresistance can be acquired by cellular mechanisms exporting glutathione conjugates and that could explain the robust synergistic effects of eprenetapopt with cytotoxic drugs in solid tumors [128].
Arsenic trioxide (ATO), although widely known for its efficacy in acute promyelocytic leukemia in combination with all-trans retinoic acid, has also been found to have mutant p53-rescuing properties [129]. There is a clinical trial testing this drug in TP53-mutated MNs (NCT06778187, currently recruiting) [130].
Lastly, while differentiation induction with ATO and ATRA has demonstrated remarkable success in patients with APL, this strategy had not exhibited promising efficacy in other types of AML so far. Very recently, the combination of decitabine with low-dose etoposide showed favorable clinical response in elderly patients with high-risk MDS or AML harboring TP53 mutations compared to TP53 wild-type disease. OS was also improved (31 months in TP53-mutated vs. 9 months in wild-type disease). This effect was confirmed in patient-derived xenograft models, while experiments with TP53 wild-type and deficient AML cell lines revealed a differentiating effect of the combination, resulting in neutrophil terminal differentiation only in TP53-mutant and knockout cells [131]. Table 3 summarizes clinical trials and the outcomes of selected treatment strategies developed for TP53-mutated MDS and AML.
Table 3.
Selected clinical trials of developed agents targeting TP53-mutated MDS/AML.
4.5. Ongoing Studies of Novel Molecular Targets
4.5.1. Tropomyosin Receptor Kinase (TRK) Inhibition
Recent studies have demonstrated that TP53 mutations increase the dependency of venetoclax-resistant AML cells on the NTRK pathway [132]. Entrectinib, an NTRK/ALK/ROS1 inhibitor, is currently evaluated in a phase 1 study in combination with oral decitabine and cedazuridine (ASTX727) in patients with TP53-mutated R/R AML (NCT05396859). Early results have reported a favorable safety profile, while one patient out of thirteen, who was enrolled following an early post-transplant relapse, achieved a CR lasting for five months [133].
4.5.2. AXL Inhibition
AXL is a tyrosine receptor kinase that is found to be overexpressed in malignant hematopoiesis compared to normal hematopoiesis and has been defined as an adverse prognostic marker in AML patients [134]. Preclinical studies of TP-0903, a multiple kinase inhibitor, have shown anti-leukemic activity in different TP53-mutated AML cell lines and prolonged survival in xenograft models, both alone and in combination with decitabine [135]. These findings led to a phase 1b/2 study of this combination as a sub-study of the Beat AML clinical trial (NCT03013998). Among patients harboring TP53 mutations, the overall composite remission rate (CR/CRi/CR with partial hematologic recovery) was 45%, and the median OS was 10 months [136].
4.5.3. Pevonedistat—An NEDD8-Activating Enzyme Inhibitor
Pevonedistat is a first-in-class inhibitor of the NEDD8-activating enzyme and induces apoptosis in AML cells via increased reactive oxygen species production, accumulation of the MYC oncoprotein, and Mcl-1 inhibition through NOXA protein upregulation [137,138]. Although preclinical and early-phase clinical study results were encouraging [137], pevonedistat combined with azacitidine did not eventually improve composite CR rates in older patients with TP53-mutated AML [138]. Moreover, the combination of azacitidine, venetoclax, and pevonedistat has been tested in patients with newly diagnosed sAML and with MDS or CMML after failure of HMAs in a phase I/II single-center study. The CR/CRi rate was the same between patients with TP53-mutated AML and the entire cohort (64% vs. 66%, respectively), but the median OS was much lower in TP53-mutated AML vs. TP53 wild-type patients (8.1 vs. 18.0 months, respectively) [139].This triplet-based regimen was also investigated in a phase 1 study in patients with R/R AML. Interestingly, a 71.4% CR rate was observed among patients not previously exposed to venetoclax therapy, and CR achievement was associated with extended median OS [140].
4.5.4. PLK4 Inhibition
Recent data revealed that polo-like kinase 4 (PLK4) expression is increased in TP53-mutated AML cell lines and primary samples, and its inhibition induces cellular senescence and defective cytokinesis, alternates histone modification, and increases cytokine and chemokine secretion via the cGAS-STING pathway. These effects were found to be mediated by the newly described PLK4/PRMT5/EZH2/H3K27m3 axis, which operated both in TP53-mutant and wild-type cells [141]. High PLK4 expression is also associated with dismal prognosis in AML [142]. Based on these findings, CFI-400945, a selective oral PLK4 inhibitor that regulates centriole duplication, was tested in preclinical studies, where it demonstrated antitumor efficacy in xenograft mouse AML models. A phase 1 study tested this drug in patients with very high-risk R/R AML and MDS, and two out of four patients with TP53-mutated disease reached CR, while another one had a >50% reduction in bone marrow blasts. Enteritis/colitis was reported as a dose-limiting toxicity [143]. Currently, a newer crystal form of this drug is tested as a single agent or in combination with azacitidine in patients with AML, MDS, or CMML, with preliminary results showing an acceptable safety profile (NCT04730258) [144,145].
4.5.5. STING Agonists
In the immunosuppressive tumor microenvironment of TP53-mutated MDS/AML, restoration and enhancement of adaptive immunity through stimulation of innate immune cells may represent an effective therapeutic strategy. Molecules that have an agonistic effect on the stimulator of interferon genes (STING) can induce the production of type 1 IFNs and thus enhance cytotoxic T lymphocyte presence and function around tumor cells (Figure 2) [146]. STING activation induces cytotoxicity in AML cells [147] and GSK3745417, a non-cyclic di-nucleotide (non-CDN) small STING agonist, was evaluated in preclinical studies and exhibited strong cell growth inhibitory effects through apoptosis induction and immune activation [148]. The safety, tolerability, and pharmacologic profile of this drug was investigated in a phase 1 trial encompassing patients with R/R AML and high-risk/very high-risk MDS, but it was terminated early due to financial reasons (NCT05424380, results posted) [149,150]. CRD3874 is another synthetic STING agonist currently in a phase 1 clinical trial (NCT06626633) [151]. Figure 3 summarizes the novel therapeutic approaches against p53 and new molecular targets.
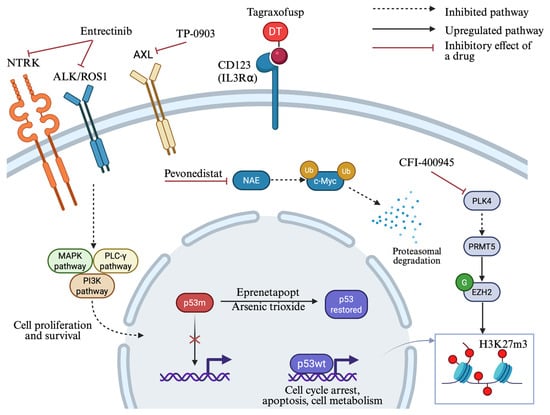
Figure 3.
Novel treatment strategies under investigation for TP53-mutated MDS/AML. Complex molecular pathways are implicated in TP53-mutated disease, giving rise to the development of new targeted agents for this poor-risk subtype. Eprenetapopt and arsenic trioxide (ATO) restore wild-type p53 conformation. Entrectinib inhibits tropomyosin receptor kinases (NTRK/ALK/ROS1), while TP-0903 inhibits the AXL tyrosine kinase receptor, suppressing signaling pathways that promote cell proliferation and survival. Tagraxofusp targets CD123 on malignant cells to induce cytotoxicity through diphtheria toxin release. Pevonedistat inhibits the NEDD8-activating enzyme (NAE), leading to c-Myc accumulation and subsequent NOXA transactivation. PLK4 inhibition by CFI-400945 activates PRMT5, resulting in histone methylation by EZH2 and increased global H3K27me3 levels. DT: truncated diphtheria toxin; p53m: mutant p53; p53wt: wild-type p53; NAE: NEDD8-activating enzyme. Created in https://BioRender.com (assessed on 25 September 2025).
Table 4 summarizes ongoing clinical trials of investigational strategies for TP53-mutated MDS/AML.
Table 4.
Ongoing clinical trials of investigational strategies for TP53-mutated MDS/AML.
5. Conclusions and Future Directions
TP53-mutated MDS/AML is an almost fatal diagnosis, since no significant improvements in therapeutic landscape have been achieved so far. Extensive research in the field continues to reveal mechanisms of treatment resistance, as well as specific molecular characteristics of the disease that could be exploited as leading lights for the development of new treatments. In this review, we aimed to summarize the current knowledge about this poor-prognosis entity and describe the research progress made toward better patient outcomes to the best of our knowledge.
The preexistence of TP53-mutated clones as clonal hemopoiesis raises the question of whether biomarkers could be found so that patients with CH could be intensively monitored or preventively treated. This is particularly important since cancer incidence continues to rise and more patients are going to undergo cytotoxic treatments that give a survival advantage to TP53-mutated clones. Pharmacological reactivation of p53 is characterized by high complexity due to the many different mutant protein variants and their different levels of functionality, the ability of p53 to be dysfunctional without mutation, and the severe toxicity affecting non-malignant cells. More individualized strategies, such as targeting specific variants, better drug combinations, or implementation of p53-targeting drugs in earlier stages of the disease treatment, could potentially improve their efficacy [122]. Despite the fact that TP53-mutated MDS and AML represent a significant unmet medical need, the majority of ongoing clinical trials are not specifically focused on this molecular subgroup. Therefore, the design and implementation of molecularly defined trials could facilitate improved patient outcomes. Additionally, more in-depth research aimed at identifying biomarkers of response beyond cytogenetics, VAF measurement, or MRD assessment, could guide precise patient selection for each therapeutic modality.
Drug repurposing is also a novel strategy investigated in this group of patients. Mevalonate pathway has been found upregulated in TP53-mutated AML and it correlates with mitochondria-dependent chemoresistance and CAR-T cell therapy failure [25,26]. Statins, widely used mevalonate pathway inhibitors, are investigated for this purpose and pitavastatin has been tested in combination with venetoclax in a phase 1 clinical trial, where it showed favorable tolerability with all patients achieving complete response [152]. Niclosamide, an oral anthelminthic medication used to treat tapeworm infections since 1960, has also been tested in vitro in TP53-mutated AML patient samples and cell lines, exhibiting potential anti-leukemic activity and restoration of sensitivity of these cells to hypomethylating therapy [153].
Finally, activation and redirection of innate and adaptive immunity into fighting this disease remains a challenge, mainly because of the different immune evasion mechanisms that AML cells use to escape. In-depth research in cancer immunology can unravel these mechanisms and find new treatments. Until better therapies are developed, allo-BMT in first remission should be attempted in all eligible patients.
Author Contributions
E.G.: writing—conceptualization and original draft preparation, T.K.: writing—review and editing and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the NHLBI Grant K08HL168777 and the Leukemia Research Foundation New Investigator Research Grant Program.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. P53 Mutations Are Associated with Resistance to Chemotherapy and Short Survival in Hematologic Malignancies. Blood 1994, 84, 3148–3157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zawacka, J.E. P53 Biology and Reactivation for Improved Therapy in MDS and AML. Biomark. Res. 2024, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Li, W. The 5th Edition of the World Health Organization Classification of Hematolymphoid Tumors. In Leukemia; Li, W., Ed.; Exon Publications: Brisbane, Australia, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK586208 (accessed on 30 October 2025).
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk Kooy, M.; et al. Molecular Characterization of Mutant TP53 Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Abel, H.J.; Oetjen, K.A.; Miller, C.A.; Ramakrishnan, S.M.; Day, R.B.; Helton, N.M.; Fronick, C.C.; Fulton, R.S.; Heath, S.E.; Tarnawsky, S.P.; et al. Genomic Landscape of TP53-Mutated Myeloid Malignancies. Blood Adv. 2023, 7, 4586–4598. [Google Scholar] [CrossRef]
- Shah, M.V.; Arber, D.A.; Hiwase, D.K. TP53-Mutated Myeloid Neoplasms: 2024 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2025, 100 (Suppl. 4), 88–115. [Google Scholar] [CrossRef]
- Singhal, D.; Kutyna, M.M.; Hahn, C.N.; Shah, M.V.; Hiwase, D.K. Therapy-Related Myeloid Neoplasms: Complex Interactions among Cytotoxic Therapies, Genetic Factors, and Aberrant Microenvironment. Blood Cancer Discov. 2024, 5, 400–416. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 Mutations in Myelodysplastic Syndromes and Secondary AML Confer an Immunosuppressive Phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Badar, T.; Knutson, K.L.; Foran, J.; Gangat, N.; Pavelko, K.D.; Kaufmann, S.H.; Litzow, M.R.; Murthy, H.; Cogen, D.; Ushman, M.; et al. T-Cell Immune Cluster Analysis Using CyTOF Identifies Unique Subgroups of Patients with Acute Myeloid Leukemia. Blood Adv. 2025, 9, 239–243. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Bewersdorf, J.P.; Hasle, V.; Shallis, R.M.; Thompson, E.; de Menezes, D.L.; Rose, S.; Boss, I.; Halene, S.; Haferlach, T.; et al. Integrated Genetic, Epigenetic, and Immune Landscape of TP53 Mutant AML and Higher Risk MDS Treated with Azacitidine. Ther. Adv. Hematol. 2024, 15, 20406207241257904. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the P53 Pathway: In Vitro Hypotheses, in Vivo Veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why Are There Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Bahaj, W.; Kewan, T.; Gurnari, C.; Durmaz, A.; Ponvilawan, B.; Pandit, I.; Kubota, Y.; Ogbue, O.D.; Zawit, M.; Madanat, Y.; et al. Novel Scheme for Defining the Clinical Implications of TP53 Mutations in Myeloid Neoplasia. J. Hematol. Oncol.J Hematol Oncol 2023, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A Dominant-Negative Effect Drives Selection of TP53 Missense Mutations in Myeloid Malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Stengel, A.; Meggendorfer, M.; Walter, W.; Baer, C.; Nadarajah, N.; Hutter, S.; Kern, W.; Haferlach, T.; Haferlach, C. Interplay of TP53 Allelic State, Blast Count, and Complex Karyotype on Survival of Patients with AML and MDS. Blood Adv. 2023, 7, 5540–5548. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556, Correction in Nat. Med. 2021, 27, 927; Correction in Nat. Med. 2021, 27, 562. [Google Scholar] [CrossRef]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis Is Linked to TP53 Alteration, Cell Cycle Impairment, and Dismal Outcome in Acute Myeloid Leukemia with Complex Karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Skuli, S.; Boocock, D.J.; Coveney, C.; Ikpo, E.G.; Wang, B.; Fenu, E.M.; Abbas, H.A.; Lai, C.E.; Carroll, M.P.; et al. Multi-Omic Analyses of TP53-Mutated Acute Myeloid Leukemia Identify Prognostic Metabolic Signatures. Blood 2024, 144, 2911. [Google Scholar] [CrossRef]
- Naji, N.S.; Pasca, S.; Chatzilygeroudi, T.; Toledano-Sanz, P.; Rimando, J.; An, Y.; Hemani, Y.; Perkins, B.; Zeng, X.; Talbot, C.; et al. C-C Motif Chemokine Receptor-like 2 Promotes the Interferon-γ Signaling Response in Myeloid Neoplasms with Erythroid Differentiation and Mutated TP53. Haematologica, 2025. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 Abnormalities Correlate with Immune Infiltration and Associate with Response to Flotetuzumab Immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, A.V.; Mahdevar, M.; Mirzaei, S.; Entezari, M.; Hashemi, M.; Hushmandi, K.; Peymani, M. Introduction of Mutant TP53 Related Genes in Metabolic Pathways and Evaluation Their Correlation with Immune Cells, Drug Resistance and Sensitivity. Life Sci. 2022, 303, 120650. [Google Scholar] [CrossRef] [PubMed]
- Skuli, S.J.; Bakayoko, A.; Kruidenier, M.; Manning, B.; Pammer, P.; Salimov, A.; Riley, O.; Brake-Sillá, G.; Dopkin, D.; Bowman, M.; et al. Chemoresistance of TP53 Mutant Acute Myeloid Leukemia Requires the Mevalonate Byproduct, Geranylgeranyl Pyrophosphate, for Induction of an Adaptive Stress Response. Leukemia 2025, 39, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.; Schimmer, R.R.; Koch, C.; Schneiter, F.; Fullin, J.; Lysenko, V.; Pellegrino, C.; Klemm, N.; Russkamp, N.; Myburgh, R.; et al. Targeting the Mevalonate or Wnt Pathways to Overcome CAR T-Cell Resistance in TP53-Mutant AML Cells. EMBO Mol. Med. 2024, 16, 445–474. [Google Scholar] [CrossRef]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.S.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The Epichaperome Is an Integrated Chaperome Network That Facilitates Tumour Survival. Nature 2016, 538, 397–401. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Muftuoglu, M.; Tao, W.; Ke, B.; Pei, J.; Bedoy, A.D.; Ostermann, L.B.; Nishida, Y.; Isgandarova, S.; et al. Epichaperome Inhibition Targets TP53-Mutant AML and AML Stem/Progenitor Cells. Blood 2023, 142, 1056–1070. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. P53 in Ferroptosis Regulation: The New Weapon for the Old Guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Y. P53 Involvement in Clonal Hematopoiesis of Indeterminate Potential. Curr. Opin. Hematol. 2019, 26, 235–240. [Google Scholar] [CrossRef]
- Warren, J.T.; Link, D.C. Clonal Hematopoiesis and Risk for Hematologic Malignancy. Blood 2020, 136, 1599–1605. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of Chemotherapy for Solid Tumors with Development of Therapy-Related Myelodysplastic Syndrome or Acute Myeloid Leukemia in the Modern Era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef]
- Leone, G.; Fianchi, L.; Pagano, L.; Voso, M.T. Incidence and Susceptibility to Therapy-Related Myeloid Neoplasms. Chem. Biol. Interact. 2010, 184, 39–45. [Google Scholar] [CrossRef]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.L.; Greipp, P.T.; Rangan, A.; Jatoi, A.; Nguyen, P.L. Myeloid Malignancies in Cancer Patients Treated with Poly(ADP-Ribose) Polymerase (PARP) Inhibitors: A Case Series. Blood Cancer J. 2022, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.E.; Khalife-Hachem, S.; Grinda, T.; Kfoury, M.; Garciaz, S.; Pasquier, F.; Vargaftig, J.; Uzunov, M.; Belhabri, A.; Bertoli, S.; et al. Therapy-Related Myeloid Neoplasms Following Treatment with PARP Inhibitors: New Molecular Insights. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, H.B.; Mohty, R.; Greipp, P.; Bansal, R.; Hathcock, M.; Rosenthal, A.; Murthy, H.; Kharfan-Dabaja, M.; Bisneto Villasboas, J.C.; Bennani, N.; et al. Therapy-Related Myeloid Neoplasms Following Chimeric Antigen Receptor T-Cell Therapy for Non-Hodgkin Lymphoma. Blood Cancer J. 2022, 12, 113. [Google Scholar] [CrossRef]
- Gazeau, N.; Beauvais, D.; Tilmont, R.; Srour, M.; Ferrant, E.; Safar, V.; Fouillet, L.; Flandrin-Gresta, P.; Gower, N.; Chauvet, P.; et al. Myeloid Neoplasms after CD19-Directed CAR T Cells Therapy in Long-Term B-Cell Lymphoma Responders, a Rising Risk over Time? Leukemia 2025, 39, 1714–1722. [Google Scholar] [CrossRef]
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 Mutations Characterize Preleukemic Stem Cells in Acute Myeloid Leukemia. Blood 2017, 129, 2587–2591. [Google Scholar] [CrossRef]
- Barakos, G.P.; Hatzimichael, E. Microenvironmental Features Driving Immune Evasion in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Dis. Basel Switz. 2022, 10, 33. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Norfo, R.; Wen, S.; Chédeville, A.L.; Rahman, H.; O’Sullivan, J.; Wang, G.; Louka, E.; Kretzschmar, W.W.; Paterson, A.; et al. Single-Cell Multi-Omics Identifies Chronic Inflammation as a Driver of TP53-Mutant Leukemic Evolution. Nat. Genet. 2023, 55, 1531–1541. [Google Scholar] [CrossRef]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-Adapted Regulation of Early Hematopoiesis in Infection and Inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 Inflammasome Functions as a Driver of the Myelodysplastic Syndrome Phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xu, Y.; Schultz, R.D.; Chen, H.; Xie, J.; Deng, M.; Liu, X.; Gui, X.; John, S.; Lu, Z.; et al. LILRB3 Supports Acute Myeloid Leukemia Development and Regulates T-Cell Antitumor Immune Responses through the TRAF2-cFLIP-NF-κB Signaling Axis. Nat. Cancer 2021, 2, 1170–1184. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.V.; Kutyna, M.; Shah, S.; Tran, E.N.H.; Baranwal, A.; Ladon, D.; Al-Kali, A.; Brown, A.; Chen, D.; Greipp, P.; et al. Comparison of World Health Organization and International Consensus Classification Guidelines for Myeloid Neoplasms Harboring TP53-Mutations Using an Independent International Cohort. Blood 2023, 142, 3243. [Google Scholar] [CrossRef]
- Hart, S.A.; Lee, L.A.; Seegmiller, A.C.; Mason, E.F. Diagnosis of TP53-Mutated Myeloid Disease by the ICC and WHO Fifth Edition Classifications. Blood Adv. 2025, 9, 445–454. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Bao, Z.; Li, B.; Qin, T.; Xu, Z.; Qu, S.; Jia, Y.; Li, C.; Pan, L.; Gao, Q.; Jiao, M.; et al. Molecular Characteristics and Clinical Implications of TP53 Mutations in Therapy-Related Myelodysplastic Syndromes. Blood Cancer J. 2025, 15, 58. [Google Scholar] [CrossRef]
- Kaur, A.; Rojek, A.E.; Symes, E.; Nawas, M.T.; Patel, A.A.; Patel, J.L.; Sojitra, P.; Aqil, B.; Sukhanova, M.; McNerney, M.E.; et al. Real World Predictors of Response and 24-Month Survival in High-Grade TP53-Mutated Myeloid Neoplasms. Blood Cancer J. 2024, 14, 99. [Google Scholar] [CrossRef]
- Ozga, M.; Nicolet, D.; Mrózek, K.; Yilmaz, A.S.; Kohlschmidt, J.; Larkin, K.T.; Blachly, J.S.; Oakes, C.C.; Buss, J.; Walker, C.J.; et al. Sex-Associated Differences in Frequencies and Prognostic Impact of Recurrent Genetic Alterations in Adult Acute Myeloid Leukemia (Alliance, AMLCG). Leukemia 2024, 38, 45–57. [Google Scholar] [CrossRef]
- Shah, M.V.; Hung, K.; Baranwal, A.; Kutyna, M.M.; Al-Kali, A.; Toop, C.; Greipp, P.; Brown, A.; Shah, S.; Khanna, S.; et al. Evidence-Based Risk Stratification of Myeloid Neoplasms Harboring TP53 Mutations. Blood Adv. 2025, 9, 3370–3380. [Google Scholar] [CrossRef]
- Jambhekar, A.; Ackerman, E.E.; Alpay, B.A.; Lahav, G.; Lovitch, S.B. Comparison of TP53 Mutations in Myelodysplasia and Acute Leukemia Suggests Divergent Roles in Initiation and Progression. Blood Neoplasia 2024, 1, 100004. [Google Scholar] [CrossRef]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef]
- Puzo, C.J.; Hager, K.M.; Rinder, H.M.; Weinberg, O.K.; Siddon, A.J. Overall Survival in TP53-Mutated AML and MDS. Ann. Hematol. 2024, 103, 5359–5369. [Google Scholar] [CrossRef] [PubMed]
- Lontos, K.; Saliba, R.M.; Kanagal-Shamanna, R.; Özcan, G.; Ramdial, J.; Chen, G.; Kadia, T.; Short, N.J.; Daver, N.G.; Kantarjian, H.; et al. TP53-Mutant Variant Allele Frequency and Cytogenetics Determine Prognostic Groups in MDS/AML for Transplantation. Blood Adv. 2025, 9, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Nawas, M.T.; Kosuri, S. Utility or Futility? A Contemporary Approach to Allogeneic Hematopoietic Cell Transplantation for TP53-Mutated MDS/AML. Blood Adv. 2024, 8, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Yoshizato, T.; Nannya, Y.; Atsuta, Y.; Shiozawa, Y.; Iijima-Yamashita, Y.; Yoshida, K.; Shiraishi, Y.; Suzuki, H.; Nagata, Y.; Sato, Y.; et al. Genetic Abnormalities in Myelodysplasia and Secondary Acute Myeloid Leukemia: Impact on Outcome of Stem Cell Transplantation. Blood 2017, 129, 2347–2358. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine Prolongs Overall Survival Compared with Conventional Care Regimens in Elderly Patients with Low Bone Marrow Blast Count Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef]
- Bories, P.; Prade, N.; Lagarde, S.; Cabarrou, B.; Largeaud, L.; Plenecassagnes, J.; Luquet, I.; De Mas, V.; Filleron, T.; Cassou, M.; et al. Impact of TP53 Mutations in Acute Myeloid Leukemia Patients Treated with Azacitidine. PLoS ONE 2020, 15, e0238795. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Döhner, H.; Pratz, K.W.; DiNardo, C.D.; Wei, A.H.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Récher, C.; Schuh, A.C.; Babu, S.; et al. Genetic Risk Stratification and Outcomes among Treatment-Naive Patients with AML Treated with Venetoclax and Azacitidine. Blood 2024, 144, 2211–2222. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53-Mutant Acute Myeloid Leukemia with Decitabine and Venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Goldfinger, M.; Mantzaris, I.; Shastri, A.; Saunthararajah, Y.; Gritsman, K.; Sica, R.A.; Kornblum, N.; Shah, N.; Levitz, D.; Rockwell, B.; et al. A Weekly Low-Dose Regimen of Decitabine and Venetoclax Is Efficacious and Less Myelotoxic in a Racially Diverse Cohort. Blood 2024, 144, 2360–2363. [Google Scholar] [CrossRef]
- Hiwase, D.; Hahn, C.; Tran, E.N.H.; Chhetri, R.; Baranwal, A.; Al-Kali, A.; Sharplin, K.; Ladon, D.; Hollins, R.; Greipp, P.; et al. TP53 Mutation in Therapy-Related Myeloid Neoplasm Defines a Distinct Molecular Subtype. Blood 2023, 141, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Montalban-Bravo, G.; Hwang, H.; Ning, J.; Franquiz, M.J.; Kanagal-Shamanna, R.; Patel, K.P.; DiNardo, C.D.; Ravandi, F.; Garcia-Manero, G.; et al. Prognostic and Therapeutic Impacts of Mutant TP53 Variant Allelic Frequency in Newly Diagnosed Acute Myeloid Leukemia. Blood Adv. 2020, 4, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Grenet, J.; Jain, A.G.; Burkart, M.; Waksal, J.; Famulare, C.; Numan, Y.; Stahl, M.; Mckinnell, Z.; Ball, B.; Ma, X.; et al. Comparing Outcomes between Liposomal Daunorubicin/Cytarabine (CPX-351) and HMA + Venetoclax As Frontline Therapy in Acute Myeloid Leukemia. Blood 2021, 138, 32. [Google Scholar] [CrossRef]
- Othman, J.; Wilhelm-Benartzi, C.; Dillon, R.; Knapper, S.; Freeman, S.D.; Batten, L.M.; Canham, J.; Hinson, E.L.; Wych, J.; Betteridge, S.; et al. A Randomized Comparison of CPX-351 and FLAG-Ida in Adverse Karyotype AML and High-Risk MDS: The UK NCRI AML19 Trial. Blood Adv. 2023, 7, 4539–4549. [Google Scholar] [CrossRef]
- Zugasti, I.; Lopez-Guerra, M.; Castaño-Díez, S.; Esteban, D.; Avendaño, A.; Pomares, H.; Perez, A.; García-Ávila, S.; Conejo, I.P.; de la Fuente Montes, C.; et al. Hypomethylating Agents plus Venetoclax for High-Risk MDS and CMML as Bridge Therapy to Transplant: A GESMD Study. Exp. Hematol. Oncol. 2025, 14, 61. [Google Scholar] [CrossRef]
- Baranwal, A.; Langer, K.J.; Gannamani, V.; Rud, D.; Cibich, A.; Saygin, C.; Nawas, M.; Badar, T.; Kharfan-Dabaja, M.A.; Ayala, E.; et al. Factors Associated with Survival after Allogeneic Transplantation for Myeloid Neoplasms Harboring TP53 Mutations. Blood Adv. 2025, 9, 3395–3407. [Google Scholar] [CrossRef]
- Shahzad, M.; Tariq, E.; Chaudhary, S.G.; Anwar, I.; Iqbal, Q.; Fatima, H.; Abdelhakim, H.; Ahmed, N.; Balusu, R.; Hematti, P.; et al. Outcomes with Allogeneic Hematopoietic Stem Cell Transplantation in TP53-Mutated Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. Leuk. Lymphoma 2022, 63, 3409–3417. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef]
- Sinanidis, I.; Hochman, M.J.; Tsai, H.-L.; Randall, M.P.; Bonilla, B.; Varadhan, R.; Ambinder, A.J.; Jones, R.J.; DeZern, A.E.; Karantanos, T. Favorable Outcomes in MDS and Oligoblastic AML-MR after Reduced-Intensity Conditioning Allogeneic Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide. Bone Marrow Transplant. 2024, 59, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Haldar, S.D.; Ambinder, A.; Webster, J.A.; Jain, T.; Dalton, W.B.; Prince, G.T.; Ghiaur, G.; DeZern, A.E.; Gojo, I.; et al. Outcome Heterogeneity of TP53-Mutated Myeloid Neoplasms and the Role of Allogeneic Hematopoietic Cell Transplantation. Haematologica 2024, 109, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Atallah, E.; Shallis, R.; Saliba, A.N.; Patel, A.; Bewersdorf, J.P.; Grenet, J.; Stahl, M.; Duvall, A.; Burkart, M.; et al. Survival of TP53-Mutated Acute Myeloid Leukemia Patients Receiving Allogeneic Stem Cell Transplantation after First Induction or Salvage Therapy: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Leukemia 2023, 37, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Komrokji, R.S.; Yun, S.; Al Ali, N.; Chan, O.; Song, J.; Hussaini, M.; Talati, C.; Sweet, K.L.; Lancet, J.E.; et al. Baseline and Serial Molecular Profiling Predicts Outcomes with Hypomethylating Agents in Myelodysplastic Syndromes. Blood Adv. 2021, 5, 1017–1028. [Google Scholar] [CrossRef]
- Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; Devine, S.M.; et al. Impact of Conditioning Intensity and Genomics on Relapse After Allogeneic Transplantation for Patients with Myelodysplastic Syndrome. JCO Precis. Oncol. 2021, 5, PO.20.00355. [Google Scholar] [CrossRef]
- Senapati, J.; Garcia-Manero, G.; DiNardo, C.D.; Deshmukh, I.; Borthakur, G.; Kadia, T.M.; Jabbour, E.; Short, N.J.; Abbas, H.A.; Pemmaraju, N.; et al. Barriers to Allogeneic Stem Cell Transplantation in Responding Patients with TP53-Mutated Acute Myeloid Leukemia. Bone Marrow Transplant. 2025, 60, 910–913. [Google Scholar] [CrossRef]
- Hourigan, C.S.; Dillon, L.W.; Gui, G.; Logan, B.R.; Fei, M.; Ghannam, J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; et al. Impact of Conditioning Intensity of Allogeneic Transplantation for Acute Myeloid Leukemia with Genomic Evidence of Residual Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1273–1283. [Google Scholar] [CrossRef]
- Byrne, M.T.; Kurian, T.J.; Patel, D.A.; Tamari, R.; Hong, S.; Abdelhakim, H.; Klein, V.; Rojas, P.; Madhavan, R.; Kent, A.; et al. Non-Relapse Mortality in TP53-Mutated MDS/AML—A Multi-Center Collaborative Study. Blood 2021, 138, 2922. [Google Scholar] [CrossRef]
- University of Birmingham. A Double-Blind, Phase III, Randomised Study to Compare the Efficacy and Safety of Oral Azacitidine (CC-486) Versus Placebo in Subjects with Acute Myeloid Leukaemia or Myelodysplastic Syndromes as Maintenance After Allogeneic Haematopoietic Stem Cell Transplantation. 2024. Available online: https://clinicaltrials.gov/study/NCT04173533 (accessed on 20 September 2025).
- AbbVie. A Randomized, Open Label Phase 3 Study Evaluating Safety and Efficacy of Venetoclax in Combination with Azacitidine After Allogeneic Stem Cell Transplantation in Subjects with Acute Myeloid Leukemia (AML) (VIALE-T). 2025. Available online: https://clinicaltrials.gov/study/NCT04161885 (accessed on 20 September 2025).
- DeFilipp, Z.M. A Phase Ib Study of Oral Decitabine/Cedazuridine as Maintenance Therapy Following Allogeneic Hematopoietic Cell Transplantation for Patients with Myeloid Neoplasms. 2025. Available online: https://clinicaltrials.gov/study/NCT04980404 (accessed on 20 September 2025).
- Garcia, J. A Phase 1 Study of Adding Venetoclax to a Reduced Intensity Conditioning Regimen and to Maintenance in Combination with a Hypomethylating Agent After Allogeneic Hematopoietic Cell Transplantation for Patients with High Risk AML, MDS, and MDS/MPN Overlap Syndromes. 2025. Available online: https://clinicaltrials.gov/study/NCT03613532 (accessed on 20 September 2025).
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, Cytarabine, and Nivolumab in Patients with Newly Diagnosed Acute Myeloid Leukaemia or High-Risk Myelodysplastic Syndrome: A Single-Arm, Phase 2 Study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Garcia, J.S.; Flamand, Y.; Penter, L.; Keng, M.; Tomlinson, B.K.; Mendez, L.M.; Koller, P.; Cullen, N.; Arihara, Y.; Pfaff, K.; et al. Ipilimumab plus Decitabine for Patients with MDS or AML in Posttransplant or Transplant-Naïve Settings. Blood 2023, 141, 1884–1888. [Google Scholar] [CrossRef]
- Bouligny, I.M.; Montalban-Bravo, G.; Sasaki, K.; Daver, N.; Jabbour, E.; Alvarado, Y.; DiNardo, C.D.; Ravandi, F.; Borthakur, G.; Bose, P.; et al. A Phase II Trial of Azacitidine with Ipilimumab, Nivolumab, or Ipilimumab and Nivolumab in Previously Untreated Myelodysplastic Syndrome. Haematologica 2025, 110, 1628–1633. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Boss, I.; Beach, C.L.; Copeland, W.B.; Thompson, E.; Fox, B.A.; Hasle, V.E.; Hellmann, A.; Taussig, D.C.; Tormo, M.; et al. A Randomized Phase 2 Trial of Azacitidine with or without Durvalumab as First-Line Therapy for Older Patients with AML. Blood Adv. 2022, 6, 2219–2229. [Google Scholar] [CrossRef]
- University of Southern California. A Phase I/II Multicenter Study Combining Guadecitabine, a DNA Methyltransferase Inhibitor, with Atezolizumab, an Immune Checkpoint Inhibitor, in Patients with Intermediate or High-Risk Myelodysplastic Syndrome or Chronic Myelomonocytic Leukemia. 2025. Available online: https://clinicaltrials.gov/study/NCT02935361 (accessed on 20 September 2025).
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.A.; Asch, A.S.; Mannis, G.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and Efficacy of the Anticluster of Differentiation 47 Antibody Magrolimab Combined with Azacitidine in Patients with Previously Untreated AML: Phase Ib Results. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 4893–4904. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Senapati, J.; Maiti, A.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Pemmaraju, N.; Jabbour, E.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (Pts) with Newly Diagnosed (ND) Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2022, 140, 141–144. [Google Scholar] [CrossRef]
- Gilead Statement on the Discontinuation of Magrolimab Study in AML with TP53 Mutations. Available online: https://www.gilead.com/company/company-statements/2023/gilead-statement-on-the-discontinuation-of-magrolimab-study-in-aml-with-tp53-mutations (accessed on 20 September 2025).
- Gilead Sciences. A Phase 3, Randomized, Open-Label Study Evaluating the Safety and Efficacy of Magrolimab in Combination with Azacitidine Versus Physician’s Choice of Venetoclax in Combination with Azacitidine or Intensive Chemotherapy in Previously Untreated Patients with TP53 Mutant Acute Myeloid Leukemia. 2025. Available online: https://clinicaltrials.gov/study/NCT04778397 (accessed on 20 September 2025).
- Gilead Statement on Discontinuation of Phase 3 ENHANCE-3 Study in AML. Available online: https://www.gilead.com/company/company-statements/2024/gilead-statement-on-discontinuation-of-phase-3-enhance-3-study-in-aml (accessed on 20 September 2025).
- Daver, N.; Vyas, P.; Huls, G.; Döhner, H.; Maury, S.; Novak, J.; Papayannidis, C.; Martínez Chamorro, C.; Montesinos, P.; Niroula, R.; et al. The ENHANCE-3 Study: Venetoclax and Azacitidine plus Magrolimab or Placebo for Untreated AML Unfit for Intensive Therapy. Blood 2025, 146, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Przespolewski, A.; Abaza, Y.; Byrne, M.; Fong, A.P.; Jin, F.; Forgie, A.J.; Tsiatis, A.C.; Guan, S.; Erba, H.P. Evorpacept (ALX148), a CD47-Blocking Myeloid Checkpoint Inhibitor, in Combination with Azacitidine and Venetoclax in Patients with Acute Myeloid Leukemia (ASPEN-05): Results from Phase 1a Dose Escalation Part. Blood 2022, 140, 9046–9047. [Google Scholar] [CrossRef]
- ALX Oncology Inc. A Phase 1/2 Study of Evorpacept (ALX148) in Combination with Venetoclax and Azacitidine in Patients with Acute Myeloid Leukemia (AML) (ASPEN-05). 2024. Available online: https://clinicaltrials.gov/study/NCT04755244 (accessed on 20 September 2025).
- ALX Oncology Inc. A Phase 1/2 Study of Evorpacept (ALX148) in Combination with Azacitidine in Patients with Higher Risk Myelodysplastic Syndrome (MDS) (ASPEN-02). 2025. Available online: https://clinicaltrials.gov/study/NCT04417517 (accessed on 20 September 2025).
- Daver, N.G.; Stevens, D.A.; Hou, J.-Z.; Yamauchi, T.; Moshe, Y.; Fong, C.Y.; Marzocchetti, A.; Adamec, R.; Patel, M.; Lambert, S.; et al. Lemzoparlimab (Lemzo) with Venetoclax (Ven) and/or Azacitidine (Aza) in Patients (Pts) with Acute Myeloid Leukemia (AML) or Myelodysplastic Syndromes (MDS): A Phase 1b Dose Escalation Study. J. Clin. Oncol. 2022, 40, TPS7067. [Google Scholar] [CrossRef]
- AbbVie. A Phase 1b Dose Escalation Study of Lemzoparlimab in Combination with Venetoclax and/or Azacitidine in Subjects with Acute Myeloid Leukemia (AML) or Myelodysplastic Syndrome (MDS). 2024. Available online: https://clinicaltrials.gov/study/NCT04912063 (accessed on 20 September 2025).
- Akeso. A Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase 2 Study of AK117 or Placebo in Combination with Azacitidine in Patients with Newly Diagnosed Higher-Risk Myelodysplastic Syndromes. 2025. Available online: https://clinicaltrials.gov/study/NCT06196203 (accessed on 20 September 2025).
- Zeidan, A.M.; Tong, H.; Xiao, Z.; Baratam, P.; Abboud, R.; Benton, C.B.; Zeidner, J.F.; Borate, U.; Chai-Ho, W.; Lu, Y.; et al. Trial in Progress: A Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase 2 Study of AK117/Placebo in Combination with Azacitidine in Patients with Newly Diagnosed Higher-Risk Myelodysplastic Syndromes (AK117-205). Blood 2024, 144, 6705. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138, 244. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Garcia-Manero, G.; Vey, N.; Wermke, M.; Janssen, J.J.W.M.; et al. Phase Ib Study of Sabatolimab (MBG453), a Novel Immunotherapy Targeting TIM-3 Antibody, in Combination with Decitabine or Azacitidine in High- or Very High-Risk Myelodysplastic Syndromes. Am. J. Hematol. 2024, 99, E32–E36. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. A Randomized, Double-Blind, Placebo-Controlled Phase II Multi-Center Study of Intravenous MBG453 Added to Hypomethylating Agents in Adult Subjects with Intermediate, High or Very High Risk Myelodysplastic Syndrome (MDS) as Per IPSS-R Criteria. 2025. Available online: https://clinicaltrials.gov/study/NCT03946670 (accessed on 20 September 2025).
- Novartis Pharmaceuticals. A Randomized, Double-Blind, Placebo-Controlled Phase III Multi-Center Study of Azacitidine with or Without MBG453 for the Treatment of Patients with Intermediate, High or Very High Risk Myelodysplastic Syndrome (MDS) as Per IPSS-R, or Chronic Myelomonocytic Leukemia-2 (CMML-2). 2025. Available online: https://clinicaltrials.gov/study/NCT04266301 (accessed on 20 September 2025).
- Novartis Pharmaceuticals. A Phase II Multi-Center, Single Arm, Safety and Efficacy Study of MBG453 in Combination with Azacitidine and Venetoclax for the Treatment of Acute Myeloid Leukemia (AML) in Adult Patients Unfit for Chemotherapy. 2025. Available online: https://clinicaltrials.gov/study/NCT04150029 (accessed on 20 September 2025).
- Novartis Pharmaceuticals. A Single-Arm, Open-Label, Phase II Study of Sabatolimab in Combination with Azacitidine and Venetoclax in Adult Participants with High or Very High Risk Myelodysplastic Syndromes (MDS) as Per IPSS-R Criteria. 2025. Available online: https://clinicaltrials.gov/study/NCT04812548 (accessed on 20 September 2025).
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.; Maris, M.; Lin, T.L.; Patel, P.; Madanat, Y.F.; Cogle, C.R.; Borthakur, G.; Huebner, D.; Khaskhely, N.; Bonham, L.; et al. Updated Results from a Phase 1 Study of APVO436, a Novel Bispecific Anti-CD123 x Anti-CD3 AdaptirTM Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2022, 140, 6204–6205. [Google Scholar] [CrossRef]
- Aptevo Therapeutics. A Phase 1b/2 Open-Label Study of APVO436 in Combination with Venetoclax and Azacitidine in Patients with Newly Diagnosed Acute Myeloid Leukemia (AML). 2025. Available online: https://clinicaltrials.gov/study/NCT06634394 (accessed on 20 September 2025).
- Lane, A.A.; Garcia, J.S.; Raulston, E.G.; Garzon, J.L.; Galinsky, I.; Baxter, E.W.; Leonard, R.; DeAngelo, D.J.; Luskin, M.R.; Reilly, C.R.; et al. Phase 1b Trial of Tagraxofusp in Combination with Azacitidine with or without Venetoclax in Acute Myeloid Leukemia. Blood Adv. 2024, 8, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Lane, A. Phase 1 Study of SL-401 in Combination with Azacitidine and Venetoclax in Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Treatment-Naive Subjects with AML Not Eligible for Standard Induction and in Subjects with Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) or SL-401 in Combination with Azacitidine in Subjects with High-Risk Myelodysplastic Syndrome (MDS). 2025. Available online: https://clinicaltrials.gov/study/NCT03113643 (accessed on 20 September 2025).
- Zhang, H.; Zhu, H.-H. Breakthroughs of CAR T-Cell Therapy in Acute Myeloid Leukemia: Updates from ASH 2024. Exp. Hematol. Oncol. 2025, 14, 57. [Google Scholar] [CrossRef]
- Liu, F.; Cao, Y.; Pinz, K.; Ma, Y.; Wada, M.; Chen, K.; Ma, G.; Shen, J.; Tse, C.O.; Su, Y.; et al. First-in-Human CLL1-CD33 Compound CAR T Cell Therapy Induces Complete Remission in Patients with Refractory Acute Myeloid Leukemia: Update on Phase 1 Clinical Trial. Blood 2018, 132, 901. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, H.; Sun, L.; Li, Y.; Zhang, S.; He, G.; Yi, H.; Wada, M.; Pinz, K.G.; Chen, K.H.; et al. First-In-Human CLL1-CD33 Compound Car (CCAR) T Cell Therapy in Relapsed and Refractory Acute Myeloid Leukemia. EHA 2020—Abstract S149. Available online: https://library.ehaweb.org/eha/2020/eha25th/294969/fang.liu.first-in-human.cll1-cd33.compound.car.28ccar29.t.cell.therapy.in.html (accessed on 20 September 2025).
- Xu, K.L. Application of Anti Tim-3/CD123 CAR-T Cell Therapy in Relapsed and Refractory Acute Myeloid Leukemia (rr/AML). 2023. Available online: https://clinicaltrials.gov/study/NCT06125652 (accessed on 20 September 2025).
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and Split CAR T Cells Targeting CD13 and TIM3 Eradicate Acute Myeloid Leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yu, K.-R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.-H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological Reactivation of P53 in the Era of Precision Anticancer Medicine. Nat. Rev. Clin. Oncol. 2024, 21, 106–120. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone Des Myélodysplasies (GFM). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Aprea Therapeutics. A Phase III Multicenter, Randomized, Open Label Study of APR-246 in Combination with Azacitidine Versus Azacitidine Alone for the Treatment of (Tumor Protein) TP53 Mutant Myelodysplastic Syndromes. 2025. Available online: https://clinicaltrials.gov/study/NCT03745716 (accessed on 20 September 2025).
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt Combined with Venetoclax and Azacitidine in TP53-Mutated Acute Myeloid Leukaemia: A Phase 1, Dose-Finding and Expansion Study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Daver, N.G. Eprenetapopt in the Post-Transplant Setting: Mechanisms and Future Directions. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 3994–3997. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Björklund, U.; Wiman, K.G. APR-246 Overcomes Resistance to Cisplatin and Doxorubicin in Ovarian Cancer Cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Song, H.; Wu, J.; Tang, Y.; Dai, Y.; Xiang, X.; Li, Y.; Wu, L.; Wu, J.; Liang, Y.; Xing, Y.; et al. Diverse Rescue Potencies of P53 Mutations to ATO Are Predetermined by Intrinsic Mutational Properties. Sci. Transl. Med. 2023, 15, eabn9155. [Google Scholar] [CrossRef]
- The University of Hong Kong. A Phase 2 Study of Oral Arsenic Trioxide (Arsenol®)-Based Low-Intensity Treatment for Previously Untreated or Relapsed/Refractory TP53-Mutated Myeloid Malignancies. 2025. Available online: https://clinicaltrials.gov/study/NCT06778187 (accessed on 20 September 2025).
- Ma, J.; Sun, S.; Xie, Y.; Zhou, S.; Wu, M.; Zuo, X.; Xie, M.; Wang, X. Decitabine with Etoposide Is Effective in TP53 Mutated Myeloid Tumors via Overcoming Differentiation Block. Blood Cancer J. 2025, 15, 19. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Tognon, C.; Mishra, S.; Kaempf, A.; Park, B.; Joshi, S.K.; Huang, A.; Kurtz, S.E.; Sax, L.; Eide, C.A.; Johnson, K.; et al. Phase 1 Trial Testing the Novel Combination Therapy of Entrectinib and ASTX727 in TP53 Mutated Relapsed/Refractory Acute Myeloid Leukemia Patients. Blood 2024, 144, 1498. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a Prognostic and Therapeutic Target in Acute Myeloid Leukemia Mediates Paracrine Crosstalk of Leukemia Cells with Bone Marrow Stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Stromatt, J.C.; Fobare, S.; Huang, K.M.; Buelow, D.R.; Orwick, S.; Jeon, J.Y.; Weber, R.H.; Larsen, B.; Mims, A.S.; et al. TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion. Cancers 2022, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, E.D.; Swords, R.; Huang, Y.; Orwick, S.; Buelow, D.; Abbott, N.; Phelps, M.; Zeidner, J.; Foster, M.C.; Lin, T.L.; et al. A Phase 1b/2 Study of TP-0903 and Decitabine Targeting Mutant TP53 and/or Complex Karyotype in Patients with Untreated Acute Myeloid Leukemia ≥Age 60 Years. Cancer Res. Commun. 2025, 5, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a First-in-Class NEDD8-Activating Enzyme Inhibitor, Combined with Azacitidine in Patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.N.; Kaufmann, S.H.; Stein, E.M.; Patel, P.A.; Baer, M.R.; Stock, W.; Deininger, M.; Blum, W.; Schiller, G.J.; Olin, R.L.; et al. Pevonedistat with Azacitidine in Older Patients with TP53-Mutated AML: A Phase 2 Study with Laboratory Correlates. Blood Adv. 2023, 7, 2360–2363. [Google Scholar] [CrossRef]
- Short, N.J.; Muftuoglu, M.; Ong, F.; Nasr, L.; Macaron, W.; Montalban-Bravo, G.; Alvarado, Y.; Basyal, M.; Daver, N.; Dinardo, C.D.; et al. A Phase 1/2 Study of Azacitidine, Venetoclax and Pevonedistat in Newly Diagnosed Secondary AML and in MDS or CMML after Failure of Hypomethylating Agents. J. Hematol. Oncol. 2023, 16, 73. [Google Scholar] [CrossRef]
- Murthy, G.S.G.; Saliba, A.N.; Szabo, A.; Harrington, A.; Abedin, S.; Carlson, K.; Michaelis, L.; Runaas, L.; Baim, A.; Hinman, A.; et al. A Phase I Study of Pevonedistat, Azacitidine, and Venetoclax in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Haematologica 2024, 109, 2864–2872. [Google Scholar] [CrossRef]
- Man, C.-H.; Lam, W.; Dang, C.-C.; Zeng, X.-Y.; Zheng, L.-C.; Chan, N.N.-M.; Ng, K.-L.; Chan, K.-C.; Kwok, T.-H.; Ng, T.C.-C.; et al. Inhibition of PLK4 Remodels Histone Methylation and Activates the Immune Response via the cGAS-STING Pathway in TP53-Mutated AML. Blood 2023, 142, 2002–2015. [Google Scholar] [CrossRef]
- Ayoub, E.; Marupudi, A.; Nishida, Y.; Heinz Montoya, R.; Mohanty, V.; Walter, W.; Bray, M.R.; Patsilevas, T.; Boettcher, S.; Issa, G.C.; et al. Polyploidy Is Necessary for Apoptotic Cell Death in TP53-Mut AML in Response to Polo-like-Kinase 4 (PLK4) Inhibition and Results in Caspase 3 Cleavage. Blood 2022, 140, 5941–5943. [Google Scholar] [CrossRef]
- Murphy, T.; Mason, J.M.; Leber, B.; Bray, M.R.; Chan, S.M.; Gupta, V.; Khalaf, D.; Maze, D.; McNamara, C.J.; Schimmer, A.D.; et al. Preclinical Characterization and Clinical Trial of CFI-400945, a Polo-like Kinase 4 Inhibitor, in Patients with Relapsed/Refractory Acute Myeloid Leukemia and Higher-Risk Myelodysplastic Neoplasms. Leukemia 2024, 38, 502–512. [Google Scholar] [CrossRef]
- Treadwell Therapeutics, Inc. Phase 1b/2 Clinical Study of the Safety, Tolerability, and Pharmacokinetic and Pharmacodynamic Profiles of CFI-400945 as a Single Agent or in Combination with Azacitidine in Patients with AML, MDS or CMML. 2025. Available online: https://clinicaltrials.gov/study/NCT04730258 (accessed on 20 September 2025).
- Jonas, B.A.; Yee, K.; Koller, P.B.; Brandwein, J.; Mims, A.S.; Michelson, G.C.; Nguyen, L.; Bray, M.R.; Roberts-Thomson, E.L.; Borthakur, G. Preliminary Results from a Phase 2 Open-Label, Multicenter, Dose Optimization Clinical Study of the Safety, Tolerability, and Pharmacokinetic (PK) and Pharmacodynamic (PD) Profiles of Cfi-400945 As a Single Agent or in Combination with Azacitidine or Decitabine in Patients with Acute Myeloid Leukemia, Myelodysplastic Syndrome or Chronic Myelomonocytic Leukemia (TWT-202). Blood 2022, 140, 9076–9077. [Google Scholar] [CrossRef]
- Song, X.; Peng, Y.; Wang, X.; Chen, Q.; Lan, X.; Shi, F. The Stimulator of Interferon Genes (STING) Agonists for Treating Acute Myeloid Leukemia (AML): Current Knowledge and Future Outlook. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2023, 25, 1545–1553. [Google Scholar] [CrossRef]
- Baba, T.; Yoshida, T.; Tanabe, Y.; Nishimura, T.; Morishita, S.; Gotoh, N.; Hirao, A.; Hanayama, R.; Mukaida, N. Cytoplasmic DNA Accumulation Preferentially Triggers Cell Death of Myeloid Leukemia Cells by Interacting with Intracellular DNA Sensing Pathway. Cell Death Dis. 2021, 12, 322. [Google Scholar] [CrossRef]
- Adam, M.; Yu, J.; Plant, R.; Shelton, C.; Schmidt, H.; Yang, J. Sting Agonist GSK3745417 Induces Apoptosis, Antiproliferation, and Cell Death in a Panel of Human AML Cell Lines and Patient Samples. Blood 2022, 140, 11829. [Google Scholar] [CrossRef]
- Montesinos, P.; Al-Ali, H.; Alonso-Dominguez, J.M.; Jentzsch, M.; Jongen-Lavrencic, M.; Martelli, M.P.; Röllig, C.; Sica, S.; Iadevaia, R.; Yablonski, K.; et al. Abstract CT124: A First-in-Clinic Phase 1 Study of GSK3745417 STING Agonist in Relapsed/Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Cancer Res. 2023, 83, CT124. [Google Scholar] [CrossRef]
- GlaxoSmithKline. A Phase 1, Open Label Study of Intravenous GSK3745417 to Evaluate Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Determine RP2D and Schedule in Participants with Relapsed or Refractory Myeloid Malignancies Including Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS). 2025. Available online: https://clinicaltrials.gov/study/NCT05424380 (accessed on 20 September 2025).
- Baer, M. Phase 1 Clinical Trial of the STING Agonist CRD3874-SI in Patients with Relapsed/Refractory Acute Myeloid Leukemia. 2024. Available online: https://clinicaltrials.gov/study/NCT06626633 (accessed on 20 September 2025).
- Brem, E.A.; Shieh, K.; Juarez, D.; Buono, R.; Jeyakumar, D.; O’Brien, S.; Taylor, T.H.; Fruman, D.A. A Phase 1 Study Adding Pitavastatin to Venetoclax Therapy in AML and CLL/SLL: A Mechanism-Based Drug Repurposing Strategy. Blood Neoplasia 2024, 1, 100036. [Google Scholar] [CrossRef]
- Maslah, N.; Rety, S.; Bonnamy, M.; Aguinaga, L.; Huynh, T.; Parietti, V.; Giraudier, S.; Fenaux, P.; Cassinat, B. Niclosamide Combined to Azacitidine to Target TP53-Mutated MDS/AML Cells. Leukemia 2024, 38, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).