
Pyrroloquinoline Quinone (PQQ) Attenuates Hydrogen Peroxide-Induced Injury Through the Enhancement of Mitochondrial Function in Human Trabecular Meshwork Cells
 , ,
, ,  , , , ,
, , , ,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results
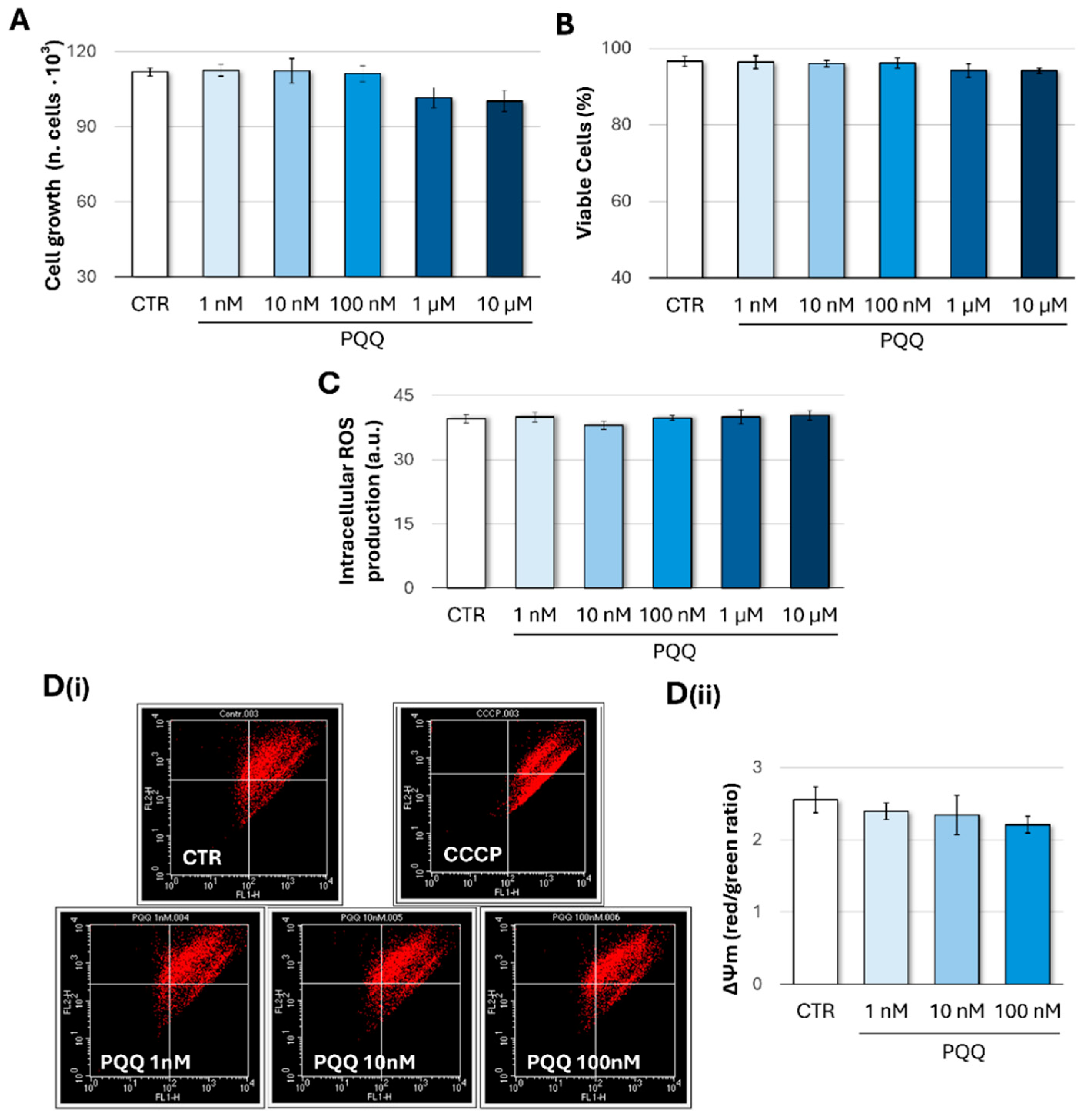
2.1. Effects of PQQ on Cell Growth and Viability, Redox Homeostasis, and Mitochondrial Membrane Potential (ΔΨm) in Human Trabecular Meshwork Cells
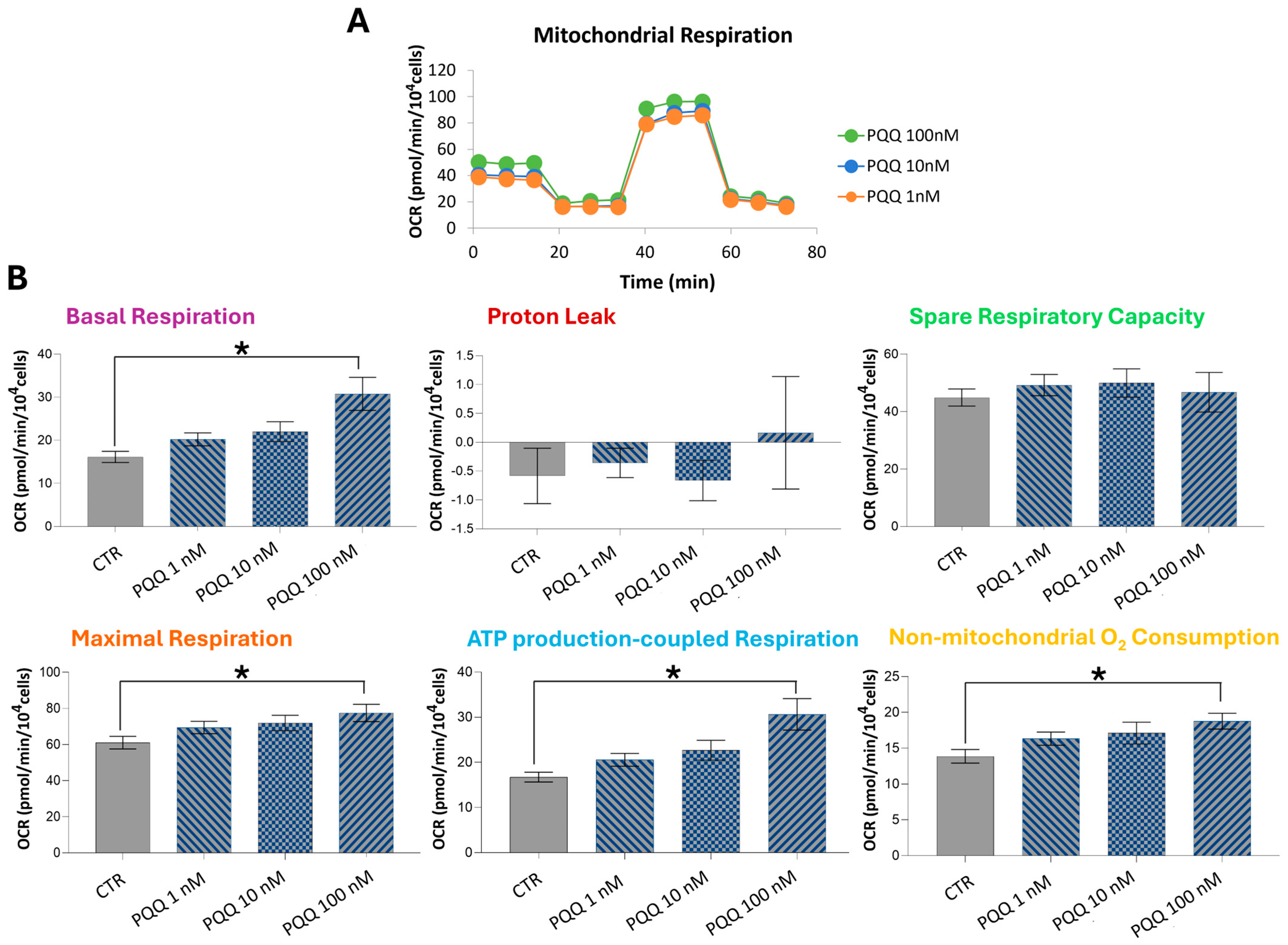
2.2. PQQ Increases Respiratory Capacity and ATP Production in HTM Cells
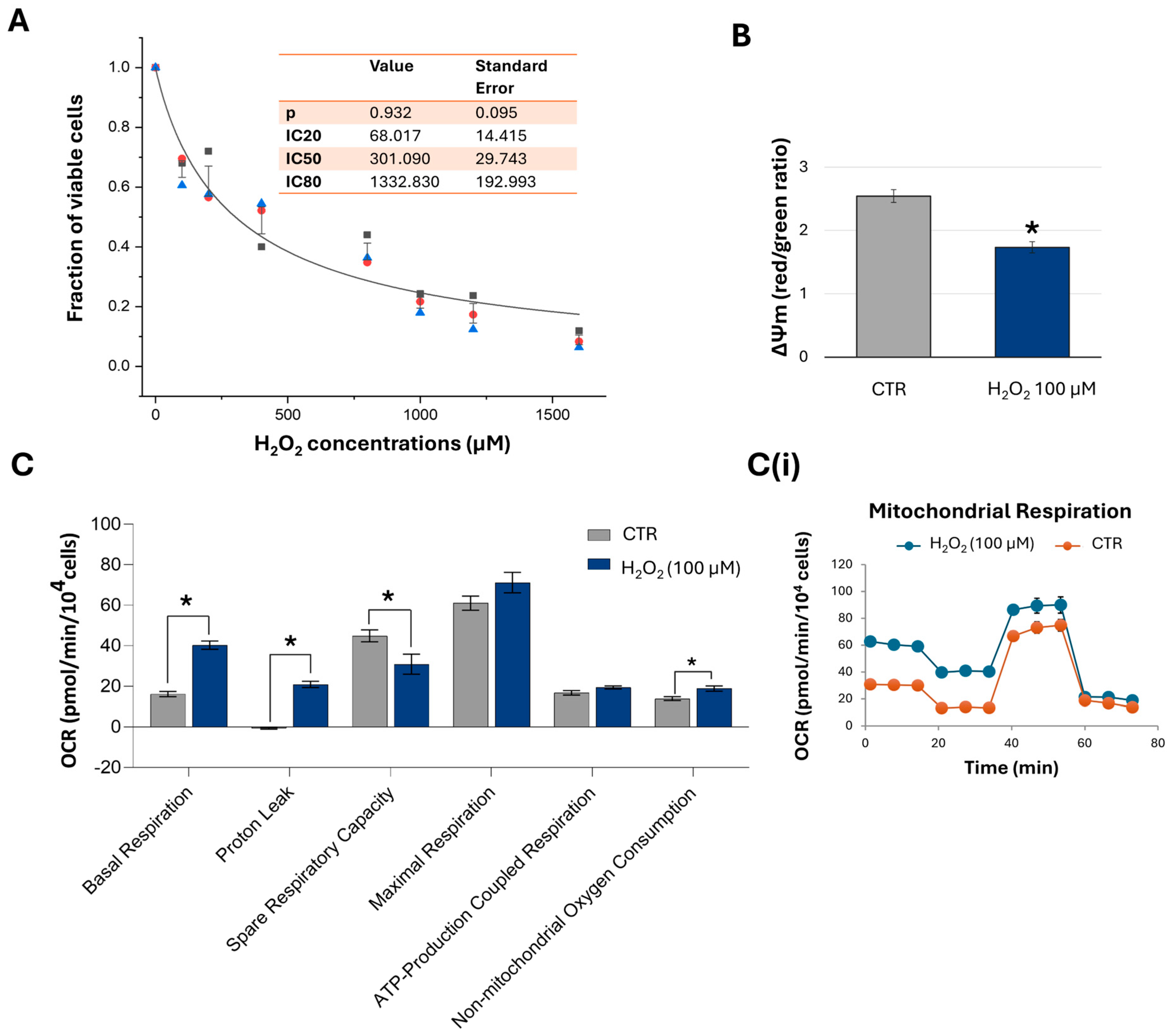
2.3. Short-Term Exposure to H2O2 Induces Concentration-Dependent Inhibitory Effects on Cell Viability and Alters the Mitochondrial Bioenergetics of HTM Cells
2.4. Protective Effects of PQQ Against H2O2-Induced Decline in Mitochondrial Respiratory Capacity in HTM Cells
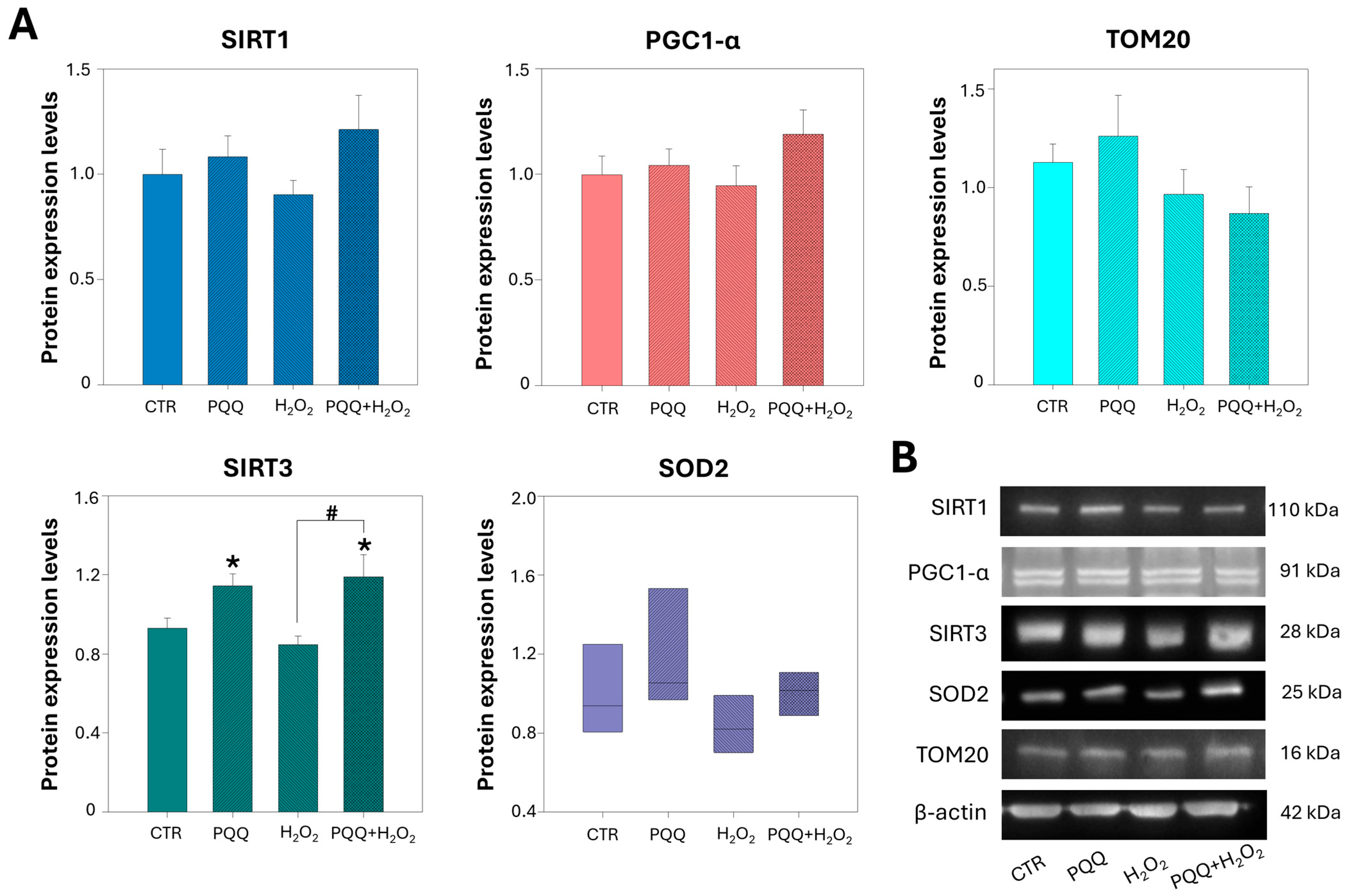
2.5. PQQ Pretreatment Upregulates the Protein Levels of SIRT3
2.6. PQQ Mitigates H2O2-Induced Morphological and Ultrastructural Damage to Mitochondria
2.7. Protective Effects of PQQ Against H2O2-Induced Cytotoxicity in HTM Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Detection of Intracellular Reactive Oxygen Species (ROS)
4.3. Mito Stress Test by Seahorse XF96 Extracellular Flux Analyser
4.4. Flow Cytometry Analyses
4.4.1. Assessment of Mitochondrial Membrane Potential (ΔΨm) by Flow Cytometry
4.4.2. Determination of Cell Death by Annexin V/Propidium Iodide Assay
4.5. Western Blot Analysis
4.6. Transmission Electron Microscopy (TEM)
4.7. Immunofluorescence
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, J.N.; Zhang, S.D.; Qu, Y.; Wang, H.L.; Tham, C.C.; Pang, C.P.; Chu, W.K. Rapamycin Removes Damaged Mitochondria and Protects Human Trabecular Meshwork (TM-1) Cells from Chronic Oxidative Stress. Mol. Neurobiol. 2019, 56, 6586–6593. [Google Scholar] [CrossRef]
- Wu, X.; Pang, Y.; Zhang, Z.; Li, X.; Wang, C.; Lei, Y.; Li, A.; Yu, L.; Ye, J. Mitochondria-targeted antioxidant peptide SS-31 mediates neuroprotection in a rat experimental glaucoma model. Acta Biochim. Biophys. Sin. 2019, 51, 411–421. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef]
- Iorio, R.; Petricca, S. Role of AMPK/ULK1 signaling in mitophagy. In Mitophagy in Health and Disease; Lemasters, J.J., Ed.; Academic Press: Cambridge, MA, USA, 2025; Chapter 3; pp. 43–70. ISBN 9780443152603. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Petricca, S.; Iorio, R.; Toniato, E.; Flati, V. Mitochondrial and metabolic alterations in cancer cells. Eur. J. Cell Biol. 2022, 101, 151225. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Petricca, S.; Mattei, V.; Delle Monache, S. Horizontal mitochondrial transfer as a novel bioenergetic tool for mesenchymal stromal/stem cells: Molecular mechanisms and therapeutic potential in a variety of diseases. J. Transl. Med. 2024, 22, 491. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Petricca, S.; Di Emidio, G.; Falone, S.; Tatone, C. Mitochondrial Extracellular Vesicles (mitoEVs): Emerging mediators of cell-to-cell communication in health, aging and age-related diseases. Ageing Res. Rev. 2024, 101, 102522. [Google Scholar] [CrossRef]
- Iorio, R.; Castellucci, A.; Rossi, G.; Cinque, B.; Cifone, M.G.; Macchiarelli, G.; Cecconi, S. Mancozeb affects mitochondrial activity, redox status and ATP production in mouse granulosa cells. Toxicol. Vitro 2015, 30, 438–445. [Google Scholar] [CrossRef]
- Petricca, S.; Flati, V.; Celenza, G.; Di Gregorio, J.; Lizzi, A.R.; Luzi, C.; Cristiano, L.; Cinque, B.; Rossi, G.; Festuccia, C.; et al. Tebuconazole and econazole act synergistically in mediating mitochondrial stress, energy imbalance, and sequential activation of autophagy and apoptosis in mouse Sertoli TM4 cells: Possible role of AMPK/ULK1 axis. Toxicol. Sci. 2019, 169, 209–223. [Google Scholar] [CrossRef]
- Petricca, S.; Carnicelli, V.; Luzi, C.; Cinque, B.; Celenza, G.; Iorio, R. Oxidative Stress, Cytotoxic and Inflammatory Effects of Azoles Combinatorial Mixtures in Sertoli TM4 Cells. Antioxidants 2023, 12, 1142. [Google Scholar] [CrossRef]
- Petricca, S.; Celenza, G.; Costagliola, C.; Tranfa, F.; Iorio, R. Cytotoxicity, Mitochondrial Functionality, and Redox Status of Human Conjunctival Cells after Short and Chronic Exposure to Preservative-Free Bimatoprost 0.03% and 0.01%: An In Vitro Comparative Study. Int. J. Mol. Sci. 2022, 23, 14113. [Google Scholar] [CrossRef]
- Petricca, S.; Celenza, G.; Luzi, C.; Cinque, B.; Lizzi, A.R.; Franceschini, N.; Festuccia, C.; Iorio, R. Synergistic Activity of Ketoconazole and Miconazole with Prochloraz in Inducing Oxidative Stress, GSH Depletion, Mitochondrial Dysfunction, and Apoptosis in Mouse Sertoli TM4 Cells. Int. J. Mol. Sci. 2022, 23, 5429. [Google Scholar] [CrossRef]
- Goyal, A.; Srivastava, A.; Sihota, R.; Kaur, J. Evaluation of oxidative stress markers in aqueous humor of primary open angle glaucoma and primary angle closure glaucoma patients. Curr. Eye Res. 2014, 39, 823–829. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch. Ophthalmol. 2010, 128, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shui, Y.-B.; Liu, Y.; Liu, X.; Siegfried, C.J. Trabecular Meshwork Mitochondrial Function and Oxidative Stress: Clues to Racial Disparities of Glaucoma. Ophthalmol. Sci. 2022, 2, 100107. [Google Scholar] [CrossRef] [PubMed]
- Benoist d’Azy, C.; Pereira, B.; Chiambaretta, F.; Dutheil, F. Oxidative and Anti-Oxidative Stress Markers in Chronic Glaucoma: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0166915. [Google Scholar] [CrossRef]
- Kennedy, S.; Williams, C.; Tsaturian, E.; Morgan, J.T. Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells. Curr. Issues Mol. Biol. 2024, 46, 9867–9880. [Google Scholar] [CrossRef]
- Venkatesan, A.; Bernstein, A.M. Protein misfolding and mitochondrial dysfunction in glaucoma. Front. Cell Dev. Biol. 2025, 13, 1595121. [Google Scholar] [CrossRef]
- Singh, P.K.; Kasetti, R.B.; Zode, G.S.; Goyal, A.; Juzych, M.S.; Kumar, A. Zika Virus Infects Trabecular Meshwork and Causes Trabeculitis and Glaucomatous Pathology in Mouse Eyes. mSphere 2019, 4, e00173-19. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Multi-Target Effects of ß-Caryophyllene and Carnosic Acid at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration: From Oxidative Stress to Microglia-Mediated Neuroinflammation. Antioxidants 2022, 11, 1199. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A.; Vernazza, S.; Tirendi, S.; Scarfì, S.; Gandolfi, S.; Bassi, A.M. Can Polyphenols in Eye Drops Be Useful for Trabecular Protection from Oxidative Damage? J. Clin. Med. 2020, 9, 3584. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Nisar, M.F.; Hu, X.; Chang, J.; Wang, Y.; Wu, Y.; Liu, Z.; Cai, Y.; Jia, J.; Xiao, Y.; et al. Pyrroloquinoline Quinone (PQQ): Its impact on human health and potential benefits: PQQ: Human health impacts and benefits. Curr. Res. food Sci. 2024, 9, 100889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, M.; Lu, J.; Wang, J.; Zhang, M.; Panichayupakaranant, P.; Chen, H. Insights into the Sources, Structure, and Action Mechanisms of Quinones on Diabetes: A Review. Molecules 2025, 30, 665. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA); Turck, D.; Bresson, J.-L.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.I.; Mangelsdorf, I.; McArdle, M.J.; et al. Safety of pyrroloquinoline quinone disodium salt as a novel food pursuant to Regulation (EC) No 258/97. EFSA J. Eur. Food Saf. Auth. 2017, 15, e05058. [Google Scholar] [CrossRef]
- Kumazawa, T.; Seno, H.; Urakami, T.; Matsumoto, T.; Suzuki, O. Trace levels of pyrroloquinoline quinone in human and rat samples detected by gas chromatography/mass spectrometry. Biochim. Biophys. Acta 1992, 1156, 62–66. [Google Scholar] [CrossRef]
- Harris, C.B.; Chowanadisai, W.; Mishchuk, D.O.; Satre, M.A.; Slupsky, C.M.; Rucker, R.B. Dietary pyrroloquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J. Nutr. Biochem. 2013, 24, 2076–2084. [Google Scholar] [CrossRef]
- Fukuda, M.; El-Maghrabey, M.H.; Kishikawa, N.; Ikemoto, K.; Kuroda, N. Ultrasensitive determination of pyrroloquinoline quinone in human plasma by HPLC with chemiluminescence detection using the redox cycle of quinone. J. Pharm. Biomed. Anal. 2017, 145, 814–820. [Google Scholar] [CrossRef]
- Xu, T.; Yang, X.; Wu, C.; Qiu, J.; Fang, Q.; Wang, L.; Yu, S.; Sun, H. Pyrroloquinoline quinone attenuates cachexia-induced muscle atrophy via suppression of reactive oxygen species. J. Thorac. Dis. 2018, 10, 2752–2759. [Google Scholar] [CrossRef]
- Zhu, B.-Q.; Zhou, H.-Z.; Teerlink, J.R.; Karliner, J.S. Pyrroloquinoline quinone (PQQ) decreases myocardial infarct size and improves cardiac function in rat models of ischemia and ischemia/reperfusion. Cardiovasc. Drugs Ther. 2004, 18, 421–431. [Google Scholar] [CrossRef]
- Pu, Z.; Ge, F.; Zhou, Y.; Liu, A.; Yang, C. Pyrroloquinoline quinone protects against murine hepatitis virus strain 3-induced fulminant hepatitis by inhibiting the Keap1/Nrf2 signaling. Cytotechnology 2024, 76, 441–452. [Google Scholar] [CrossRef]
- Xu, X.; Chen, C.; Lu, W.-J.; Su, Y.-L.; Shi, J.-Y.; Liu, Y.-C.; Wang, L.; Xiao, C.-X.; Wu, X.; Lu, Q. Pyrroloquinoline quinone can prevent chronic heart failure by regulating mitochondrial function. Cardiovasc. Diagn. Ther. 2020, 10, 453–469. [Google Scholar] [CrossRef]
- Yang, L.; Rong, Z.; Zeng, M.; Cao, Y.; Gong, X.; Lin, L.; Chen, Y.; Cao, W.; Zhu, L.; Dong, W. Pyrroloquinoline quinone protects nucleus pulposus cells from hydrogen peroxide-induced apoptosis by inhibiting the mitochondria-mediated pathway. Eur. Spine J. 2015, 24, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Meruvu, S.; Bedi, Y.S.; Chau, J.; Arguelles, A.; Rucker, R.; Choudhury, M. Pyrroloquinoline quinone increases the expression and activity of Sirt1 and -3 genes in HepG2 cells. Nutr. Res. 2015, 35, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Saihara, K.; Kamikubo, R.; Ikemoto, K.; Uchida, K.; Akagawa, M. Pyrroloquinoline Quinone, a Redox-Active o-Quinone, Stimulates Mitochondrial Biogenesis by Activating the SIRT1/PGC-1α Signaling Pathway. Biochemistry 2017, 56, 6615–6625. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J.; Guo, H.; Lu, J.-L.; Zhou, J.; Guo, X.-Y.; Shi, Y.; Zhang, Y.; Yu, S.; Zhang, Q.; et al. Pyrroloquinoline quinone promotes mitochondrial biogenesis in rotenone-induced Parkinson’s disease model via AMPK activation. Acta Pharmacol. Sin. 2021, 42, 665–678. [Google Scholar] [CrossRef]
- Gao, Y.; Kamogashira, T.; Fujimoto, C.; Iwasaki, S.; Yamasoba, T. Pyrroloquinoline quinone (PQQ) protects mitochondrial function of HEI-OC1 cells under premature senescence. Npj Aging 2022, 8, 3. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Yang, H.; Tan, J.; Zhang, J.; Zi, D. Pyrroloquinoline quinone promotes human mesenchymal stem cell-derived mitochondria to improve premature ovarian insufficiency in mice through the SIRT1/ATM/p53 pathway. Stem Cell Res. Ther. 2024, 15, 97. [Google Scholar] [CrossRef]
- Chowanadisai, W.; Bauerly, K.A.; Tchaparian, E.; Wong, A.; Cortopassi, G.A.; Rucker, R.B. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J. Biol. Chem. 2010, 285, 142–152. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Wang, Y.; Zhao, K.; Chi, Y.; Wang, B. Pyrroloquinoline quinine protects HK-2 cells against high glucose-induced oxidative stress and apoptosis through Sirt3 and PI3K/Akt/FoxO3a signaling pathway. Biochem. Biophys. Res. Commun. 2019, 508, 398–404. [Google Scholar] [CrossRef]
- Canovai, A.; Tribble, J.R.; Jöe, M.; Westerlund, D.Y.; Amato, R.; Trounce, I.A.; Dal Monte, M.; Williams, P.A. Pyrroloquinoline quinone drives ATP synthesis in vitro and in vivo and provides retinal ganglion cell neuroprotection. Acta Neuropathol. Commun. 2023, 11, 146. [Google Scholar] [CrossRef]
- Rossi, G.C.M.; Rinaldi, M.; Matarazzo, F.; Strianese, D.; Campagna, G.; La Ragione, M.; Esposito Veneruso, P.; Scapagnini, G.; Costagliola, C. Randomized, Cross over, Multicenter, Single-Blind Study Comparing Citicoline 500 mg/Homotaurine 50 mg/Vitamin B3 54 mg/Pyrroloquinoline Quinone 5 mg (Neuprozin Mito®) and Citicoline 800 mg (Cebrolux®) on Pattern Electroretinogram (PERG) and Quality of Lif. J. Clin. Med. 2025, 14, 3774. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Abdulkader, F. How and when to measure mitochondrial inner membrane potentials. Biophys. J. 2024, 123, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Lionnard, L.; Singh, I.; Karim, M.A.; Chajra, H.; Frechet, M.; Kissa, K.; Racine, V.; Ammanamanchi, A.; McCarty, P.J.; et al. Mitochondrial morphology is associated with respiratory chain uncoupling in autism spectrum disorder. Transl. Psychiatry 2021, 11, 527. [Google Scholar] [CrossRef] [PubMed]
- Lhuissier, C.; Desquiret-Dumas, V.; Girona, A.; Alban, J.; Faure, J.; Cassereau, J.; Codron, P.; Lenaers, G.; Baris, O.R.; Gueguen, N.; et al. Mitochondrial F0F1-ATP synthase governs the induction of mitochondrial fission. iScience 2024, 27, 109808. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef]
- Wang, M.; Zheng, Y. Oxidative stress and antioxidants in the trabecular meshwork. PeerJ 2019, 7, e8121. [Google Scholar] [CrossRef]
- Deng, X.; Zhu, M.; Liu, Y.; Zhang, N.; Zhang, P.; Zeng, W.; Ke, M. Suppression of CDK1/Drp1-Mediated Mitochondrial Fission Attenuates Dexamethasone-Induced Extracellular Matrix Deposition in the Trabecular Meshwork. Antioxid. Redox Signal. 2025, 42, 249–264. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, N.; Hu, Y.; Deng, X.; Zhu, M.; Lai, C.; Zeng, W.; Ke, M. Role of PI3K/AKT/MAOA in glucocorticoid-induced oxidative stress and associated premature senescence of the trabecular meshwork. Aging Cell 2025, 24, e14452. [Google Scholar] [CrossRef]
- Liu, Y.; Bu, Q.; Hu, D.; Chen, C.; Zhu, J.; Zhou, Q.; Li, Z.; Pan, X. NAD+ supplementation improves mitochondrial functions and normalizes glaucomatous trabecular meshwork features. Exp. Cell Res. 2024, 440, 114137. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, P.; Deng, X.; Zhu, M.; Hu, Y.; Ji, D.; Li, L.; Liu, Y.; Zeng, W.; Ke, M. Protective Effect of Nicotinamide Riboside on Glucocorticoid-Induced Glaucoma: Mitigating Mitochondrial Damage and Extracellular Matrix Deposition. Investg. Ophthalmol. Vis. Sci. 2024, 65, 1. [Google Scholar] [CrossRef]
- Cordell, G.A.; Daley, S.-K. Pyrroloquinoline Quinone Chemistry, Biology, and Biosynthesis. Chem. Res. Toxicol. 2022, 35, 355–377. [Google Scholar] [CrossRef]
- He, K.; Nukada, H.; Urakami, T.; Murphy, M.P. Antioxidant and pro-oxidant properties of pyrroloquinoline quinone (PQQ): Implications for its function in biological systems. Biochem. Pharmacol. 2003, 65, 67–74. [Google Scholar] [CrossRef]
- Peng, Y.; Xu, D.; Mao, S.; Zhou, X. Neurotoxicity and apoptosis induced by pyrroloquinoline quinone and its ester derivative on primary cortical neurons. Neurotoxicology 2020, 78, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shen, M.; Ding, M.; Shen, D.; Ding, F. The neuroprotective action of pyrroloquinoline quinone against glutamate-induced apoptosis in hippocampal neurons is mediated through the activation of PI3K/Akt pathway. Toxicol. Appl. Pharmacol. 2011, 252, 62–72. [Google Scholar] [CrossRef]
- Nunome, K.; Miyazaki, S.; Nakano, M.; Iguchi-Ariga, S.; Ariga, H. Pyrroloquinoline quinone prevents oxidative stress-induced neuronal death probably through changes in oxidative status of DJ-1. Biol. Pharm. Bull. 2008, 31, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xu, Y.; Sun, J.; Li, X.; Wang, L.; Jin, L. Protection of pyrroloquinoline quinone against methylmercury-induced neurotoxicity via reducing oxidative stress. Free Radic. Res. 2009, 43, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.; Wang, L.; Jin, J.; Wang, X.; Zhu, B.; Chen, H.; Cheng, Y. Pyrroloquinoline Quinone Induces Cancer Cell Apoptosis via Mitochondrial-Dependent Pathway and Down-Regulating Cellular Bcl-2 Protein Expression. J. Cancer 2014, 5, 609–624. [Google Scholar] [CrossRef]
- Vernazza, S.; Passalacqua, M.; Tirendi, S.; Marengo, B.; Domenicotti, C.; Sbardella, D.; Oddone, F.; Bassi, A.M. Citicoline Eye Drops Protect Trabecular Meshwork Cells from Oxidative Stress Injury in a 3D In Vitro Glaucoma Model. Int. J. Mol. Sci. 2022, 23, 11375. [Google Scholar] [CrossRef]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Sabouny, R.; Shutt, T.E. Reciprocal Regulation of Mitochondrial Fission and Fusion. Trends Biochem. Sci. 2020, 45, 564–577. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Hou, M.; Xia, X.; Liu, J.; Xu, Y.; Shi, Q.; Zhang, Z.; Wang, L.; Shen, Y.; et al. Reprogramming of Mitochondrial Respiratory Chain Complex by Targeting SIRT3-COX4I2 Axis Attenuates Osteoarthritis Progression. Adv. Sci. Ger. 2023, 10, e2206144. [Google Scholar] [CrossRef]
- Minnelli, C.; Piva, F.; Cecati, M.; Armeni, T.; Mobbili, G.; Galeazzi, R.; Melecchi, A.; Cristaldi, M.; Corsaro, R.; Rusciano, D. Meldonium Inhibits Cell Motility and Wound-Healing in Trabecular Meshwork Cells and Scleral Fibroblasts: Possible Applications in Glaucoma. Pharmaceuticals 2023, 16, 594. [Google Scholar] [CrossRef]
- Ragusa, A.; Cristiano, L.; Di Vinci, P.; Familiari, G.; Nottola, S.A.; Macchiarelli, G.; Svelato, A.; De Luca, C.; Rinaldo, D.; Neri, I.; et al. Artificial plasticenta: How polystyrene nanoplastics affect in-vitro cultured human trophoblast cells. Front. Cell Dev. Biol. 2025, 13, 1539600. [Google Scholar] [CrossRef] [PubMed]
- Faitg, J.; Davey, T.; Turnbull, D.M.; White, K.; Vincent, A.E. Mitochondrial morphology and function: Two for the price of one! J. Microsc. 2020, 278, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Katti, P.; Biete, M.; Mungai, M.; AshShareef, S.; Neikirk, K.; Garza Lopez, E.; Vue, Z.; Christensen, T.A.; Beasley, H.K.; et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells 2021, 10, 2177. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Bakare, A.B.; Daniel, J.; Stabach, J.; Rojas, A.; Bell, A.; Henry, B.; Iyer, S. Quantifying Mitochondrial Dynamics in Patient Fibroblasts with Multiple Developmental Defects and Mitochondrial Disorders. Int. J. Mol. Sci. 2021, 22, 6263. [Google Scholar] [CrossRef]
- Kritskaya, K.A.; Fedotova, E.I.; Berezhnov, A.V. Impaired Mitochondrial Network Morphology and Reactive Oxygen Species Production in Fibroblasts from Parkinson’s Disease Patients. Biomedicines 2024, 12, 282. [Google Scholar] [CrossRef]


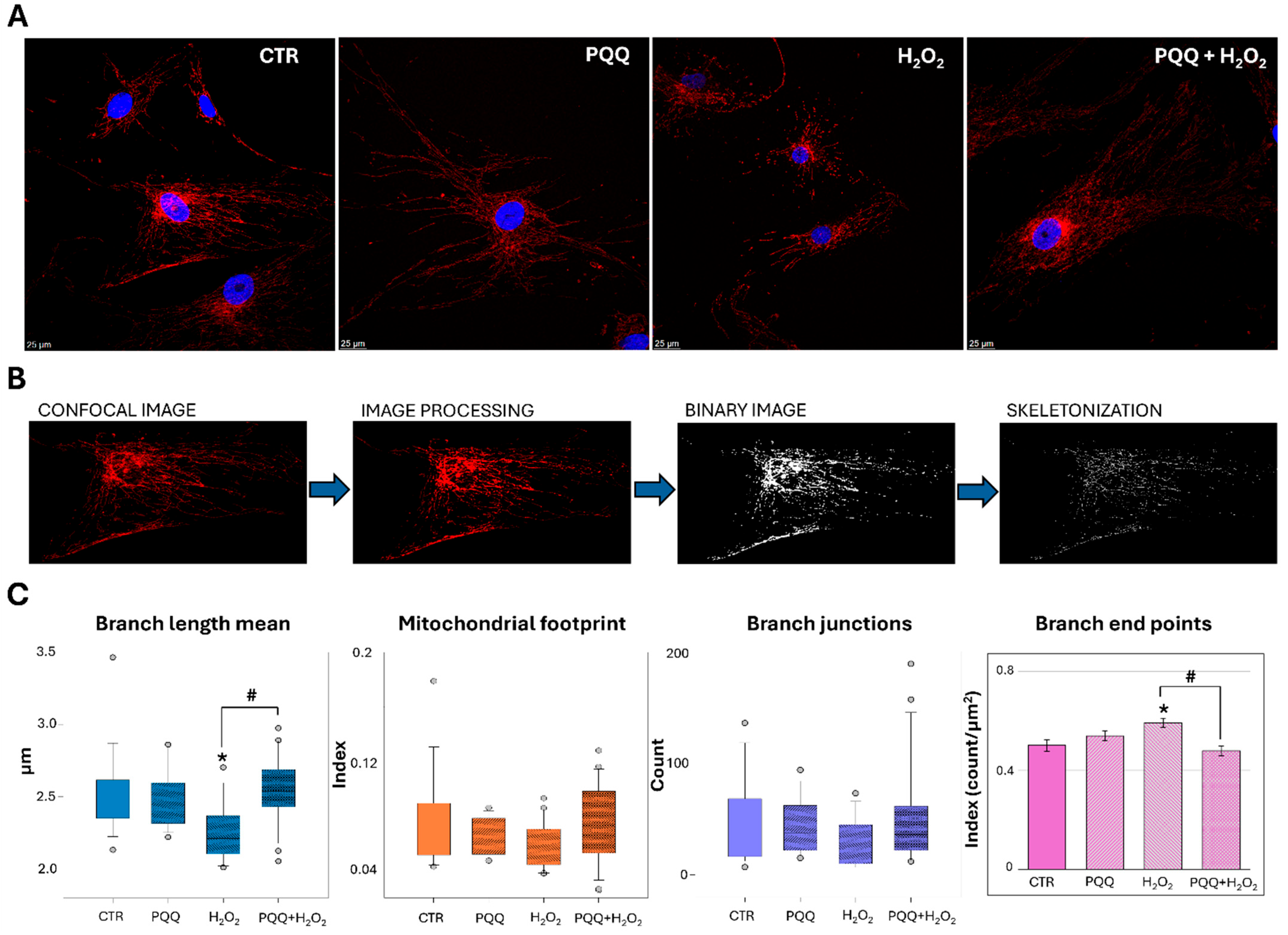
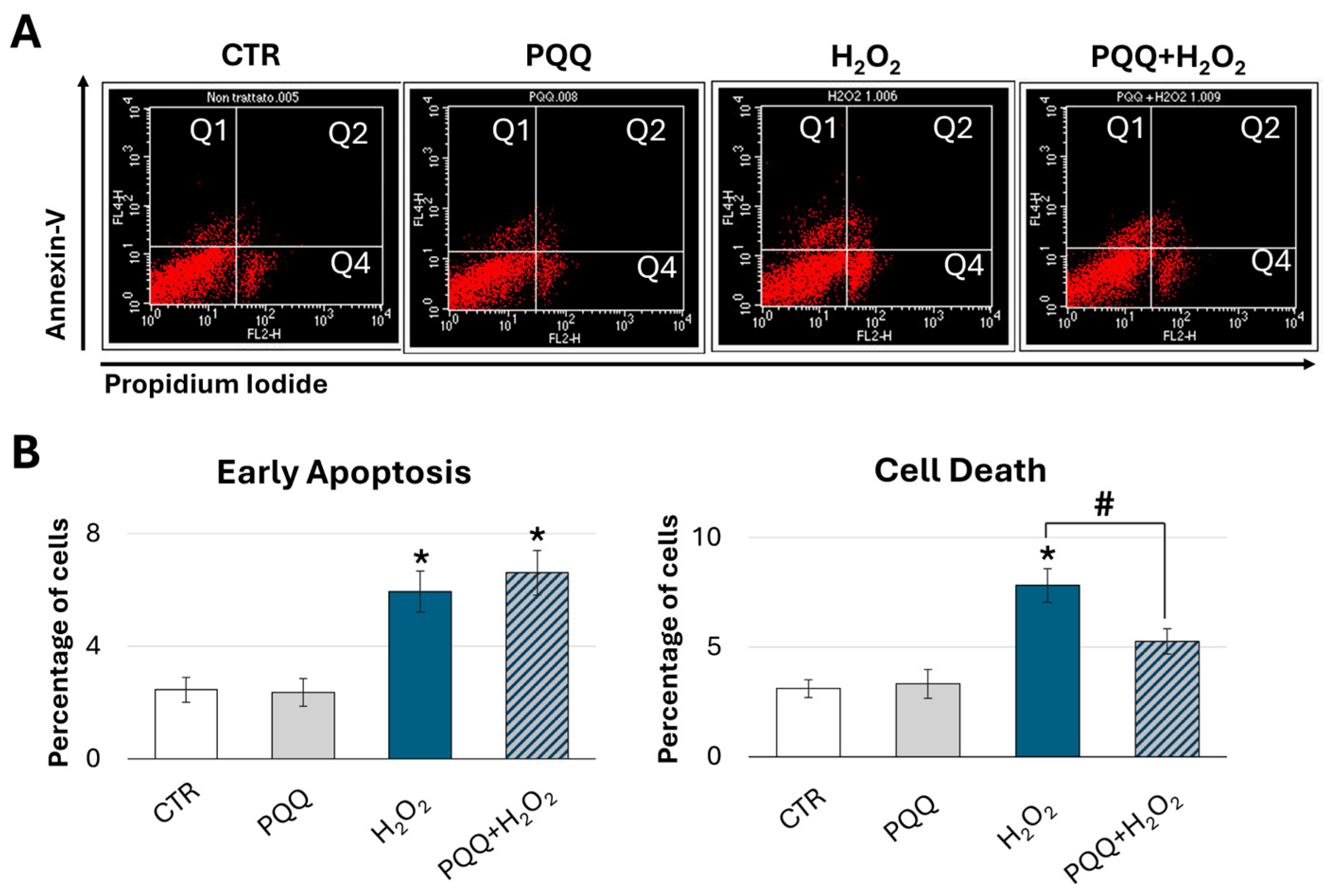

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTR | PQQ | H2O2 | PQQ + H2O2 | |
|---|---|---|---|---|
| Area (µm2) | 0.081 [0.054–0.110] | 0.088 [0.060–0.125] | 0.042 * [0.030–0.066] | 0.072 # [0.044–0.115] |
| Perimeter (µm) | 1.125 [0.913–1.449] | 1.212 [0.954–1.487] | 0.786 * [0.665–1.006] | 1.081 # [0.789–1.491] |
| Circularity | 0.801 [0.657–0.855] | 0.785 [0.652–0.855] | 0.855 * [0.798–0.888] | 0.800 # [0.647–0.871] |
| Aspect ratio | 1.733 [1.416–2.390] | 1.798 [1.430–2.583] | 1.392 * [1.194–1.765] | 1.678 # [1.321–2.528] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petricca, S.; Matrone, A.; Capece, D.; Flati, I.; Flati, V.; Ricevuto, E.; Celenza, G.; Franceschini, N.; Mastrangelo, M.; Pellegrini, C.; et al. Pyrroloquinoline Quinone (PQQ) Attenuates Hydrogen Peroxide-Induced Injury Through the Enhancement of Mitochondrial Function in Human Trabecular Meshwork Cells. Int. J. Mol. Sci. 2025, 26, 6938. https://doi.org/10.3390/ijms26146938
Petricca S, Matrone A, Capece D, Flati I, Flati V, Ricevuto E, Celenza G, Franceschini N, Mastrangelo M, Pellegrini C, et al. Pyrroloquinoline Quinone (PQQ) Attenuates Hydrogen Peroxide-Induced Injury Through the Enhancement of Mitochondrial Function in Human Trabecular Meshwork Cells. International Journal of Molecular Sciences. 2025; 26(14):6938. https://doi.org/10.3390/ijms26146938
Chicago/Turabian StylePetricca, Sabrina, Antonio Matrone, Daria Capece, Irene Flati, Vincenzo Flati, Enrico Ricevuto, Giuseppe Celenza, Nicola Franceschini, Mirco Mastrangelo, Cristina Pellegrini, and et al. 2025. "Pyrroloquinoline Quinone (PQQ) Attenuates Hydrogen Peroxide-Induced Injury Through the Enhancement of Mitochondrial Function in Human Trabecular Meshwork Cells" International Journal of Molecular Sciences 26, no. 14: 6938. https://doi.org/10.3390/ijms26146938
APA StylePetricca, S., Matrone, A., Capece, D., Flati, I., Flati, V., Ricevuto, E., Celenza, G., Franceschini, N., Mastrangelo, M., Pellegrini, C., Cristiano, L., Familiari, G., Cinque, B., Di Emidio, G., Tatone, C., & Iorio, R. (2025). Pyrroloquinoline Quinone (PQQ) Attenuates Hydrogen Peroxide-Induced Injury Through the Enhancement of Mitochondrial Function in Human Trabecular Meshwork Cells. International Journal of Molecular Sciences, 26(14), 6938. https://doi.org/10.3390/ijms26146938