
Protecting the Brain: Novel Strategies for Preventing Breast Cancer Brain Metastases through Selective Estrogen Receptor β Agonists and In Vitro Blood–Brain Barrier Models


 , and
, and 
Abstract

1. Introduction
2. Results
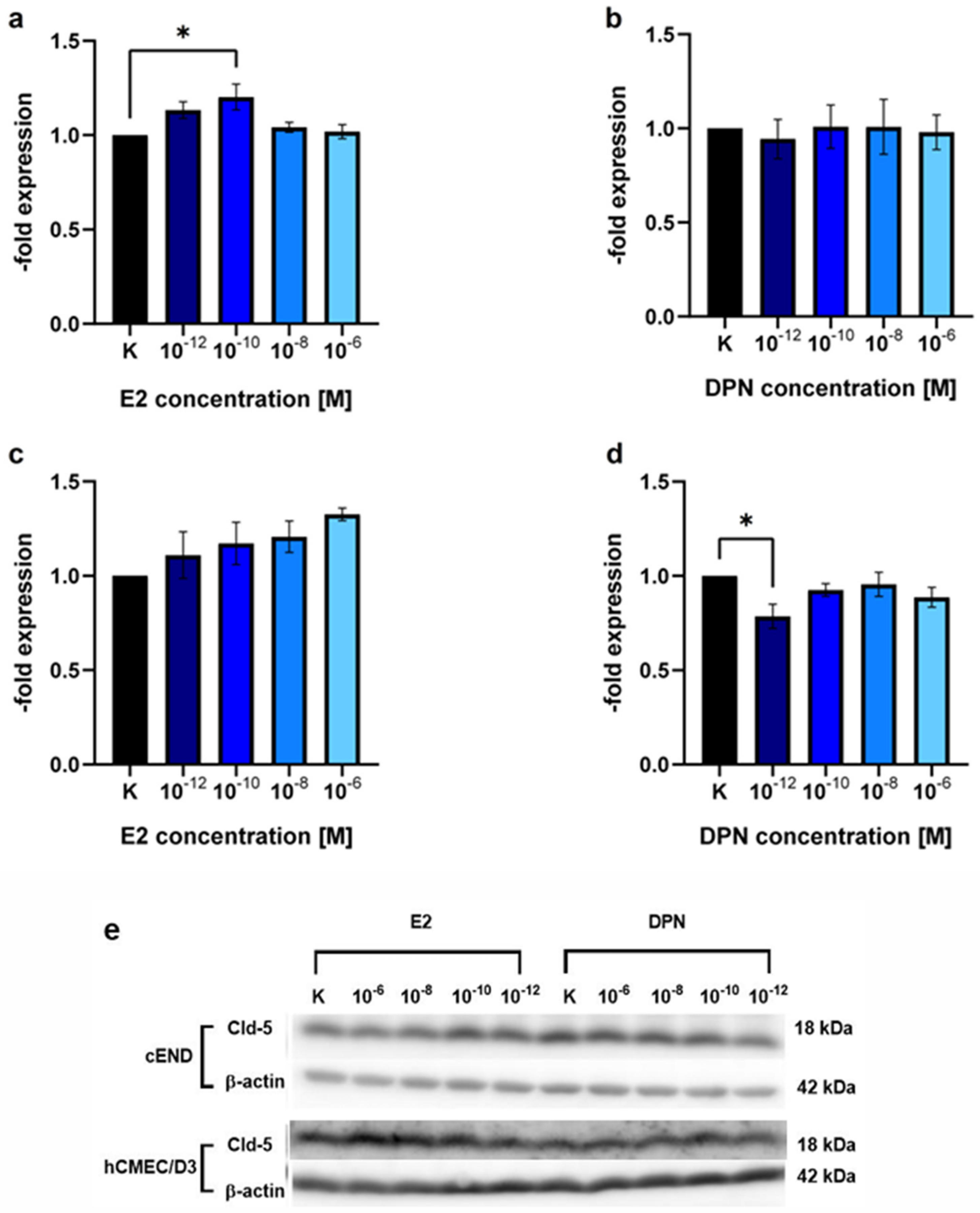
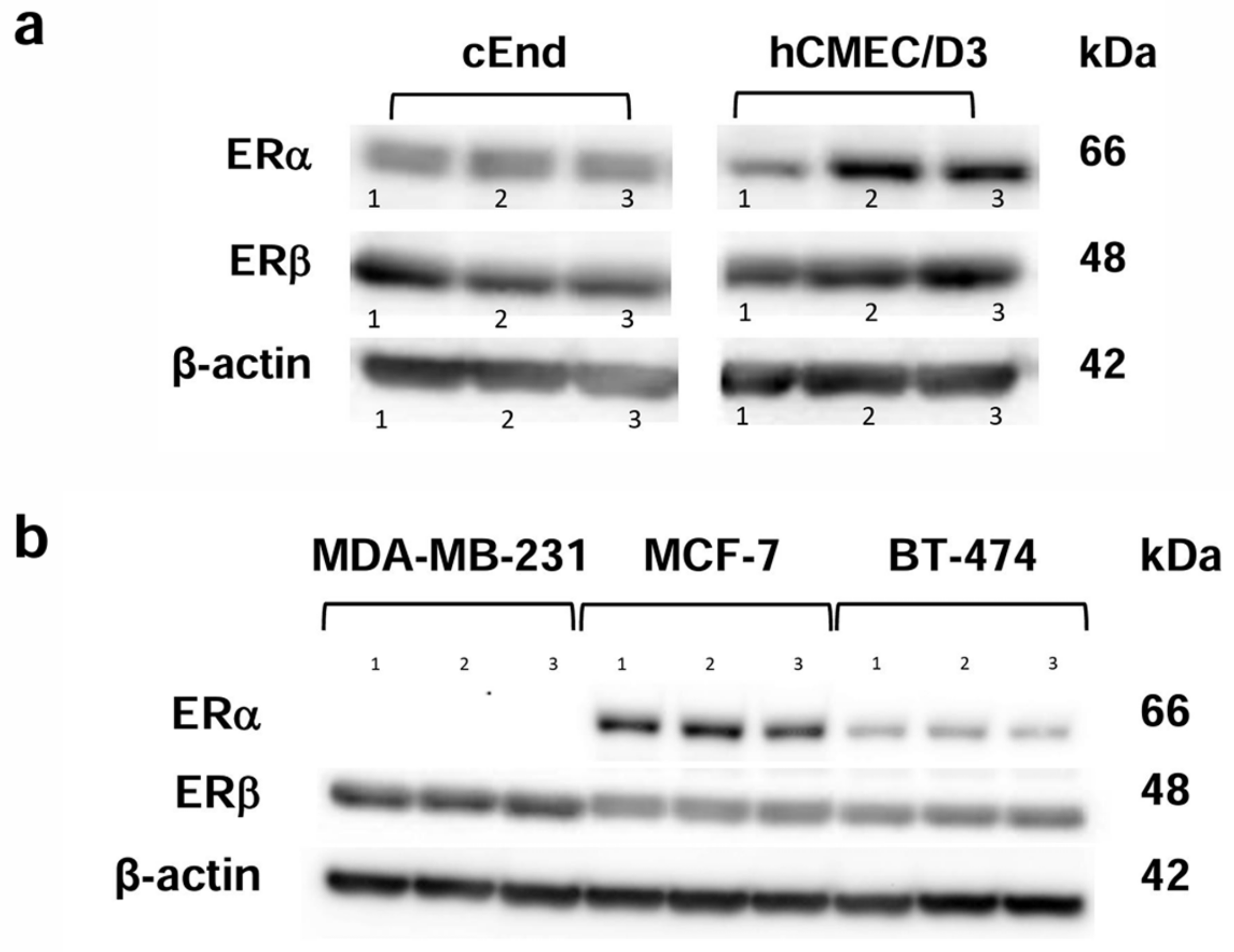
2.1. Western Blot
2.1.1. Characterization of the ER Status of the Cell Lines
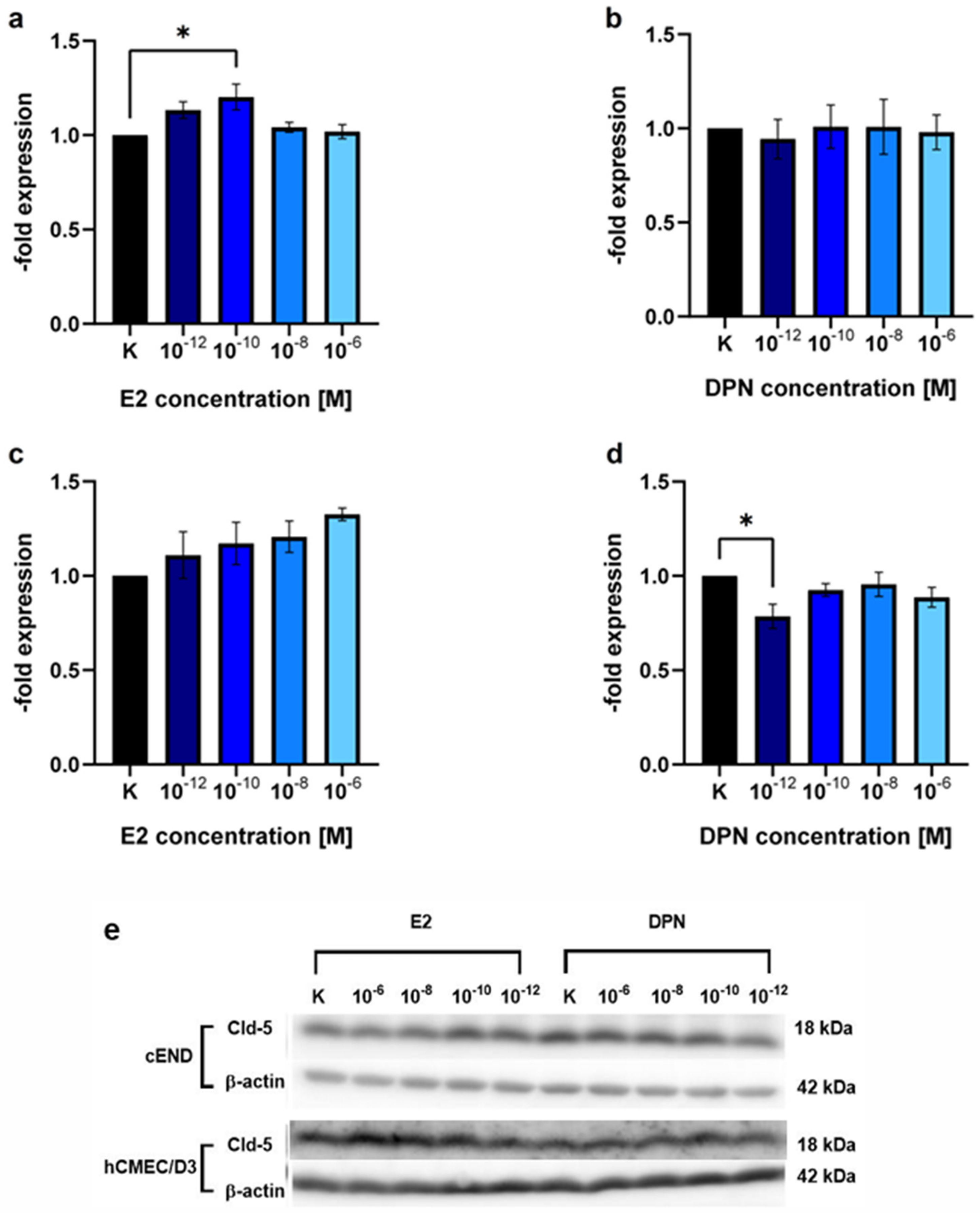
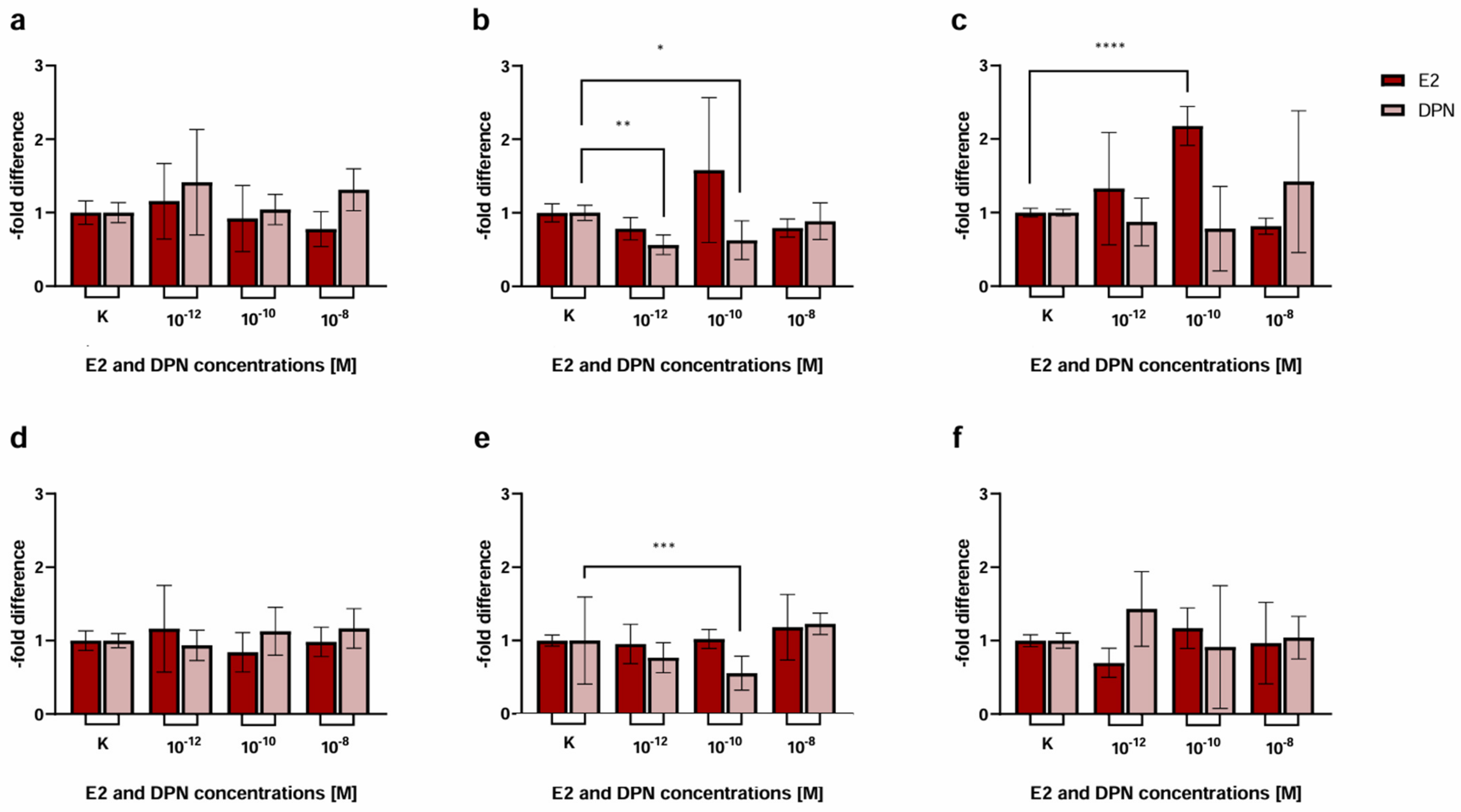
2.1.2. Concentration-Dependent Induction of Cld-5 Expression by E2 and DPN
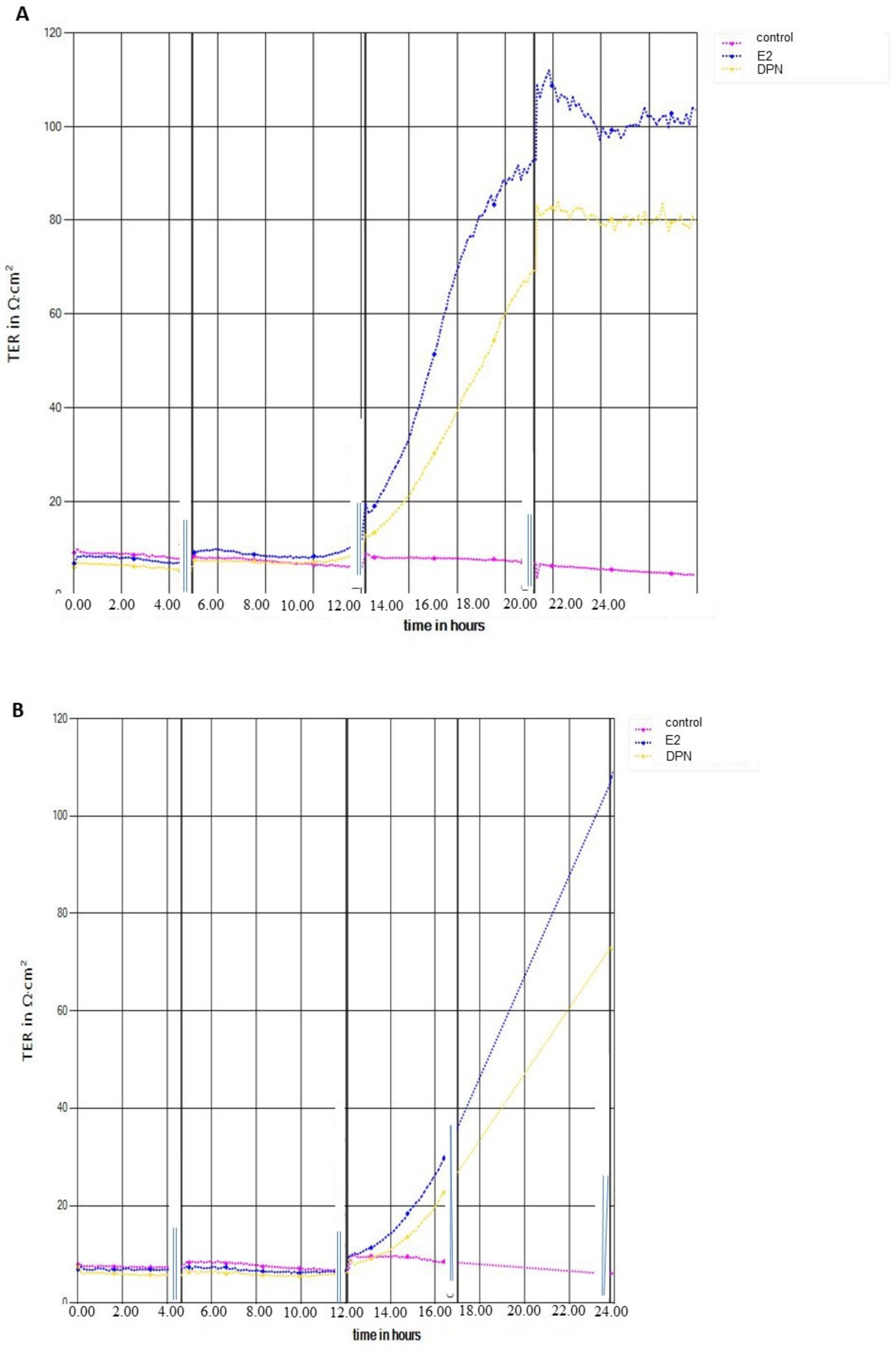
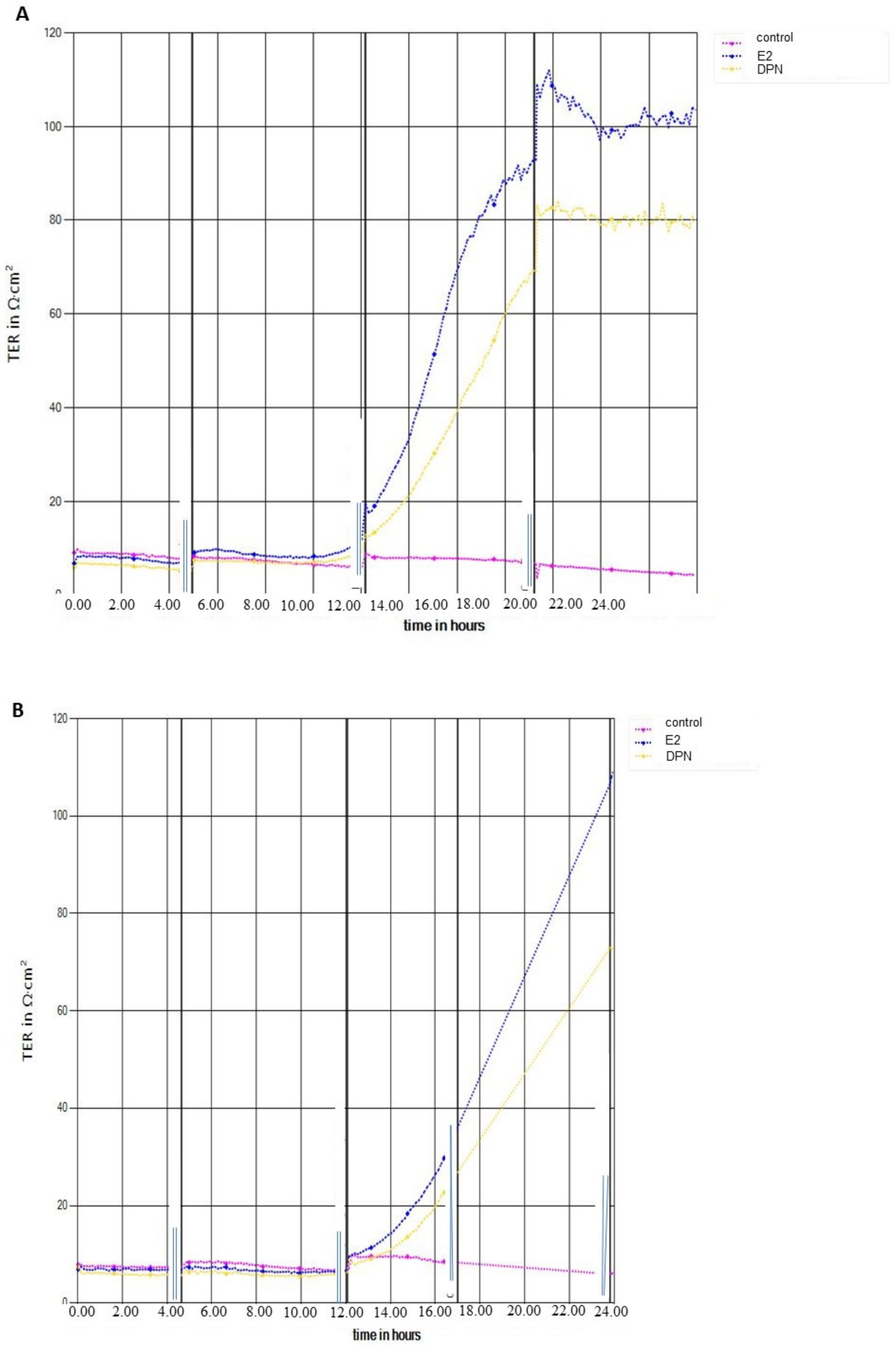
2.2. Transendothelial Electrical Resistance Measurement (TEER)
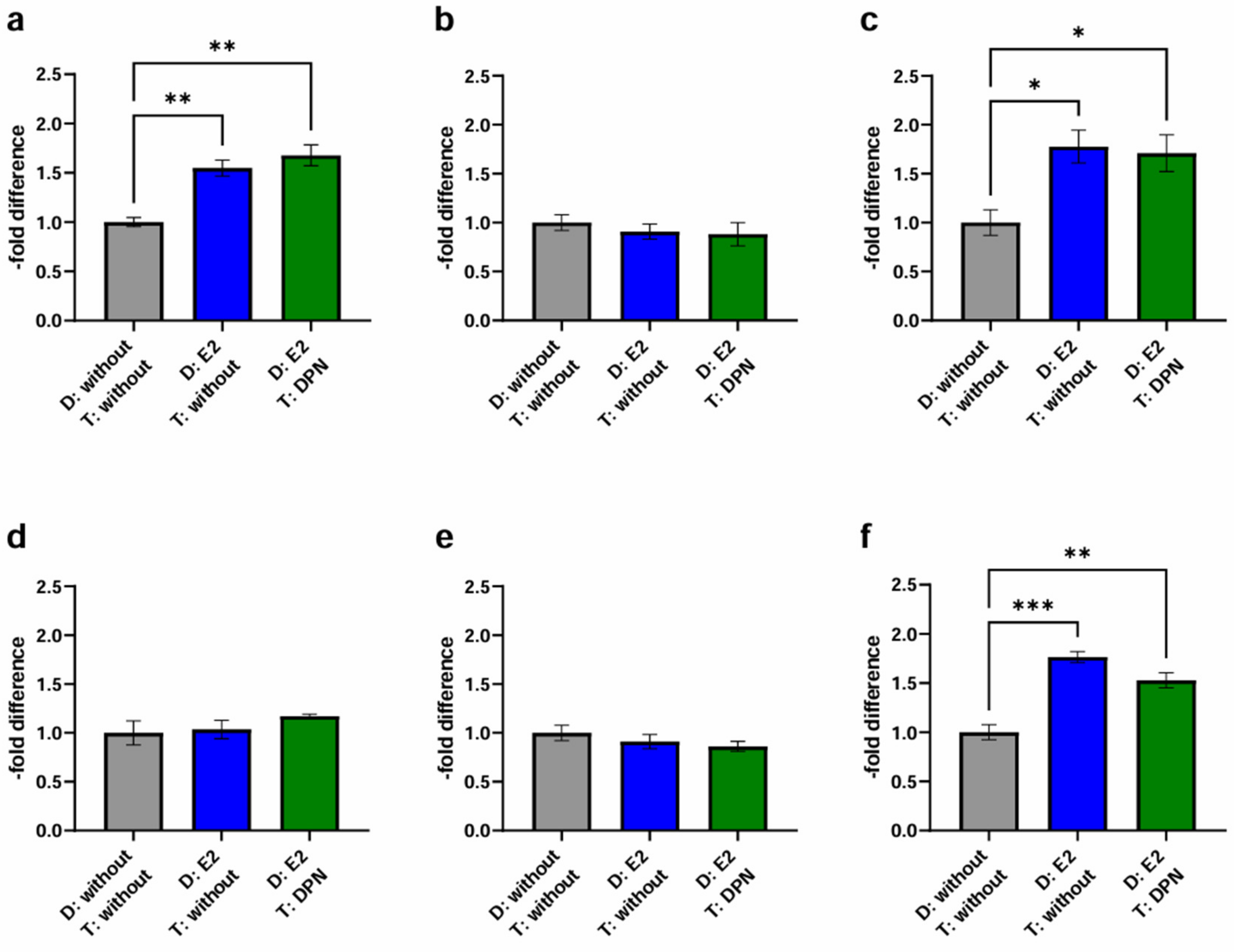
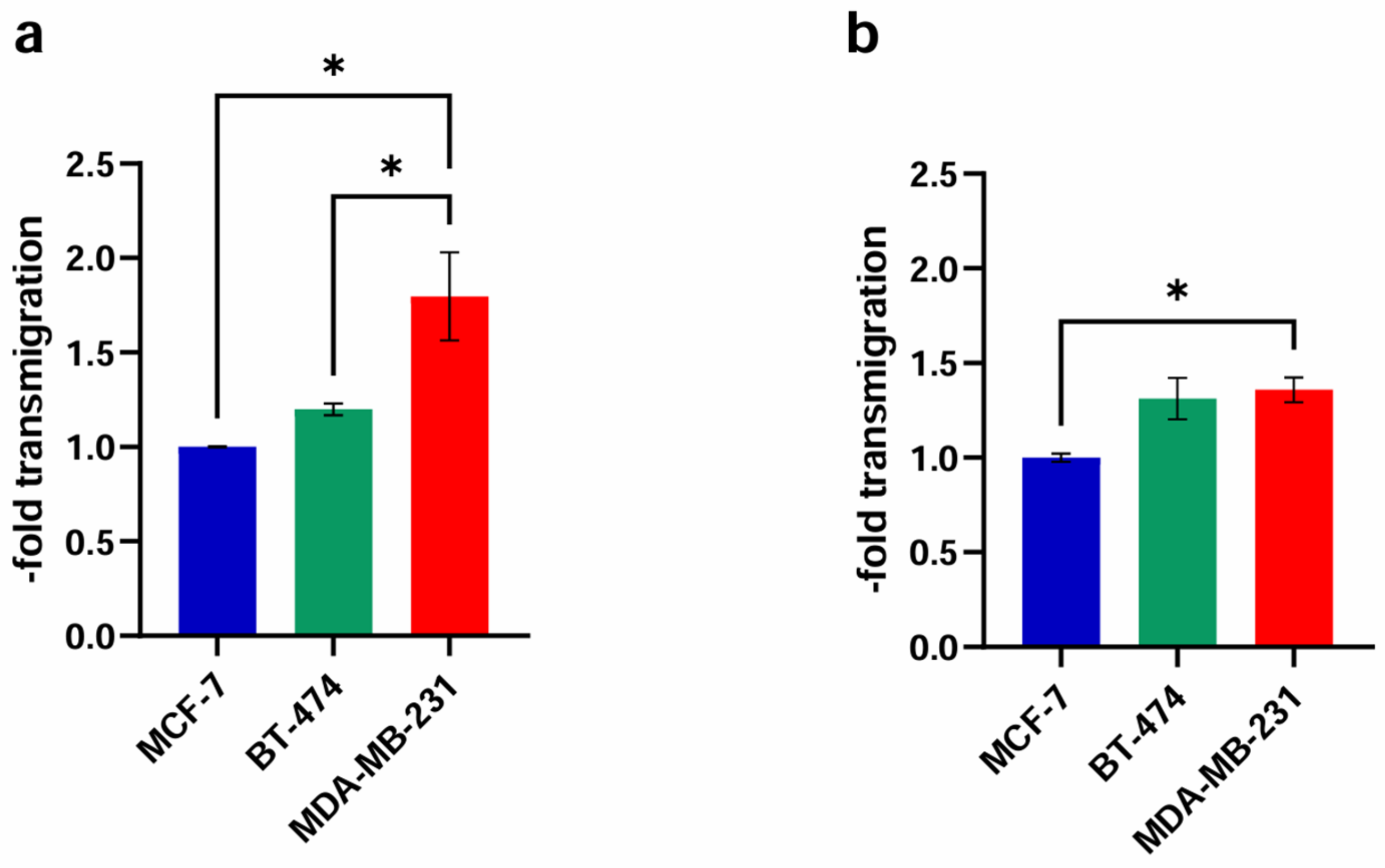
2.3. Transendothelial Migration
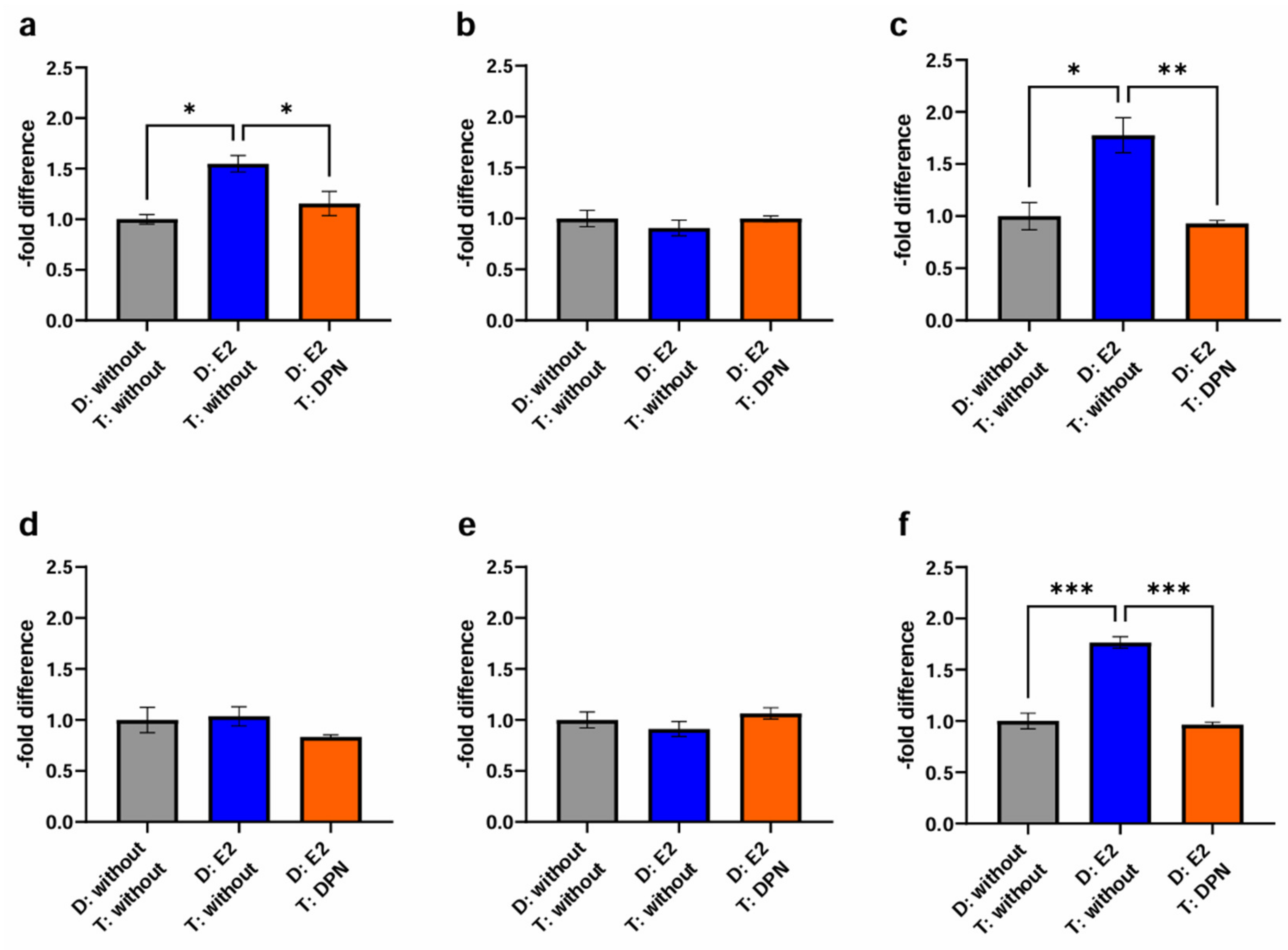
2.3.1. Basic Experiment
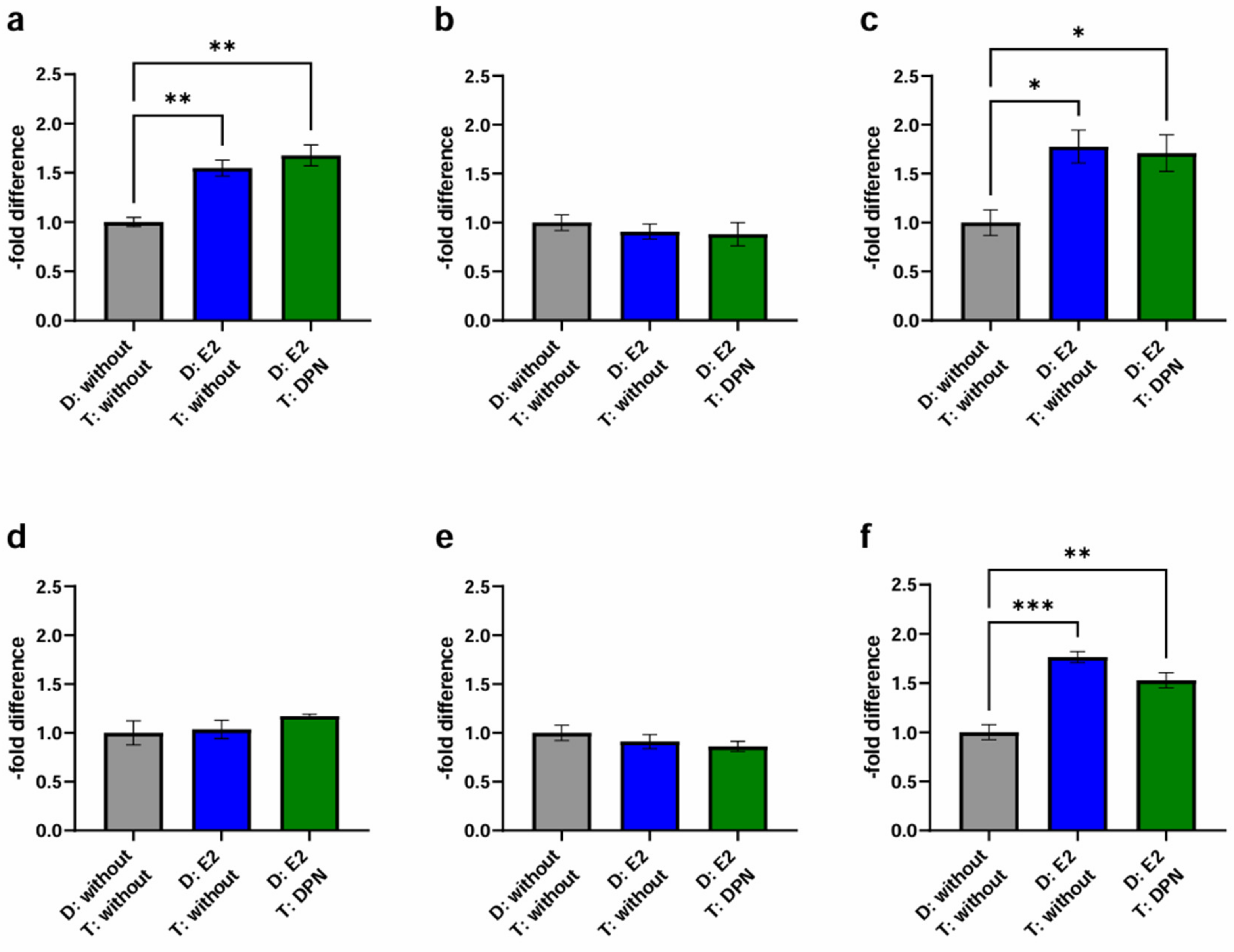
2.3.2. Physiological Stimulation Experiment
3. Discussion
3.1. ER Expression Status
3.2. Modulation of TJ Protein Claudin-5 by ER Agonists
3.3. Effect of ER Ligands on TEER in cEND and hCMEC/D3 Cells
3.4. Transmigration Experiment—Effect of Different ER Agonist Administrations
3.4.1. Basic Experiment
3.4.2. Physiological Stimulation Experiment
4. Materials and Methods
4.1. Chemicals
4.2. Cell Cultures
4.3. Transendothelial Electrical Resistance Measurement
4.4. Western Blot
4.5. Transendothelial Migration
4.5.1. Basic Experiment
4.5.2. Physiological Stimulation Experiment
4.6. Analysis and Statistics
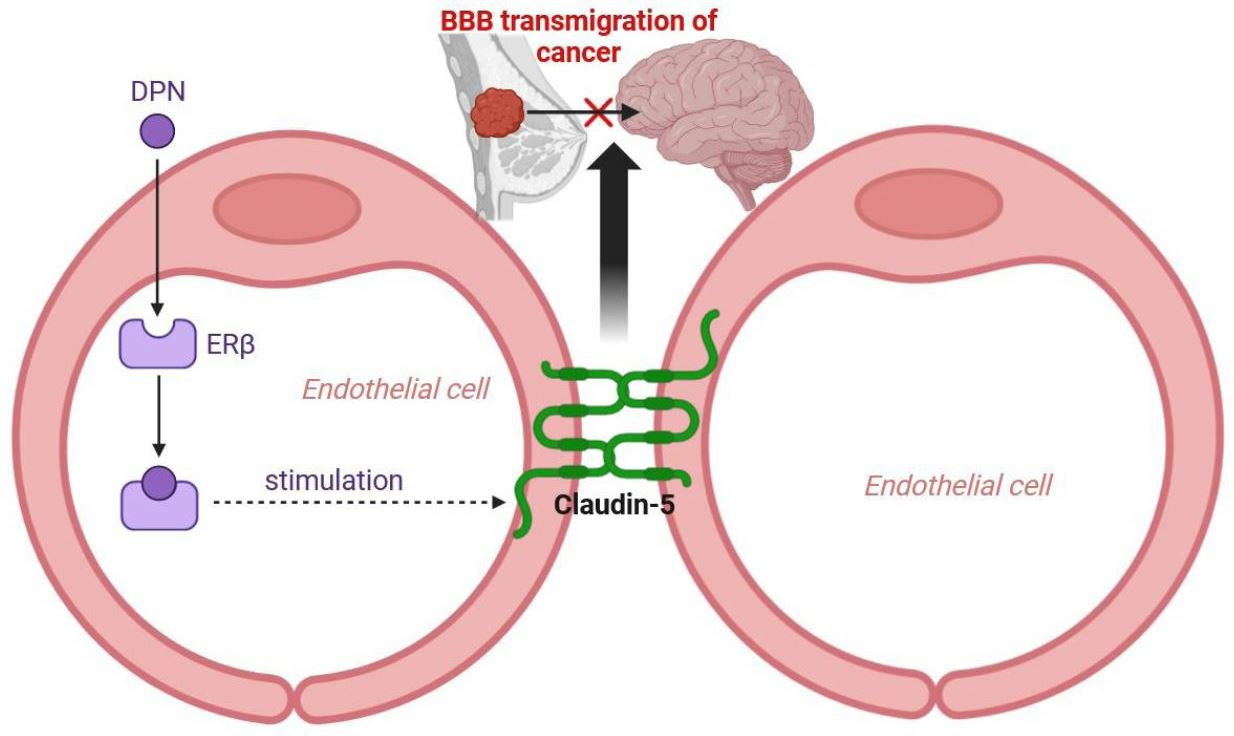
5. Conclusions
Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pedrosa, R.; Mustafa, D.A.; Soffietti, R.; Kros, J.M. Breast cancer brain metastasis: Molecular mechanisms and directions for treatment. Neuro Oncol. 2018, 20, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rüger, R. Dissection of the Process of Brain Metastasis Reveals Targets and Mechanisms for Molecular-based Intervention. Cancer Genom. Proteom. 2016, 13, 245–258. [Google Scholar]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 2011, 71, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Molnár, J.; Fazakas, C.; Haskó, J.; Krizbai, I.A. Role of the Blood-Brain Barrier in the Formation of Brain Metastases. Int. J. Mol. Sci. 2013, 14, 1383–1411. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V. Mechanisms for transcellular diapedesis: Probing and pathfinding by ‘invadosome-like protrusions’. J. Cell Sci. 2009, 122, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Kanojia, D.; Rashidi, A.; Ulasov, I.; Lesniak, M.S. Genes that Mediate Metastasis across the Blood-Brain Barrier. Trends Cancer 2020, 6, 660–676. [Google Scholar] [CrossRef] [PubMed]
- Tzanakakis, G.; Kavasi, R.M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Hosonaga, M.; Saya, H.; Arima, Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer Metastasis Rev. 2020, 39, 711–720. [Google Scholar] [CrossRef]
- Izraely, S.; Witz, I.P. Site-specific metastasis: A cooperation between cancer cells and the metastatic microenvironment. Int. J. Cancer 2021, 148, 1308–1322. [Google Scholar] [CrossRef]
- Warner, M.; Huang, B.; Gustafsson, J.A. Estrogen Receptor β as a Pharmaceutical Target. Trends Pharmacol. Sci. 2017, 38, 92–99. [Google Scholar] [CrossRef]
- Sandoval, K.E.; Witt, K.A. Age and 17β-estradiol effects on blood-brain barrier tight junction and estrogen receptor proteins in ovariectomized rats. Microvasc. Res. 2011, 81, 198–205. [Google Scholar] [CrossRef]
- Förster, C.; Mäkela, S.; Wärri, A.; Kietz, S.; Becker, D.; Hultenby, K.; Warner, M.; Gustafsson, J.A. Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 15578–15583. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Sharma, S.; Wu, K.; Tyagi, A.; Zhao, D.; Deshpande, R.P.; Watabe, K. Tamoxifen suppresses brain metastasis of estrogen receptor-deficient breast cancer by skewing microglia polarization and enhancing their immune functions. Breast Cancer Res. 2021, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Srivastava, A.; Pateriya, A.; Tomar, M.S.; Mishra, A.K.; Shrivastava, A. Metabolic reprograming confers tamoxifen resistance in breast cancer. Chem. Biol. Interact. 2021, 347, 109602. [Google Scholar] [CrossRef]
- Burek, M.; Arias-Loza, P.A.; Roewer, N.; Förster, C.Y. Claudin-5 as a novel estrogen target in vascular endothelium. Arter. Thromb. Vasc. Biol. 2010, 30, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Burek, M.; Steinberg, K.; Förster, C.Y. Mechanisms of transcriptional activation of the mouse claudin-5 promoter by estrogen receptor alpha and beta. Mol. Cell. Endocrinol. 2014, 392, 144–151. [Google Scholar] [CrossRef]
- Contreras-Zárate, M.J.; Cittelly, D.M. Sex steroid hormone function in the brain niche: Implications for brain metastatic colonization and progression. Cancer Rep. 2022, 5, e1241. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Yuhanna, I.S.; Mineo, C.; Liu, P.; German, Z.; Sherman, T.S.; Mendelsohn, M.E.; Anderson, R.G.W.; Shaul, P.W. Estrogen Receptor α and Endothelial Nitric Oxide Synthase Are Organized Into a Functional Signaling Module in Caveolae. Circ. Res. 2000, 87, e44–e52. [Google Scholar] [CrossRef]
- Haynes, M.P.; Li, L.; Sinha, D.; Russell, K.S.; Hisamoto, K.; Baron, R.; Collinge, M.; Sessa, W.C.; Bender, J.R. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J. Biol. Chem. 2003, 278, 2118–2123. [Google Scholar] [CrossRef]
- Lei, S.; Fan, P.; Wang, M.; Zhang, C.; Jiang, Y.; Huang, S.; Fang, M.; He, Z.; Wu, A. Elevated estrogen receptor β expression in triple negative breast cancer cells is associated with sensitivity to doxorubicin by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2020, 20, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Förster, C.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Ittner, C.; Burek, M.; Stork, S.; Nagai, M.; Forster, C.Y. Increased Catecholamine Levels and Inflammatory Mediators Alter Barrier Properties of Brain Microvascular Endothelial Cells in vitro. Front. Cardiovasc. Med. 2020, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Blecharz, K.; Kahles, T.; Schwarz, T.; Kraft, P.; Gobel, K.; Meuth, S.G.; Burek, M.; Thum, T.; Stoll, G.; et al. Glucocorticoid insensitivity at the hypoxic blood-brain barrier can be reversed by inhibition of the proteasome. Stroke 2011, 42, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Smetak, M.; Bär, C.; Schlegel, N.; Thum, T.; Nagai, M.; Förster, C.Y. Blood biomarkers in Takotsubo syndrome and their role in diagnosis and management. Front. Cardiovasc. Med. 2022. submitted. [Google Scholar]
- Kuruca, S.E.; Karadenizli, S.; Akgun-Dar, K.; Kapucu, A.; Kaptan, Z.; Uzum, G. The effects of 17β-estradiol on blood brain barrier integrity in the absence of the estrogen receptor alpha; an in-vitro model. Acta Histochem. 2017, 119, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Segura-Bautista, D.; Olivares, A.; Casas-González, P.; Bonilla, E.; Salazar, Z.; Pérez-Solis, M.A. GPR30 expression and function in breast cancer cells are induced through a cis-acting element targeted by ETS factors. Oncol. Rep. 2020, 43, 1669–1682. [Google Scholar] [CrossRef]
- Al-Bader, M.; Ford, C.; Al-Ayadhy, B.; Francis, I. Analysis of estrogen receptor isoforms and variants in breast cancer cell lines. Exp. Ther. Med. 2011, 2, 537–544. [Google Scholar] [CrossRef]
- Liu, M.M.; Albanese, C.; Anderson, C.M.; Hilty, K.; Webb, P.; Uht, R.M.; Price, R.H., Jr.; Pestell, R.G.; Kushner, P.J. Opposing action of estrogen receptors alpha and beta on cyclin D1 gene expression. J. Biol. Chem. 2002, 277, 24353–24360. [Google Scholar] [CrossRef]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, N.S. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef]
- Harati, R.; Mohammad, M.G.; Tlili, A.; El-Awady, R.A.; Hamoudi, R. Loss of miR-101-3p Promotes Transmigration of Metastatic Breast Cancer Cells through the Brain Endothelium by Inducing COX-2/MMP1 Signaling. Pharmaceuticals 2020, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Molnár, J.; Fazakas, C.; Haskó, J.; Sipos, O.; Nagy, K.; Nyúl-Tóth, Á.; Farkas, A.E.; Végh, A.G.; Váró, G.; Galajda, P.; et al. Transmigration characteristics of breast cancer and melanoma cells through the brain endothelium: Role of Rac and PI3K. Cell Adh. Migr. 2016, 10, 269–281. [Google Scholar] [CrossRef]
- Harati, R.; Hafezi, S.; Mabondzo, A.; Tlili, A. Silencing miR-202-3p increases MMP-1 and promotes a brain invasive phenotype in metastatic breast cancer cells. PLoS ONE 2020, 15, e0239292. [Google Scholar] [CrossRef] [PubMed]
- Tourovskaia, A.; Fauver, M.; Kramer, G.; Simonson, S.; Neumann, T. Tissue-engineered microenvironment systems for modeling human vasculature. Exp. Biol. Med. 2014, 239, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Drolez, A.; Vandenhaute, E.; Julien, S.; Gosselet, F.; Burchell, J.; Cecchelli, R.; Delannoy, P.; Dehouck, M.P.; Mysiorek, C. Selection of a Relevant In Vitro Blood-Brain Barrier Model to Investigate Pro-Metastatic Features of Human Breast Cancer Cell Lines. PLoS ONE 2016, 11, e0151155. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, B.M. Quantification of Malignant Breast Cancer Cell MDA-MB-231 Transmigration Across Brain and Lung Microvascular Endothelium. Ann. Biomed. Eng. 2016, 44, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.G.; Iturriaga, M.P.; Cruz, P. Hormonal Carcinogenesis in Canine Mammary Cancer: Molecular Mechanisms of Estradiol Involved in Malignant Progression. Animals 2021, 11, 608. [Google Scholar] [CrossRef]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of Estrogen Receptor β on Gene Networks Regulated by Estrogen Receptor α in Breast Cancer Cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef]
- Ström, A.; Hartman, J.; Foster, J.S.; Kietz, S.; Wimalasena, J.; Gustafsson, J.A. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad. Sci. USA 2004, 101, 1566–1571. [Google Scholar] [CrossRef]
- Datta, J.; Willingham, N.; Manouchehri, J.M.; Schnell, P.; Sheth, M.; David, J.J.; Kassem, M.; Wilson, T.A.; Radomska, H.S.; Coss, C.C.; et al. Activity of Estrogen Receptor beta Agonists in Therapy-Resistant Estrogen Receptor-Positive Breast Cancer. Front. Oncol. 2022, 12, 857590. [Google Scholar] [CrossRef]
- Motylewska, E.; Stasikowska, O.; Melen-Mucha, G. The inhibitory effect of diarylpropionitrile, a selective agonist of estrogen receptor beta, on the growth of MC38 colon cancer line. Cancer Lett. 2009, 276, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Mahringer, A.; Miller, D.S.; Bauer, B. 17-beta-Estradiol: A powerful modulator of blood-brain barrier BCRP activity. J. Cereb. Blood Flow Metab. 2010, 30, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Burek, M.; Salvador, E.; Förster, C.Y. Generation of an immortalized murine brain microvascular endothelial cell line as an in vitro blood brain barrier model. J. Vis. Exp. 2012, 7, e4022. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Vazguez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Cailleau, R.; Young, R.; Olive, M.; Reeves, W.J., Jr. Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef]
- Lasfargues, E.Y.; Coutinho, W.G.; Redfield, E.S. Isolation of two human tumor epithelial cell lines from solid breast carcinomas. J. Natl. Cancer Inst. 1978, 61, 967–978. [Google Scholar]
- Reznik, R.R.; Kotlyar, P.K.; Gridchin, V.O.; Ubyivovk, E.V.; Federov, V.V.; Khrebtov, A.I.; Shevchuk, D.S.; Cirlin, G.E. Low-Temperature In-Induced Holes Formation in Native-SiOx/Si(111) Substrates for Self-Catalyzed MBE Growth of GaAs Nanowires. Materials 2020, 13, 3449. [Google Scholar] [CrossRef]
- Tarasov, S.A.; Gorbunov, E.A.; Don, E.S.; Emelyanova, A.G.; Kovalchuk, A.L.; Yanamala, N.; AS, S.S.; Klein-Seetharaman, J.; Groenestein, R.; Tafani, J.P.; et al. Insights into the Mechanism of Action of Highly Diluted Biologics. J. Immunol. 2020, 205, 1345–1354. [Google Scholar] [CrossRef]
- Linville, R.M.; Searson, P.C. Next-generation in vitro blood-brain barrier models: Benchmarking and improving model accuracy. Fluids Barriers CNS 2021, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Haddad-Tovolli, R.; Dragano, N.R.V.; Ramalho, A.F.S.; Velloso, L.A. Development and Function of the Blood-Brain Barrier in the Context of Metabolic Control. Front. Neurosci. 2017, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Williams-Medina, A.; Deblock, M.; Janigro, D. In vitro Models of the Blood-Brain Barrier: Tools in Translational Medicine. Front. Med. Technol. 2020, 2, 623950. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Salvador, E.; Pastorin, G.; Förster, C. Blood-brain barrier transport studies, aggregation, and molecular dynamics simulation of multiwalled carbon nanotube functionalized with fluorescein isothiocyanate. Int. J. Nanomed. 2015, 10, 1703–1713. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E2 (Mean ± SD) | p Value (Dunnett’s Post hoc Multiple Comparisons Test) | DPN (Mean ± SD) | p Value (Dunnett’s Post hoc Multiple Comparisons Test) | ||
|---|---|---|---|---|---|
| cEND | 10−12 M | 1.13 ± 0.07 | 0.1386 | 0.94 ± 0.18 | 0.9848 |
| 10−10 M | 1.20 ± 0.12 | 0.0205 (*) | 1.01 ± 0.20 | >0.9999 | |
| 10−8 M | 1.04 ± 0.05 | 0.8850 | 1.01 ± 0.25 | >0.9999 | |
| 10−6 M | 1.02 ± 0.06 | 0.9922 | 0.98 ± 0.16 | 0.9998 | |
| hCMEC/D3 | 10−12 M | 1.11 ± 0.21 | 0.7695 | 0.79 ± 0.11 | 0.0372 (*) |
| 10−10 M | 1.17 ± 0.19 | 0.4495 | 0.93 ± 0.06 | 0.6782 | |
| 10−8 M | 1.21 ± 0.14 | 0.3014 | 0.95 ± 0.11 | 0.9153 | |
| 10−6 M | 1.32 ± 0.06 | 0.0653 | 0.88 ± 0.09 | 0.3548 |
| Endothelial Cell Lines | BC Cell Lines | E2 | DPN |
|---|---|---|---|
| cEND | MCF-7 | ||
| BT-474 | −− | ||
| MDA-MB-231 | ++++ | ||
| hCMEC/D3 | MCF-7 | ||
| BT-474 | −−− | ||
| MDA-MB-231 |
| D: without T: without | D: E2 T: without | D: E2 T: DPN | D: E2 T: DPN | |
|---|---|---|---|---|
| MCF-7 | ||||
| cEND | 1181.10 | 1638.71 | 1465.37 | 1731.47 |
| 1047.11 | 1844.56 | 1015.82 | 2041.55 | |
| 1017.45 | 1546.68 | 1270.75 | 1674.19 | |
| hCMEC/D3 | 1190.88 | 1416.22 | 1197.07 | 1805.76 |
| 1503.52 | 1850.85 | 1272.05 | 1713.36 | |
| 1836.36 | 1426.94 | 1307.59 | 1788.39 | |
| BT-474 | ||||
| cEND | 1707.45 | 1796.22 | 1958.11 | 1220.80 |
| 1712.58 | 1404.17 | 1795.11 | 1727.16 | |
| 2160.75 | 1863.79 | 1819.01 | 1973.94 | |
| hCMEC/D3 | 1432.61 | 1618.21 | 1589.70 | 1338.38 |
| 1784.99 | 1377.08 | 1810.65 | 1190.43 | |
| 1416.93 | 1228.84 | 1531.83 | 1465.79 | |
| MDA-MB-231 | ||||
| cEND | 9728.15 | 21,198.07 | 10,635.70 | 14,973.33 |
| 9613.24 | 16,039.96 | 9644.18 | 20,085.65 | |
| 13,996.28 | 21,984.58 | 10,703.47 | 21,914.55 | |
| hCMEC/D3 | 12,145.83 | 17,893.11 | 9928.52 | 14,924.84 |
| 9342.15 | 19,821.68 | 10,868.48 | 17,599.52 | |
| 10,992.24 | 19,614.28 | 10,483.95 | 17,120.07 | |
| Endothelial Cell Lines | BC Cell Lines | D: E2 (C+E) T: without (2) | D: E2 (E) T: DPN (C+E) (3) | D: E2 (C+E) T: DPN (C+E) (4) |
|---|---|---|---|---|
| cEND | MCF-7 | + | − | ++ |
| BT-474 | ||||
| MDA-MB-231 | + | −− | + | |
| hCMEC/D3 | MCF-7 | |||
| BT-474 | ||||
| MDA-MB-231 | +++ | −−− | ++ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchner, J.; Völker, E.; Shityakov, S.; Saji, S.; Förster, C.Y. Protecting the Brain: Novel Strategies for Preventing Breast Cancer Brain Metastases through Selective Estrogen Receptor β Agonists and In Vitro Blood–Brain Barrier Models. Int. J. Mol. Sci. 2024, 25, 3379. https://doi.org/10.3390/ijms25063379
Kirchner J, Völker E, Shityakov S, Saji S, Förster CY. Protecting the Brain: Novel Strategies for Preventing Breast Cancer Brain Metastases through Selective Estrogen Receptor β Agonists and In Vitro Blood–Brain Barrier Models. International Journal of Molecular Sciences. 2024; 25(6):3379. https://doi.org/10.3390/ijms25063379
Chicago/Turabian StyleKirchner, Janine, Elisabeth Völker, Sergey Shityakov, Shigehira Saji, and Carola Y. Förster. 2024. "Protecting the Brain: Novel Strategies for Preventing Breast Cancer Brain Metastases through Selective Estrogen Receptor β Agonists and In Vitro Blood–Brain Barrier Models" International Journal of Molecular Sciences 25, no. 6: 3379. https://doi.org/10.3390/ijms25063379
APA StyleKirchner, J., Völker, E., Shityakov, S., Saji, S., & Förster, C. Y. (2024). Protecting the Brain: Novel Strategies for Preventing Breast Cancer Brain Metastases through Selective Estrogen Receptor β Agonists and In Vitro Blood–Brain Barrier Models. International Journal of Molecular Sciences, 25(6), 3379. https://doi.org/10.3390/ijms25063379