
Connexin43, A Promising Target to Reduce Cardiac Arrhythmia Burden in Pulmonary Arterial Hypertension
 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Electrical Instability and Incidence of Cardiac Arrhythmias in PAH
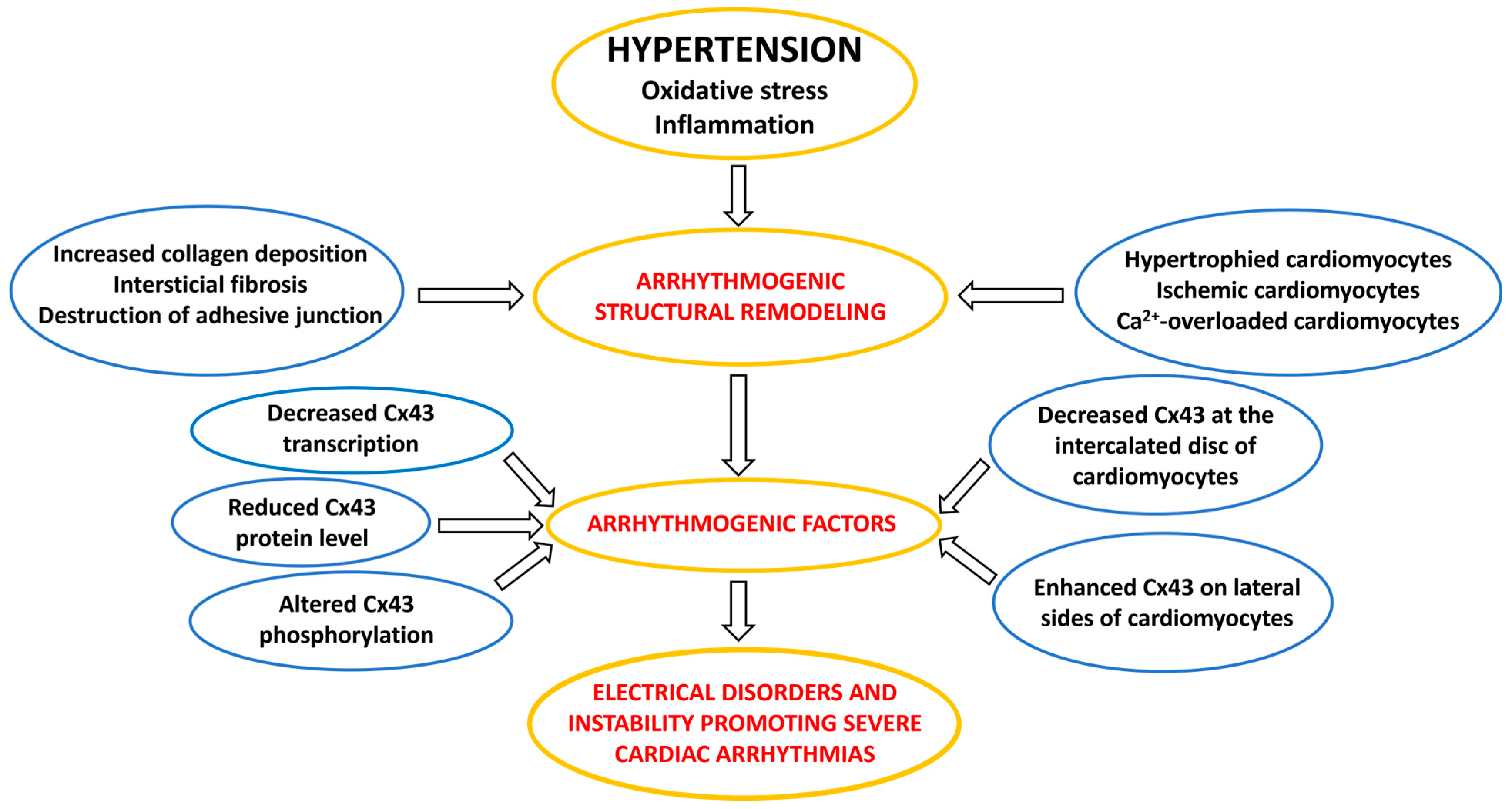
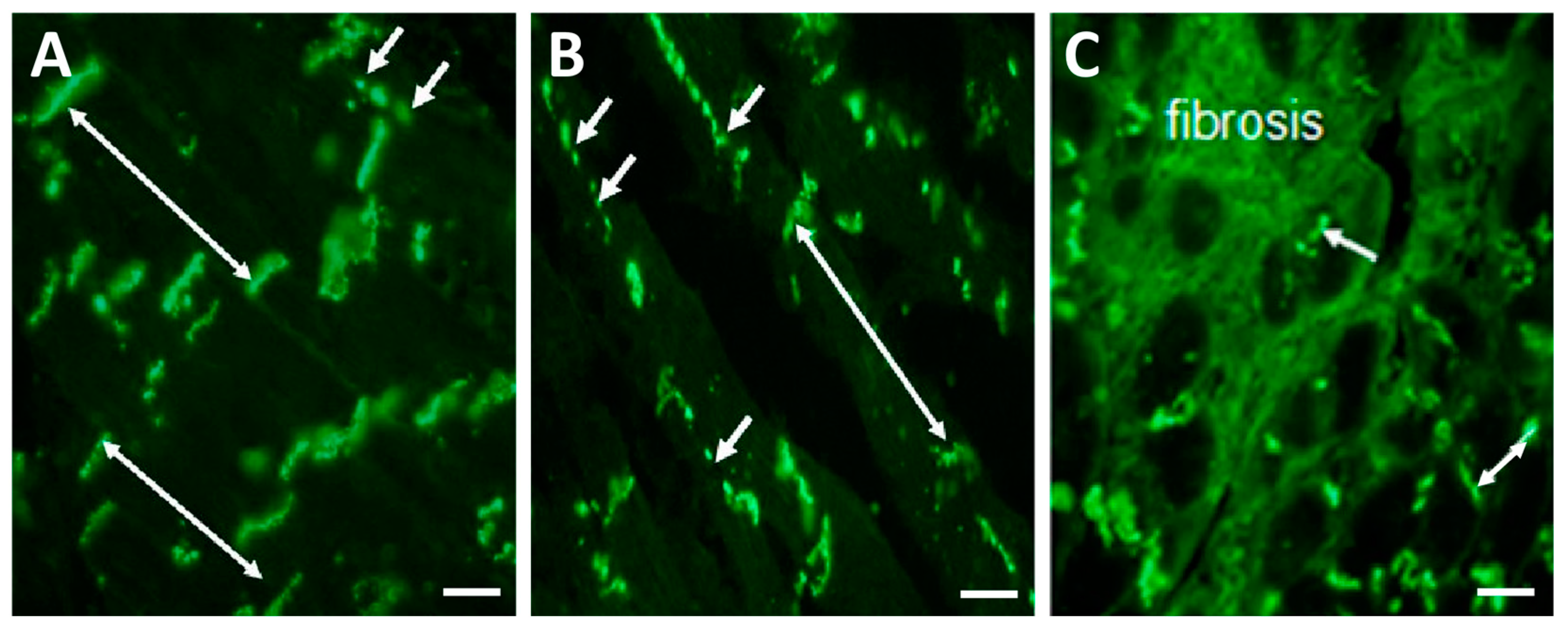
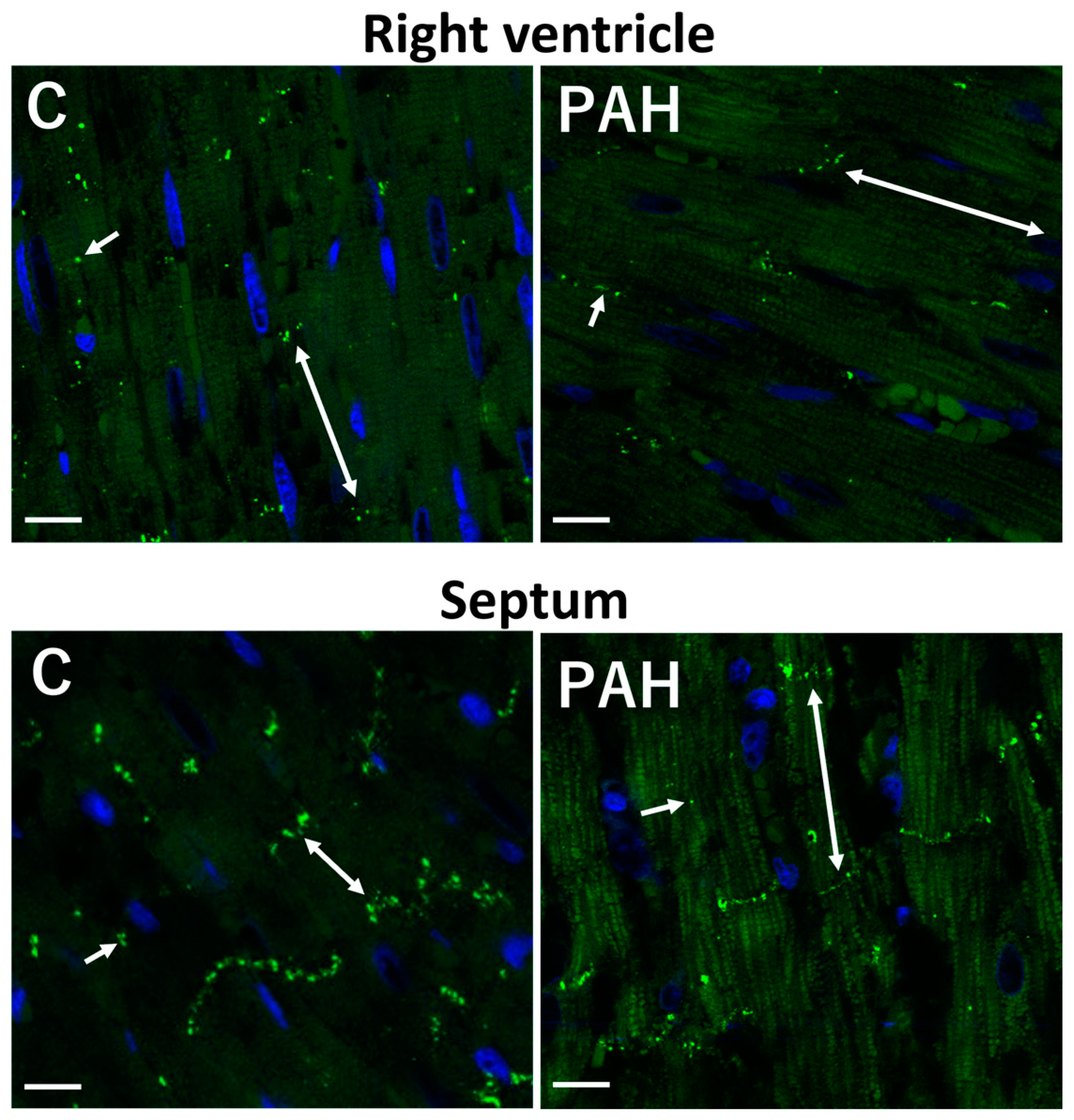
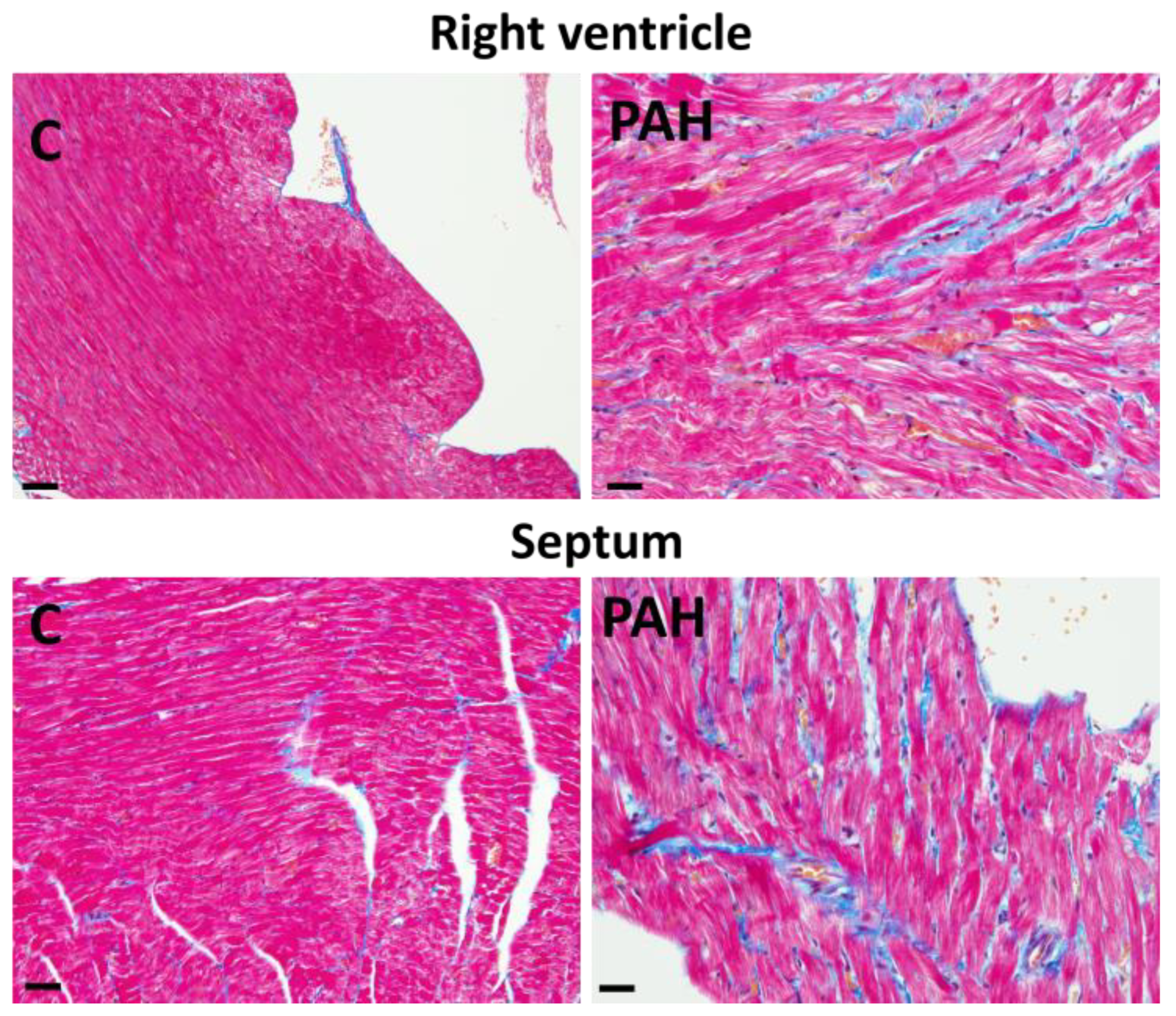
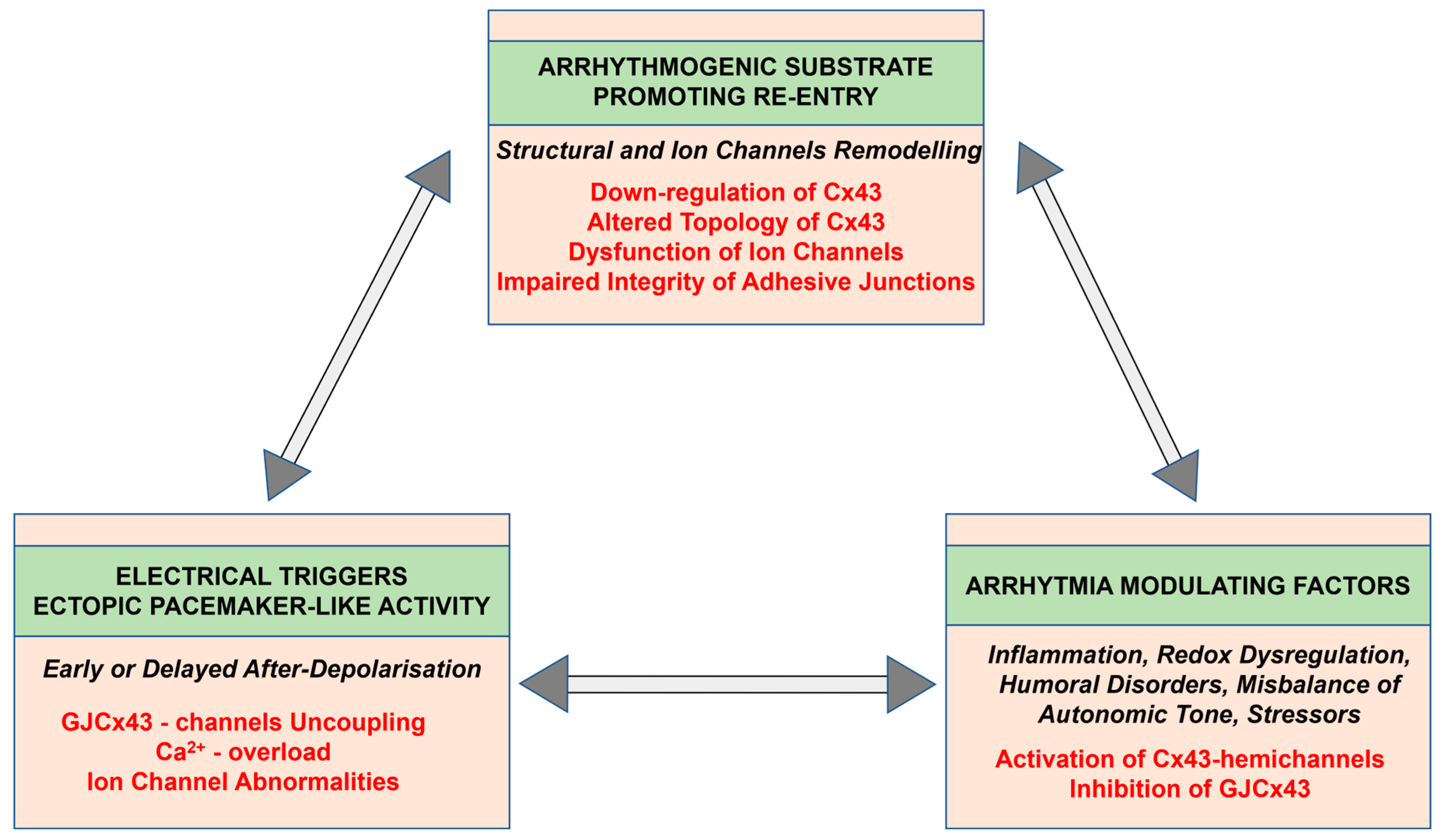
3. Factors and Mechanisms Involved in the Occurrence of Cardiac Arrhythmias: Cx43 as a Key Player
4. Progress in Research and Treatment in PAH with the Potential to Prevent Cardiac Arrhythmias
4.1. Benefits of SGLT2i Therapy
4.2. Targeted Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wits, M.; Becher, C.; De Man, F.; Sanchez-Duffhues, G.; Goumans, M.J. Sex-biased TGFβ signalling in pulmonary arterial hypertension. Cardiovasc. Res. 2023, 119, 2262–2277. [Google Scholar] [CrossRef]
- Bernardo, R.J.; Haddad, F.; Couture, E.J.; Hansmann, G.; de Jesus Perez, V.A.; Denault, A.Y.; de Man, F.S.; Amsallem, M. Mechanics of right ventricular dysfunction in pulmonary arterial hypertension and heart failure with preserved ejection fraction. Cardiovasc. Diagn. Ther. 2020, 10, 1580–1603. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Mayer, E.; Nagavci, B.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension Developed by the Task Force for the Diagnosis and Treatment of (ESC) and the European Respiratory Society (ERS). Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.R.; Lawrie, A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol. Med. 2017, 23, 31–45. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, P.P.; Zhou, R.L.; Zhang, Y.; Tian, Z.; Zhang, S.Y. Pathological mechanisms and potential therapeutic targets of pulmonary arterial hypertension: A review. Aging Dis. 2020, 11, 1623–1629. [Google Scholar] [CrossRef]
- Van Der Bruggen, C.E.E.; Tedford, R.J.; Handoko, M.L.; Van Der Velden, J.; De Man, F.S. RV pressure overload: From hypertrophy to failure. Cardiovasc. Res. 2017, 113, 1423–1432. [Google Scholar] [CrossRef]
- van Wezenbeek, J.; Groeneveldt, J.A.; Llucià-Valldeperas, A.; van der Bruggen, C.E.; Jansen, S.M.A.; Smits, A.J.; Smal, R.; van Leeuwen, J.W.; dos Remedios, C.; Keogh, A.; et al. Interplay of sex hormones and long-term right ventricular adaptation in a Dutch PAH-cohort. J. Heart Lung Transplant. 2022, 41, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Ashek, A.; Spruijt, O.A.; Harms, H.J.; Lammertsma, A.A.; Cupitt, J.; Dubois, O.; Wharton, J.; Dabral, S.; Pullamsetti, S.S.; Huisman, M.C.; et al. 3’-Deoxy-3’-[18F]Fluorothymidine Positron Emission Tomography Depicts Heterogeneous Proliferation Pathology in Idiopathic Pulmonary Arterial Hypertension Patient Lung. Circ. Cardiovasc. Imaging 2018, 11, e007402. [Google Scholar] [CrossRef] [PubMed]
- Jarabicová, I.; Horváth, C.; Veľasová, E.; Bies Piváčková, L.; Vetešková, J.; Klimas, J.; Křenek, P.; Adameová, A. Analysis of necroptosis and its association with pyroptosis in organ damage in experimental pulmonary arterial hypertension. J. Cell. Mol. Med. 2022, 26, 2633–2645. [Google Scholar] [CrossRef]
- Suen, C.M.; Chaudhary, K.R.; Deng, Y.; Jiang, B.; Stewart, D.J. Fischer rats exhibit maladaptive structural and molecular right ventricular remodelling in severe pulmonary hypertension: A genetically prone model for right heart failure. Cardiovasc. Res. 2019, 115, 788–799. [Google Scholar] [CrossRef]
- Andersen, M.Ø.; Diederichsen, S.Z.; Svendsen, J.H.; Carlsen, J. Assessment of cardiac arrhythmias using long-term continuous monitoring in patients with pulmonary hypertension. Int. J. Cardiol. 2021, 334, 110–115. [Google Scholar] [CrossRef]
- Celant, L.R.; Wessels, J.N.; Kianzad, A.; Marcus, J.T.; Meijboom, L.J.; Bogaard, H.J.; De Man, F.S.; Vonk Noordegraaf, A. Restoration of right ventricular function in the treatment of pulmonary arterial hypertension. Heart 2023, 109, 1844–1850. [Google Scholar] [CrossRef]
- Tomaszewski, M.; Mertowska, P.; Janczewska, M.; Styczeń, A.; Mertowski, S.; Jonas, K.; Grywalska, E.; Kopeć, G. In the Search for Biomarkers of Pulmonary Arterial Hypertension, Are Cytokines IL-2, IL-4, IL-6, IL-10, and IFN-Gamma the Right Indicators to Use? Int. J. Mol. Sci. 2023, 24, 13694. [Google Scholar] [CrossRef]
- Bandorski, D.; Erkapic, D.; Stempfl, J.; Höltgen, R.; Grünig, E.; Schmitt, J.; Chasan, R.; Grimminger, J.; Neumann, T.; Hamm, C.W.; et al. Ventrikuläre Tachykardien bei Patienten mit pulmonaler Hypertonie—Wird die Prävalenz unterschätzt? Eine prospektive klinische Studie. Herzschrittmachertherapie und Elektrophysiologie 2015, 26, 155–162. [Google Scholar] [CrossRef]
- Ruiz-Cano, M.J.; Gonzalez-Mansilla, A.; Escribano, P.; Delgado, J.; Arribas, F.; Torres, J.; Flox, A.; Riva, M.; Gomez, M.A.; Saenz, C. Clinical implications of supraventricular arrhythmias in patients with severe pulmonary arterial hypertension. Int. J. Cardiol. 2011, 146, 105–106. [Google Scholar] [CrossRef]
- Ley, L.; Höltgen, R.; Bogossian, H.; Ghofrani, H.A.; Bandorski, D. Electrocardiogram in patients with pulmonary hypertension. J. Electrocardiol. 2023, 79, 24–29. [Google Scholar] [CrossRef]
- Crisan, S.; Baghina, R.M.; Luca, S.A.; Cozlac, A.R.; Negru, A.G.; Vacarescu, C.; Lazar, M.A.; Luca, C.T.; Gaita, D. Comprehensive imaging in patients with suspected pulmonary arterial hypertension. Heart 2023, 110, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.J.; Arkani, M.; In’t Veld, S.G.J.G.; Huis in ‘t Veld, A.E.; Sol, N.; Groeneveldt, J.A.; Botros, L.; Braams, N.J.; Jansen, S.M.A.; Ramaker, J.; et al. Distinct Platelet Ribonucleic Acid Signatures in Patients with Pulmonary Hypertension. Ann. Am. Thorac. Soc. 2022, 19, 1650–1660. [Google Scholar] [CrossRef]
- Sykora, M.; Andelova, K.; Szeiffova Bacova, B.; Egan Benova, T.; Martiskova, A.; Knezl, V.; Tribulova, N. Hypertension Induces Pro-arrhythmic Cardiac Connexome Disorders: Protective Effects of Treatment. Biomolecules 2023, 13, 330. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Bacova, B.S.; Sykora, M.; Hlivak, P.; Barancik, M.; Tribulova, N. Mechanisms Underlying Antiarrhythmic Properties of Cardioprotective Agents Impacting Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1416. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging role of the inflammasome and pyroptosis in hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef]
- Andersen, S.; Birkmose Axelsen, J.; Ringgaard, S.; Randel Nyengaard, J.; Holm Nielsen, S.; Genovese, F.; Asser Karsdal, M.; Adler Hyldebrandt, J.; Brandt Sørensen, C.; de Man, F.S.; et al. Pressure overload induced right ventricular remodeling is not attenuated by the anti-fibrotic agent pirfenidone. Pulm. Circ. 2019, 9, 2045894019848659. [Google Scholar] [CrossRef]
- Andersen, S.; Nielsen-Kudsk, J.E.; Vonk Noordegraaf, A.; De Man, F.S. Right Ventricular Fibrosis: A Pathophysiological Factor in Pulmonary Hypertension? Circulation 2019, 139, 269–285. [Google Scholar] [CrossRef]
- Andelova, K.; Benova, T.E.; Bacova, B.S.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac connexin-43 hemichannels and pannexin1 channels: Provocative antiarrhythmic targets. Int. J. Mol. Sci. 2021, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Radosinska, J.; Bacova, B.; Knezl, V.; Benova, T.; Zurmanova, J.; Soukup, T.; Arnostova, P.; Slezak, J.; Goncalvesova, E.; Tribulova, N. Dietary omega-3 fatty acids attenuate myocardial arrhythmogenic factors and propensity of the heart to lethal arrhythmias in a rodent model of human essential hypertension. J. Hypertens. 2013, 31, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Salameh, A. Remodeling of cardiac gap junctional cell–cell coupling. Cells 2021, 10, 2422. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, M.; Honjo, H.; Takagishi, Y.; Emdad, L.; Magee, A.I.; Severs, N.J.; Kodama, I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ. Res. 2000, 86, 871–878. [Google Scholar] [CrossRef]
- Liu, F.; Gutstein, D.E. The cardiac gap junction: A potential therapeutic target in the treatment of heart disease. Mt. Sinai J. Med. 2002, 69, 421–424. [Google Scholar]
- Htet, M.; Nally, J.E.; Shaw, A.; Foote, B.E.; Martin, P.E.; Dempsie, Y. Connexin 43 plays a role in pulmonary vascular reactivity in mice. Int. J. Mol. Sci. 2018, 19, 1891. [Google Scholar] [CrossRef]
- Htet, M.; Nally, J.E.; Martin, P.E.; Dempsie, Y. New insights into pulmonary hypertension: A role for connexin-mediated signalling. Int. J. Mol. Sci. 2022, 23, 379. [Google Scholar] [CrossRef]
- de Man, F.S.; Noordegraaf, A.V. The magic of communication: The need to study organ and cell communication in pulmonary arterial hypertension-induced right heart failure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L634–L636. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Piaditis, G.; Bacopoulou, F.; Chrousos, G.P. Cardiac Remodeling in Hypertension: Clinical Impact on Brain, Heart, and Kidney Function. Horm. Metab. Res. 2022, 54, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Liles, J.T.; Hoyer, K.; Oliver, J.; Chi, L.; Dhalla, A.K.; Belardinelli, L. Ranolazine reduces remodeling of the right ventricle and provoked arrhythmias in rats with pulmonary hypertensions. J. Pharmacol. Exp. Ther. 2015, 353, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Tateno, S.; Niwa, K.; Nakazawa, M.; Iwamoto, M.; Yokota, M.; Nagashima, M.; Echigo, S.; Kado, H.; Shima, M.; Gatzoulis, M.A. Risk factors for arrhythmia and late death in patients with right ventricle to pulmonary artery conduit repair—Japanese multicenter study. Int. J. Cardiol. 2006, 106, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Rajdev, A.; Garan, H.; Biviano, A. Arrhythmias in Pulmonary Arterial Hypertension. Prog. Cardiovasc. Dis. 2012, 55, 180–186. [Google Scholar] [CrossRef]
- Cirulis, M.M.; Ryan, J.J.; Archer, S.L. Pathophysiology, incidence, management, and consequences of cardiac arrhythmia in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2019, 9, 2045894019834890. [Google Scholar] [CrossRef]
- Mercurio, V.; Peloquin, G.; Bourji, K.I.; Diab, N.; Sato, T.; Enobun, B.; Housten-Harris, T.; Damico, R.; Kolb, T.M.; Mathai, S.C.; et al. Pulmonary arterial hypertension and atrial arrhythmias: Incidence, risk factors, and clinical impact. Pulm. Circ. 2018, 8, 4–11. [Google Scholar] [CrossRef]
- Cannillo, M.; Grosso Marra, W.; Gili, S.; D’Ascenzo, F.; Morello, M.; Mercante, L.; Mistretta, E.; Salera, D.; Zema, D.; Bissolino, A.; et al. Supraventricular Arrhythmias in Patients with Pulmonary Arterial Hypertension. Am. J. Cardiol. 2015, 116, 1883–1889. [Google Scholar] [CrossRef]
- Małaczyńska-Rajpold, K.; Komosa, A.; Błaszyk, K.; Araszkiewicz, A.; Janus, M.; Olasińska-Wiśniewska, A.; Jankiewicz, S.; Maczyński, M.; Mularek-Kubzdela, T. The Management of Supraventricular Tachyarrhythmias in Patients with Pulmonary Arterial Hypertension. Heart Lung Circ. 2016, 25, 442–450. [Google Scholar] [CrossRef]
- Rottlaender, D.; Motloch, L.J.; Schmidt, D.; Reda, S.; Larbig, R.; Wolny, M.; Dumitrescu, D.; Rosenkranz, S.; Erdmann, E.; Hoppe, U.C. Clinical impact of atrial fibrillation in patients with pulmonary hypertension. PLoS ONE 2012, 7, e33902. [Google Scholar] [CrossRef] [PubMed]
- Wessels, J.N.; Mouratoglou, S.A.; van Wezenbeek, J.; Louis Handoko, M.; Marcus, J.T.; Meijboom, L.J.; Westerhof, B.E.; Bogaard, H.J.; Strijkers, G.J.; Vonk Noordegraaf, A.; et al. Right atrial function is associated with right venticular diastolic stiffness: RA-RV interaction in pulmonary arterial hypertension. Eur. Respir. J. 2022, 59, 2101454. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Nickel, N.P.; Tongers, J.; Hoeper, M.M. Atrial flutter and fibrillation in patients with pulmonary hypertension. Int. J. Cardiol. 2013, 167, 2300–2305. [Google Scholar] [CrossRef]
- Hiram, R.; Naud, P.; Xiong, F.; Al-u’datt, D.; Algalarrondo, V.; Sirois, M.G.; Tanguay, J.F.; Tardif, J.C.; Nattel, S. Right Atrial Mechanisms of Atrial Fibrillation in a Rat Model of Right Heart Disease. J. Am. Coll. Cardiol. 2019, 74, 1332–1347. [Google Scholar] [CrossRef]
- Smith, B.; Genuardi, M.V.; Koczo, A.; Zou, R.H.; Thoma, F.W.; Handen, A.; Craig, E.; Hogan, C.M.; Girard, T.; Althouse, A.D.; et al. Atrial arrhythmias are associated with increased mortality in pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045894018790316. [Google Scholar] [CrossRef] [PubMed]
- Folino, A.F.; Bobbo, F.; Schiraldi, C.; Tona, F.; Romano, S.; Buja, G.; Bellotto, F. Ventricular Arrhythmias and Autonomic Profile in Patients with Primary Pulmonary Hypertension. Lung 2003, 181, 321–328. [Google Scholar] [CrossRef]
- Fazelifar, A.F.; Talebian, F.; Ghaffarinejad, Z.; Habibi, M.A.; Pasebani, Y.; Mazloomi, A.A.; Fazelifar, A.F.; Khajali, Z. Electrocardiographic manifestations of pulmonary stenosis versus pulmonary hypertension. J. Electrocardiol. 2023, 81, 117–122. [Google Scholar] [CrossRef]
- Hakacova, N.; Wagner, G.; Bacharova, L. Right and left ventricular pressure overload as imaged by electrocardiogram. J. Electrocardiol. 2014, 47, 273. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Q.; Liu, Z.; Zhao, Z.; Xiong, C.; Ni, X.; He, J.; Wei, Y.; Zhang, S. Heart rate-corrected QT interval and QT dispersion in patients with pulmonary hypertension. Wien. Klin. Wochenschr. 2009, 121, 330–333. [Google Scholar] [CrossRef]
- Luo, J.; Sun, J.; Xu, L.; Chen, J.; Chen, Y.; Chen, W.; Qiu, H.; Luo, X.; Chen, S.; Li, J. Analysis of relationship between P wave dispersion and diagnosis of pulmonary arterial hypertension and risk stratification. J. Electrocardiol. 2023, 81, 94–100. [Google Scholar] [CrossRef]
- Zeltser, I.; Gaynor, J.W.; Petko, M.; Myung, R.J.; Birbach, M.; Waibel, R.; Ittenbach, R.F.; Tanel, R.E.; Vetter, V.L.; Rhodes, L.A. The roles of chronic pressure and volume overload states in induction of arrhythmias: An animal model of physiologic sequelae after repair of tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2005, 130, 1542–1548. [Google Scholar] [CrossRef][Green Version]
- Hardziyenka, M.; Campian, M.E.; Verkerk, A.O.; Surie, S.; Van Ginneken, A.C.G.; Hakim, S.; Linnenbank, A.C.; De Bruin-Bon, H.A.C.M.R.; Beekman, L.; Van Der Plas, M.N.; et al. Electrophysiologic remodeling of the left ventricle in pressure overload-induced right ventricular failure. J. Am. Coll. Cardiol. 2012, 59, 2193–2202. [Google Scholar] [CrossRef]
- Manders, E.; Bogaard, H.J.; Handoko, M.L.; Van De Veerdonk, M.C.; Keogh, A.; Westerhof, N.; Stienen, G.J.M.; Dos Remedios, C.G.; Humbert, M.; Dorfmüller, P.; et al. Contractile dysfunction of left ventricular cardiomyocytes in patients with pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2014, 64, 28–37. [Google Scholar] [CrossRef]
- Manders, E.; Rain, S.; Bogaard, H.J.; Handoko, M.L.; Stienen, G.J.M.; Vonk-Noordegraaf, A.; Ottenheijm, C.A.C.; De Man, F.S. The striated muscles in pulmonary arterial hypertension: Adaptations beyond the right ventricle. Eur. Respir. J. 2015, 46, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Rain, S.; Goncalves Bos, D.S.; Handoko, M.L.; Westerhof, N.; Stienen, G.; Ottenheijm, C.; Goebel, M.; Dorfmüller, P.; Guignabert, C.; Humbert, M.; et al. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J. Am. Heart Assoc. 2014, 3, e000716. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Seki, S.; Radosinska, J.; Kaplan, P.; Babusikova, E.; Knezl, V.; Mochizuki, S. Myocardial Ca2+ handling and cell-to-cell coupling, key factors in prevention of sudden cardiac death1. Can. J. Physiol. Pharmacol. 2009, 87, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Knezl, V.; Szeiffova Bacova, B.; Egan Benova, T.; Viczenczova, C.; Gonçalvesova, E.; Slezak, J. Disordered myocardial Ca2+ homeostasis results in substructural alterations that may promote occurrence of malignant arrhythmias. Physiol. Res. 2016, 65, S139–S148. [Google Scholar] [CrossRef] [PubMed]
- Bacharova, L.; Plandorova, J.; Klimas, J.; Krenek, P.; Kyselovic, J. Discrepancy between increased left ventricular mass and “normal” QRS voltage is associated with decreased connexin 43 expression in early stage of left ventricular hypertrophy in spontaneously hypertensive rats. J. Electrocardiol. 2008, 41, 730–734. [Google Scholar] [CrossRef]
- Benoist, D.; Dubes, V.; Roubertie, F.; Gilbert, S.H.; Charron, S.; Constantin, M.; Elbes, D.; Vieillot, D.; Quesson, B.; Cochet, H.; et al. Proarrhythmic remodelling of the right ventricle in a porcine model of repaired tetralogy of Fallot. Heart 2017, 103, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Nakagawa, M.; Kajimoto, M.; Nobe, S.; Ooie, T.; Ichinose, M.; Yonemochi, H.; Ono, N.; Shimada, T.; Saikawa, T. Heterogeneous expression of connexin 43 in the myocardium of rabbit right ventricular outflow tract. Life Sci. 2005, 77, 52–59. [Google Scholar] [CrossRef]
- Boengler, K.; Rohrbach, S.; Weissmann, N.; Schulz, R. Importance of CX43 for right ventricular function. Int. J. Mol. Sci. 2021, 22, 987. [Google Scholar] [CrossRef]
- Bouvard, C.; Genet, N.; Phan, C.; Rode, B.; Thuillet, R.; Tu, L.; Robillard, P.; Campagnac, M.; Soleti, R.; de la Roque, E.D.; et al. Connexin-43 is a promising target for pulmonary hypertension due to hypoxaemic lung disease. Eur. Respir. J. 2020, 55, 1900169. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Okruhlicova, L.; Imanaga, I.; Hirosawa, N.; Ogawa, K.; Weismann, P. Factors involved in the susceptibility of spontaneously hypertensive rats to low K+-induced arrhythmias. Gen. Physiol. Biophys. 2003, 22, 369–382. [Google Scholar] [PubMed]
- Nguyen, T.P.; Sovari, A.A.; Pezhouman, A.; Iyer, S.; Cao, H.; Ko, C.Y.; Bapat, A.; Vahdani, N.; Ghanim, M.; Fishbein, M.C.; et al. Increased susceptibility of spontaneously hypertensive rats to ventricular tachyarrhythmias in early hypertension. J. Physiol. 2016, 594, 1689–1707. [Google Scholar] [CrossRef]
- Nagibin, V.; Egan Benova, T.; Viczenczova, C.; Szeiffova Bacova, B.; Dovinova, I.; Barancik, M.; Tribulova, N. Ageing related down-regulation of myocardial connexin-43 and up-regulation of MMP-2 may predict propensity to atrial fibrillation in experimental animals. Physiol. Res. 2016, 65, S91–S100. [Google Scholar] [CrossRef] [PubMed]
- Sasano, C.; Honjo, H.; Takagishi, Y.; Uzzaman, M.; Emdad, L.; Shimizu, A.; Murata, Y.; Kamiya, K.; Kodama, I. Internalization and dephosphorylation of Connexin43 in hypertrophied right ventricles of rats with pulmonary hypertension. Circ. J. 2007, 71, 382–389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nayakanti, S.R.; Friedrich, A.; Sarode, P.; Jafari, L.; Maroli, G.; Boehm, M.; Bourgeois, A.; Grobs, Y.; Khassafi, F.; Kuenne, C.; et al. Targeting Wnt-ß-Catenin-FOSL Signaling Ameliorates Right Ventricular Remodeling. Circ. Res. 2023, 132, 1468–1485. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.S.C.; Van Veen, T.A.B.; De Bakker, J.M.T.; Van Rijen, H.V.M. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2020–2029. [Google Scholar] [CrossRef]
- Dhein, S. Gap junction channels in the cardiovascular system: Pharmacological and physiological modulation. Trends Pharmacol. Sci. 1998, 19, 229–241. [Google Scholar] [CrossRef]
- Salameh, A.; Dhein, S. Pharmacology of Gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer? Biochim. Biophys. Acta Biomembr. 2005, 1719, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.; Qu, Z.; Weiss, J.N. Cardiac fibrosis and arrhythmogenesis: The road to repair is paved with perils. J. Mol. Cell. Cardiol. 2014, 70, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Dumotier, B.M. A straightforward guide to the basic science behind arrhythmogenesis. Heart 2014, 100, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Christ, T.; Fabritz, L.; Goette, A.; Hammwöhner, M.; Heijman, J.; Kockskämper, J.; Linz, D.; Odening, K.E.; Schweizer, P.A.; et al. German Cardiac Society Working Group on Cellular Electrophysiology state-of-the-art paper: Impact of molecular mechanisms on clinical arrhythmia management. Clin. Res. Cardiol. 2019, 108, 577–599. [Google Scholar] [CrossRef]
- Brown, J.M.; Zhou, W.; Weber, B.; Divakaran, S.; Barrett, L.; Bibbo, C.F.; Hainer, J.; Taqueti, V.R.; Dorbala, S.; Blankstein, R.; et al. Low coronary flow relative to myocardial mass predicts heart failure in symptomatic hypertensive patients with no obstructive coronary artery disease. Eur. Heart J. 2022, 43, 3323–3331. [Google Scholar] [CrossRef]
- Dobrev, D.; Heijman, J.; Hiram, R.; Li, N.; Nattel, S. Inflammatory signalling in atrial cardiomyocytes: A novel unifying principle in atrial fibrillation pathophysiology. Nat. Rev. Cardiol. 2023, 20, 145–167. [Google Scholar] [CrossRef]
- Adameova, A.; Shah, A.K.; Dhalla, N.S. Role of oxidative stress in the genesis of ventricular arrhythmias. Int. J. Mol. Sci. 2020, 21, 4200. [Google Scholar] [CrossRef]
- De Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; François, C.; Schalij, I.; Dorfmüller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef]
- Molina, C.E.; Heijman, J.; Dobrev, D. Differences in left versus right ventricular electrophysiological properties in cardiac dysfunction and arrhythmogenesis. Arrhythmia Electrophysiol. Rev. 2016, 5, 14–19. [Google Scholar] [CrossRef]
- Benova, T.; Viczenczova, C.; Radosinska, J.; Bacova, B.; Knezl, V.; Dosenko, V.; Weismann, P.; Zeman, M.; Navarova, J.; Tribulova, N. Melatonin attenuates hypertension-related proarrhythmic myocardial maladaptation of connexin-43 and propensity of the heart to lethal arrhythmias. Can. J. Physiol. Pharmacol. 2013, 91, 633–639. [Google Scholar] [CrossRef]
- Bacova, B.S.; Viczenczova, C.; Andelova, K.; Sykora, M.; Chaudagar, K.; Barancik, M.; Adamcova, M.; Knezl, V.; Benova, T.E.; Weismann, P.; et al. Antiarrhythmic effects of melatonin and omega-3 are linked with protection of myocardial cx43 topology and suppression of fibrosis in catecholamine stressed normotensive and hypertensive rats. Antioxidants 2020, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Tomos, I.; Roussis, I.; Matthaiou, A.M.; Dimakou, K. Molecular and Genetic Biomarkers in Idiopathic Pulmonary Fibrosis: Where Are We Now? Biomedicines 2023, 11, 2796. [Google Scholar] [CrossRef]
- Strauss, B.; Bisserier, M.; Obus, E.; Katz, M.G.; Fargnoli, A.; Cacheux, M.; Akar, J.G.; Hummel, J.P.; Hadri, L.; Sassi, Y.; et al. Right predominant electrical remodeling in a pure model of pulmonary hypertension promotes reentrant arrhythmias. Heart Rhythm 2022, 19, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Dubes, V.; Benoist, D.; Roubertie, F.; Gilbert, S.H.; Constantin, M.; Charron, S.; Elbes, D.; Vieillot, D.; Quesson, B.; Cochet, H.; et al. Arrhythmogenic Remodeling of the Left Ventricle in a Porcine Model of Repaired Tetralogy of Fallot. Circ. Arrhythm. Electrophysiol. 2018, 11, e006059. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Electrophysiological effects of remodeling cardiac gap junctions and cell size. Experimental and model studies of normal cardiac growth. Circ. Res. 2000, 86, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, B.E.J.; Jongsma, H.J.; Bierhuizen, M.F.A. Regulation of myocardial connexins during hypertrophic remodelling. Eur. Heart J. 2004, 25, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Valderrábano, M. Influence of anisotropic conduction properties in the propagation of the cardiac action potential. Prog. Biophys. Mol. Biol. 2008, 94, 144–168. [Google Scholar] [CrossRef]
- Duffy, H.S. The molecular mechanisms of gap junction remodeling. Heart. Rhythm 2012, 9, 1331–1334. [Google Scholar] [CrossRef]
- Gutstein, D.E.; Morley, G.E.; Vaidya, D.; Liu, F.; Chen, F.L.; Stuhlmann, H.; Fishman, G.I. Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation 2001, 104, 1194–1199. [Google Scholar] [CrossRef]
- Handa, B.S.; Li, X.; Baxan, N.; Roney, C.H.; Shchendrygina, A.; Mansfield, C.A.; Jabbour, R.J.; Pitcher, D.S.; Chowdhury, R.A.; Peters, N.S.; et al. Ventricular fibrillation mechanism and global fibrillatory organization are determined by gap junction coupling and fibrosis pattern. Cardiovasc. Res. 2021, 117, 1078–1090. [Google Scholar] [CrossRef]
- Antigny, F.; Mercier, O.; Humbert, M.; Sabourin, J. Excitation-contraction coupling and relaxation alteration in right ventricular remodelling caused by pulmonary arterial hypertension. Arch. Cardiovasc. Dis. 2020, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Peracchia, C. Gap Junction Channel Regulation: A Tale of Two Gates—Voltage Sensitivity of the Chemical Gate and Chemical Sensitivity of the Fast Voltage Gate. Int. J. Mol. Sci. 2024, 25, 98. [Google Scholar] [CrossRef] [PubMed]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Remo, B.F.; Qu, J.; Volpicelli, F.M.; Giovannone, S.; Shin, D.; Lader, J.; Liu, F.Y.; Zhang, J.; Lent, D.S.; Morley, G.E.; et al. Phosphatase-Resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 2011, 108, 1459–1466. [Google Scholar] [CrossRef]
- De Groot, J.R.; Veenstra, T.; Verkerk, A.O.; Wilders, R.; Smits, J.P.P.; Wilms-Schopman, F.J.G.; Wiegerinck, R.F.; Bourier, J.; Belterman, C.N.W.; Coronel, R.; et al. Conduction slowing by the gap junctional uncoupler carbenoxolone. Cardiovasc. Res. 2003, 60, 288–297. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta Biomembr. 2018, 1860, 83–90. [Google Scholar] [CrossRef]
- Pun, R.; Kim, M.H.; North, B.J. Role of Connexin 43 phosphorylation on Serine-368 by PKC in cardiac function and disease. Front. Cardiovasc. Med. 2023, 9, 1080131. [Google Scholar] [CrossRef]
- Saffitz, J.E. Dependence of electrical coupling on mechanical coupling in cardiac myocytes: Insights gained from cardiomyopathies caused by defects in cell-cell connections. Ann. N. Y. Acad. Sci. 2005, 1047, 336–344. [Google Scholar] [CrossRef]
- Noorman, M.; van der Heyden, M.A.G.; van Veen, T.A.B.; Cox, M.G.P.J.; Hauer, R.N.W.; de Bakker, J.M.T.; van Rijen, H.V.M. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J. Mol. Cell. Cardiol. 2009, 47, 23–31. [Google Scholar] [CrossRef]
- Chkourko, H.S.; Guerrero-Serna, G.; Lin, X.; Darwish, N.; Pohlmann, J.R.; Cook, K.E.; Martens, J.R.; Rothenberg, E.; Musa, H.; Delmar, M. Remodeling of mechanical junctions and of microtubule-associated proteins accompany cardiac connexin43 lateralization. Heart Rhythm 2012, 9, 1133–1140.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Levin, M.D.; Xiong, Y.; Petrenko, N.; Patel, V.V.; Radice, G.L. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J. Mol. Cell. Cardiol. 2008, 44, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Saleem, K.; Pandey, J.; Charania, A.N.; Zhou, Y.; He, C. Cell Adhesion Molecules in Fibrotic Diseases. Biomedicines 2023, 11, 1995. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Andre, G.K.; Hong, T.T.; et al. Auxiliary trafficking subunit GJA1-20k protects connexin-43 from degradation and limits ventricular arrhythmias. J. Clin. Investig. 2020, 130, 4858–4870. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Grailer, J.J.; Wang, N.; Wang, M.; Yao, J.; Zhong, R.; Gao, G.F.; Ward, P.A.; Tan, D.X.; et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J. Pineal Res. 2016, 60, 405–414. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, X.; Liu, M.; Fan, H.; Zheng, H.; Zhang, S.; Rahman, N.; Wołczyński, S.; Kretowski, A.; Li, X. Melatonin inhibits inflammasome-associated activation of endothelium and macrophages attenuating pulmonary arterial hypertension. Cardiovasc. Res. 2020, 116, 2156–2169. [Google Scholar] [CrossRef]
- Yao, C.; Veleva, T.; Scott, L.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Majzunova, M.; Dovinova, I.; Barancik, M.; Chan, J.Y.H. Redox signaling in pathophysiology of hypertension. J. Biomed. Sci. 2013, 20, 69. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Adameova, A.; Kaur, M. Role of catecholamine oxidation in sudden cardiac death. Fundam. Clin. Pharmacol. 2010, 24, 539–546. [Google Scholar] [CrossRef]
- Ghosh, A.; Shcherbik, N. Effects of oxidative stress on protein translation: Implications for cardiovascular diseases. Int. J. Mol. Sci. 2020, 21, 2661. [Google Scholar] [CrossRef]
- Leybaert, L.; De Smet, M.A.J.; Lissoni, A.; Allewaert, R.; Roderick, H.L.; Bultynck, G.; Delmar, M.; Sipido, K.R.; Witschas, K. Connexin hemichannels as candidate targets for cardioprotective and anti-arrhythmic treatments. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Tello, K.; Naeije, R.; De Man, F.; Guazzi, M. Pathophysiology of the right ventricle in health and disease: An update. Cardiovasc. Res. 2023, 119, 1891–1904. [Google Scholar] [CrossRef]
- de Smet, M.A.J.; Lissoni, A.; Nezlobinsky, T.; Wang, N.; Dries, E.; Pérez-Hernández, M.; Lin, X.; Amoni, M.; Vervliet, T.; Witschas, K.; et al. Cx43 hemichannel microdomain signaling at the intercalated disc enhances cardiac excitability. J. Clin. Investig. 2021, 131, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schlüter, K.D.; Schermuly, R.T.; Schulz, R. Cardioprotection in right heart failure. Br. J. Pharmacol. 2020, 177, 5413–5431. [Google Scholar] [CrossRef] [PubMed]
- Pokorski, M. Pulmonary Dysfunction and Disease; Springer: Cham, Switzerland, 2016; ISBN 9783319420097. [Google Scholar]
- Bekedam, F.T.; Goumans, M.J.; Bogaard, H.J.; de Man, F.S.; Llucià-Valldeperas, A. Molecular mechanisms and targets of right ventricular fibrosis in pulmonary hypertension. Pharmacol. Ther. 2023, 244, 108389. [Google Scholar] [CrossRef]
- Assayag, P.; Carré, F.; Chevalier, B.; Delcayre, C.; Mansier, P.; Swynghedauw, B. Compensated cardiac hypertrophy: Arrhythmogenicity and the new myocardial phenotype. I. Fibrosis. Cardiovasc. Res. 1997, 34, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; Van Rijen, H.V.M.; Van Veen, T.A.B.; De Bakker, J.M.T.; Stein, M. Differences in distribution of fibrosis in the ventricles underlie dominant arrhythmia vulnerability of the right ventricle in senescent mice. Neth. Heart J. 2008, 16, 356–358. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; Van Veen, T.A.B.; Van Rijen, H.V.M.; De Bakker, J.M.T. Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 2011, 57, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Kawara, T.; Derksen, R.; De Groot, J.R.; Coronel, R.; Tasseron, S.; Linnenbank, A.C.; Hauer, R.N.W.; Kirkels, H.; Janse, M.J.; De Bakker, J.M.T. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation 2001, 104, 3069–3075. [Google Scholar] [CrossRef]
- Borgdorff, M.A.J.; Dickinson, M.G.; Berger, R.M.F.; Bartelds, B. Right ventricular failure due to chronic pressure load: What have we learned in animal models since the NIH working group statement? Heart Fail. Rev. 2015, 20, 475–491. [Google Scholar] [CrossRef][Green Version]
- Voelkel, N.F.; Quaife, R.A.; Leinwand, L.A.; Barst, R.J.; McGoon, M.D.; Meldrum, D.R.; Dupuis, J.; Long, C.S.; Rubin, L.J.; Smart, F.W.; et al. Right ventricular function and failure: Report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006, 114, 1883–1891. [Google Scholar] [CrossRef]
- Perna, F.; Telesca, A.; Scacciavillani, R.; Narducci, M.L.; Bencardino, G.; Pinnacchio, G.; Spera, F.R.; Sabarese, R.; Comerci, G.; Pelargonio, G. Clinical Impact of Cardiac Fibrosis on Arrhythmia Recurrence after Ablation in Adults with Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2023, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-β1 (Transforming Growth Factor-β1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythmia Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Are myocardial fibrosis and diastolic dysfunction reversible in hypertensive heart disease? Congest. Heart Fail. 2005, 11, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F. Oxidant stress derails the cardiac connexon connection. J. Clin. Investig. 2010, 120, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Bonda, T.A.; Szynaka, B.; Sokołowska, M.; Dziemidowicz, M.; Winnicka, M.M.; Chyczewski, L.; Kamiński, K.A. Remodeling of the intercalated disc related to aging in the mouse heart. J. Cardiol. 2016, 68, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Bonini, M.G.; Dudley, S.C. Effective antioxidant therapy for the management of arrhythmia. Expert Rev. Cardiovasc. Ther. 2011, 9, 797–800. [Google Scholar] [CrossRef]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.W. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: The action of antiarrhythmic peptides. Naunyn. Schmiedebergs Arch. Pharmacol. 2010, 381, 221–234. [Google Scholar] [CrossRef]
- Wang, Z.; Schreier, D.A.; Hacker, T.A.; Chesler, N.C. Progressive right ventricular functional and structural changes in a mouse model of pulmonary arterial hypertension. Physiol. Rep. 2013, 1, e00184. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef]
- Castillo, E.C.; Vázquez-Garza, E.; Yee-Trejo, D.; García-Rivas, G.; Torre-Amione, G. What Is the Role of the Inflammation in the Pathogenesis of Heart Failure? Curr. Cardiol. Rep. 2020, 22, 139. [Google Scholar] [CrossRef]
- Chang, L.T.; Sun, C.K.; Sheu, J.J.; Chiang, C.H.; Youssef, A.A.; Lee, F.Y.; Wu, C.J.; Yip, H.K. Cilostazol therapy attenuates monocrotaline-induced pulmonary arterial hypertension in rat model. Circ. J. 2008, 72, 825–831. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef]
- Marsh, S.R.; Williams, Z.J.; Pridham, K.J.; Gourdie, R.G. Peptidic connexin43 therapeutics in cardiac reparative medicine. J. Cardiovasc. Dev. Dis. 2021, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Llucià-Valldeperas, A.; de Man, F.S.; Bogaard, H.J. Adaptation and Maladaptation of the Right Ventricle in Pulmonary Vascular Diseases. Clin. Chest Med. 2021, 42, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Benes, J.; Kroupova, K.; Kotrc, M.; Petrak, J.; Jarolim, P.; Novosadova, V.; Kautzner, J.; Melenovsky, V. FGF-23 is a biomarker of RV dysfunction and congestion in patients with HFrEF. Sci. Rep. 2023, 13, 16004. [Google Scholar] [CrossRef]
- Ambrosino, P.; Bachetti, T.; D’anna, S.E.; Galloway, B.; Bianco, A.; D’agnano, V.; Papa, A.; Motta, A.; Perrotta, F.; Maniscalco, M. Mechanisms and Clinical Implications of Endothelial Dysfunction in Arterial Hypertension. J. Cardiovasc. Dev. Dis. 2022, 9, 136. [Google Scholar] [CrossRef]
- Handoko, M.L.; De Man, F.S.; Allaart, C.P.; Paulus, W.J.; Westerhof, N.; Vonk-Noordegraaf, A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: Lessons from the left heart. Eur. Respir. Rev. 2010, 19, 72–82. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef]
- Lu, G.; Du, R.; Liu, Y.; Zhang, S.; Li, J.; Pei, J. RGS5 as a Biomarker of Pericytes, Involvement in Vascular Remodeling and Pulmonary Arterial Hypertension. Vasc. Health Risk Manag. 2023, 19, 673–688. [Google Scholar] [CrossRef] [PubMed]
- McQuaid, C.; Montagne, A. SARS-CoV-2 and vascular dysfunction: A growing role for pericytes. Cardiovasc. Res. 2023, 119, 2591–2593. [Google Scholar] [CrossRef]
- Montani, D.; Chaumais, M.C.; Guignabert, C.; Günther, S.; Girerd, B.; Jaïs, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- Groeneveldt, J.A.; De Man, F.S.; Westerhof, B.E. The right treatment for the right ventricle. Curr. Opin. Pulm. Med. 2019, 25, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bao, G.; Chen, W.; Qiang, X.; Zhu, S.; Wang, S.; He, M.; Ma, G.; Ochani, M.; Al-Abed, Y.; et al. Connexin 43 Hemichannel as a Novel Mediator of Sterile and Infectious Inflammatory Diseases. Sci. Rep. 2018, 8, 166. [Google Scholar] [CrossRef]
- Dunaway, L.S.; Billaud, M.; Macal, E.; Good, M.E.; Medina, C.B.; Lorenz, U.; Ravichandran, K.; Koval, M.; Isakson, B.E. Amount of Pannexin 1 in Smooth Muscle Cells Regulates Sympathetic Nerve–Induced Vasoconstriction. Hypertension 2023, 80, 416–425. [Google Scholar] [CrossRef]
- Grimmer, B.; Krauszman, A.; Hu, X.; Kabir, G.; Connelly, K.A.; Li, M.; Grune, J.; Madry, C.; Isakson, B.E.; Kuebler, W.M. Pannexin 1: A novel regulator of acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2022, 118, 2535–2547. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.R. Melatonin: Shining some light on pulmonary hypertension. Cardiovasc. Res. 2020, 116, 2036–2037. [Google Scholar] [CrossRef] [PubMed]
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The Uncoupling Proteins: A Systematic Review on the Mechanism Used in the Prevention of Oxidative Stress. Antioxidants 2022, 11, 322. [Google Scholar] [CrossRef]
- Dimitriadis, K.; Adamopoulou, E.; Pyrpyris, N.; Sakalidis, A.; Leontsinis, I.; Manta, E.; Mantzouranis, E.; Beneki, E.; Soulaidopoulos, S.; Konstantinidis, D.; et al. The effect of SGLT2 inhibitors on the endothelium and the microcirculation: From bench to bedside and beyond. Eur. Hear. J. Cardiovasc. Pharmacother. 2023, 9, 741–757. [Google Scholar] [CrossRef]
- Li, H.L.; Lip, G.Y.H.; Feng, Q.; Fei, Y.; Tse, Y.K.; Wu, M.Z.; Ren, Q.-W.; Tse, H.F.; Cheung, B.M.Y.; Yiu, K.H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Turner, M.E.; Aikawa, E. Updating the paradigm: Inflammation as a targetable modulator of medial vascular calcification. Cardiovasc. Res. 2023, 119, 2259–2261. [Google Scholar] [CrossRef]
- Chen, A.; Lan, Z.; Li, L.; Xie, L.; Liu, X.; Yang, X.; Wang, S.; Liang, Q.; Dong, Q.; Feng, L.; et al. Sodium-glucose cotransporter 2 inhibitor canagliflozin alleviates vascular calcification through suppression of nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 inflammasome. Cardiovasc. Res. 2023, 119, 2368–2381. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.C.; Lin, Y.K.; Chen, Y.C.; Kao, Y.H.; Yeh, Y.H.; Trang, N.N.; Chen, Y.J. Empagliflozin suppressed cardiac fibrogenesis through sodium-hydrogen exchanger inhibition and modulation of the calcium homeostasis. Cardiovasc. Diabetol. 2023, 22, 27. [Google Scholar] [CrossRef] [PubMed]
- Curtain, J.P.; Docherty, K.F.; Jhund, P.S.; Petrie, M.C.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; et al. Effect of dapagliflozin on ventricular arrhythmias, resuscitated cardiac arrest, or sudden death in DAPA-HF. Eur. Heart J. 2021, 42, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ju, F.; Du, L.; Abbott, G.W. Empagliflozin protects the heart against ischemia/reperfusion-induced sudden cardiac death. Cardiovasc. Diabetol. 2021, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Chen, W.T.; Chen, S.y.; Lee, T.M. Dapagliflozin attenuates arrhythmic vulnerabilities by regulating connexin43 expression via the AMPK pathway in post-infarcted rat hearts. Biochem. Pharmacol. 2021, 192, 114674. [Google Scholar] [CrossRef]
- Donniacuo, M.; De Angelis, A.; Telesca, M.; Bellocchio, G.; Riemma, M.A.; Paolisso, P.; Scisciola, L.; Cianflone, E.; Torella, D.; Castaldo, G.; et al. Atrial fibrillation: Epigenetic aspects and role of sodium-glucose cotransporter 2 inhibitors. Pharmacol. Res. 2023, 188, 106591. [Google Scholar] [CrossRef]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef]
- Jhuo, S.J.; Liu, I.H.; Tasi, W.C.; Chou, T.W.; Lin, Y.H.; Wu, B.N.; Lee, K.T.; Lai, W. Ter Characteristics of ventricular electrophysiological substrates in metabolic mice treated with empagliflozin. Int. J. Mol. Sci. 2021, 22, 6105. [Google Scholar] [CrossRef]
- Ilyas, F.; Jones, L.; Tee, S.L.; Horsfall, M.; Swan, A.; Wollaston, F.; Hecker, T.; De Pasquale, C.; Thomas, S.; Chong, W.; et al. Acute pleiotropic effects of dapagliflozin in type 2 diabetic patients with heart failure with reduced ejection fraction: A crossover trial. ESC Heart Fail. 2021, 8, 4346–4352. [Google Scholar] [CrossRef]
- Desai, A.S.; Jhund, P.S.; Claggett, B.L.; Vaduganathan, M.; Miao, Z.M.; Kondo, T.; Barkoudah, E.; Brahimi, A.; Connolly, E.; Finn, P.; et al. Effect of Dapagliflozin on Cause-Specific Mortality in Patients with Heart Failure Across the Spectrum of Ejection Fraction: A Participant-Level Pooled Analysis of DAPA-HF and DELIVER. JAMA Cardiol. 2022, 7, 1227–1234. [Google Scholar] [CrossRef]
- Kopeć, G. Sotatercept as a next-generation therapy for pulmonary arterial hypertension: Insights from the STELLAR trial. Cardiovasc. Res. 2023, 119, E155–E157. [Google Scholar] [CrossRef]
- Omura, J.; Habbout, K.; Shimauchi, T.; Wu, W.H.; Breuils-Bonnet, S.; Tremblay, E.; Martineau, S.; Nadeau, V.; Gagnon, K.; Mazoyer, F.; et al. Identification of Long Noncoding RNA H19 as a New Biomarker and Therapeutic Target in Right Ventricular Failure in Pulmonary Arterial Hypertension. Circulation 2020, 142, 1464–1484. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef] [PubMed]
- Sykora, M.; Kratky, V.; Kopkan, L.; Tribulova, N.; Szeiffova Bacova, B. Anti-Fibrotic Potential of Angiotensin (1-7) in Hemodynamically Overloaded Rat Heart. Int. J. Mol. Sci. 2023, 24, 3490. [Google Scholar] [CrossRef] [PubMed]
- Sykora, M.; Kratky, V.; Cervenka, L.; Kopkan, L.; Tribulova, N.; Szeiffova Bacova, B. The treatment with trandolapril and losartan attenuates pressure and volume overload alternations of cardiac connexin-43 and extracellular matrix in Ren-2 transgenic rats. Sci. Rep. 2023, 13, 20923. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Pellicori, P.; González, A. A novel treatment for heart failure targets myocardial fibrosis. Nat. Med. 2021, 27, 1343–1344. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; De Raaf, M.A.; Sun, X.Q.; Kuiper, V.P.; Da Silva Gonçalves Bos, D.; Happé, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib improves cardiac fibrosis but leaves pulmonary vascular remodelling unaltered in experimental pulmonary hypertension. Cardiovasc. Res. 2019, 115, 432–439. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, A.; Blanco, I.; García-Lunar, I.; Jordà, P.; Rodriguez-Arias, J.J.; Fernández-Friera, L.; Zegri, I.; Nuche, J.; Gomez-Bueno, M.; Prat, S.; et al. β3 adrenergic agonist treatment in chronic pulmonary hypertension associated with heart failure (SPHERE-HF): A double blind, placebo-controlled, randomized clinical trial. Eur. J. Heart Fail. 2023, 25, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Gonçalves Bós, D.; Van Der Bruggen, C.E.E.; Kurakula, K.; Sun, X.Q.; Casali, K.R.; Casali, A.G.; Rol, N.; Szulcek, R.; Dos Remedios, C.; Guignabert, C.; et al. Contribution of impaired parasympathetic activity to right ventricular dysfunction and pulmonary vascular remodeling in pulmonary arterial hypertension. Circulation 2018, 137, 910–924. [Google Scholar] [CrossRef] [PubMed]
- da Silva Gonçalves Bos, D.; Happé, C.; Schalij, I.; Pijacka, W.; Paton, J.F.R.; Guignabert, C.; Tu, L.; Thuillet, R.; Bogaard, H.J.; van Rossum, A.C.; et al. Renal Denervation Reduces Pulmonary Vascular Remodeling and Right Ventricular Diastolic Stiffness in Experimental Pulmonary Hypertension. JACC Basic Transl. Sci. 2017, 2, 22–35. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, Y.; Du, L. Epigenetic Modulation in the Initiation and Progression of Pulmonary Hypertension. Hypertension 2019, 74, 733–739. [Google Scholar] [CrossRef]
- Guo, X.; Olajuyin, A.; Tucker, T.A.; Idell, S.; Qian, G. BRD4 as a Therapeutic Target in Pulmonary Diseases. Int. J. Mol. Sci. 2023, 24, 13231. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, T.; Feng, Q.; Yang, J.; Cao, J.; Xu, X.; Yang, C. Inhibition of MicroRNA-206 Ameliorates Ischemia–Reperfusion Arrhythmia in a Mouse Model by Targeting Connexin43. J. Cardiovasc. Transl. Res. 2020, 13, 584–592. [Google Scholar] [CrossRef]
- Botros, L.; Szulcek, R.; Jansen, S.M.A.; Kurakula, K.; Goumans, M.J.T.H.; Van Kuilenburg, A.B.P.; Noordegraaf, A.V.; De Man, F.S.; Aman, J.; Bogaard, H.J. The effects of mercaptopurine on pulmonary vascular resistance and bmpr2 expression in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2020, 202, 296–299. [Google Scholar] [CrossRef]
- Maki, H.; Hara, T.; Tsuji, M.; Saito, A.; Minatsuki, S.; Inaba, T.; Amiya, E.; Hosoya, Y.; Hatano, M.; Morita, H.; et al. The clinical efficacy of endothelin receptor antagonists in patients with pulmonary arterial hypertension comparison between each generation. Int. Heart J. 2020, 61, 799–805. [Google Scholar] [CrossRef]
- Peters, E.L.; Bogaard, H.J.; Noordegraaf, A.V.; de Man, F.S. Neurohormonal modulation in pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2004633. [Google Scholar] [CrossRef]
- Grünig, E.; Eichstaedt, C.; Barberà, J.A.; Benjamin, N.; Blanco, I.; Bossone, E.; Cittadini, A.; Coghlan, G.; Corris, P.; D’Alto, M.; et al. ERS statement on exercise training and rehabilitation in patients with severe chronic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1800332. [Google Scholar] [CrossRef]
- Rodríguez-Chiaradía, D.A.; Khilzi, K.; Blanco, I.; Rodó-Pin, A.; Martin-Ontiyuelo, C.; Herranz Blasco, A.; Garcia-Lucio, J.; Molina, L.; Marco, E.; Barreiro, E.; et al. Effects of Exercise Training on Circulating Biomarkers of Endothelial Function in Pulmonary Arterial Hypertension. Biomedicines 2023, 11, 1822. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Förster, C.Y. Exercise in treatment-resistant hypertension. A natural neuromodulation therapy? Hypertens. Res. 2023, 46, 2231–2234. [Google Scholar] [CrossRef] [PubMed]
- Kwant, C.T.; van der Horst, F.A.L.; Bogaard, H.J.; de Man, F.S.; Vonk Noordegraaf, A. Nutritional status in pulmonary arterial hypertension. Pulm. Circ. 2022, 12, e12173. [Google Scholar] [CrossRef] [PubMed]
- Kwant, C.T.; de Man, F.; van der Horst, F.A.L.; Bogaard, H.J.; Vonk Noordegraaf, A. The UPHILL study: A nutrition and lifestyle intervention to improve quality of life for patients with pulmonary arterial hypertension. Pulm. Circ. 2023, 13, e12243. [Google Scholar] [CrossRef]
- Jang, A.Y.; Chung, W.J. Current status of pulmonary arterial hypertension in Korea. Korean J. Intern. Med. 2019, 34, 696–707. [Google Scholar] [CrossRef]
- Temple, I.P. Arrhythmias in pulmonary arterial hypertension. J. Congenit. Cardiol. 2017, 1, 2. [Google Scholar] [CrossRef]
- Swisher, J.W.; Weaver, E. The Evolving Management and Treatment Options for Patients with Pulmonary Hypertension: Current Evidence and Challenges. Vasc. Health Risk Manag. 2023, 19, 103–126. [Google Scholar] [CrossRef]
- Mirna, M.; Rohm, I.; Jirak, P.; Wernly, B.; Bäz, L.; Paar, V.; Kretzschmar, D.; Hoppe, U.C.; Schulze, P.C.; Lichtenauer, M.; et al. Analysis of Novel Cardiovascular Biomarkers in Patients With Pulmonary Hypertension (PH). Heart Lung Circ. 2020, 29, 337–344. [Google Scholar] [CrossRef]
- Jansen, S.M.A.; van de Heuvel, L.M.; Houweling, A.C.; Peter van Tintelen, J.; de Man, F.S.; Noordegraaf, A.V.; Bogaard, H.J. Uptake and patient perspectives on additional testing for novel disease-associated genes: Lessons from a pah cohort. Genes 2021, 12, 1540. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sykora, M.; Szeiffova Bacova, B.; Andelova, K.; Egan Benova, T.; Martiskova, A.; Kurahara, L.-H.; Hirano, K.; Tribulova, N. Connexin43, A Promising Target to Reduce Cardiac Arrhythmia Burden in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2024, 25, 3275. https://doi.org/10.3390/ijms25063275
Sykora M, Szeiffova Bacova B, Andelova K, Egan Benova T, Martiskova A, Kurahara L-H, Hirano K, Tribulova N. Connexin43, A Promising Target to Reduce Cardiac Arrhythmia Burden in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2024; 25(6):3275. https://doi.org/10.3390/ijms25063275
Chicago/Turabian StyleSykora, Matus, Barbara Szeiffova Bacova, Katarina Andelova, Tamara Egan Benova, Adriana Martiskova, Lin-Hai Kurahara, Katsuya Hirano, and Narcis Tribulova. 2024. "Connexin43, A Promising Target to Reduce Cardiac Arrhythmia Burden in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 25, no. 6: 3275. https://doi.org/10.3390/ijms25063275
APA StyleSykora, M., Szeiffova Bacova, B., Andelova, K., Egan Benova, T., Martiskova, A., Kurahara, L.-H., Hirano, K., & Tribulova, N. (2024). Connexin43, A Promising Target to Reduce Cardiac Arrhythmia Burden in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 25(6), 3275. https://doi.org/10.3390/ijms25063275